1. Introduction
As interest in metabolomics grows, so does the number of data sets. For example, during the past three years (December 2015–2018), the available studies deposited in Metabolomics WorkBench [
1] have increased from 161 to 816. Once a set of metabolites influenced by a treatment has been identified, a next step in understanding is interpreting the changes in the context of the organism’s complete metabolic network.
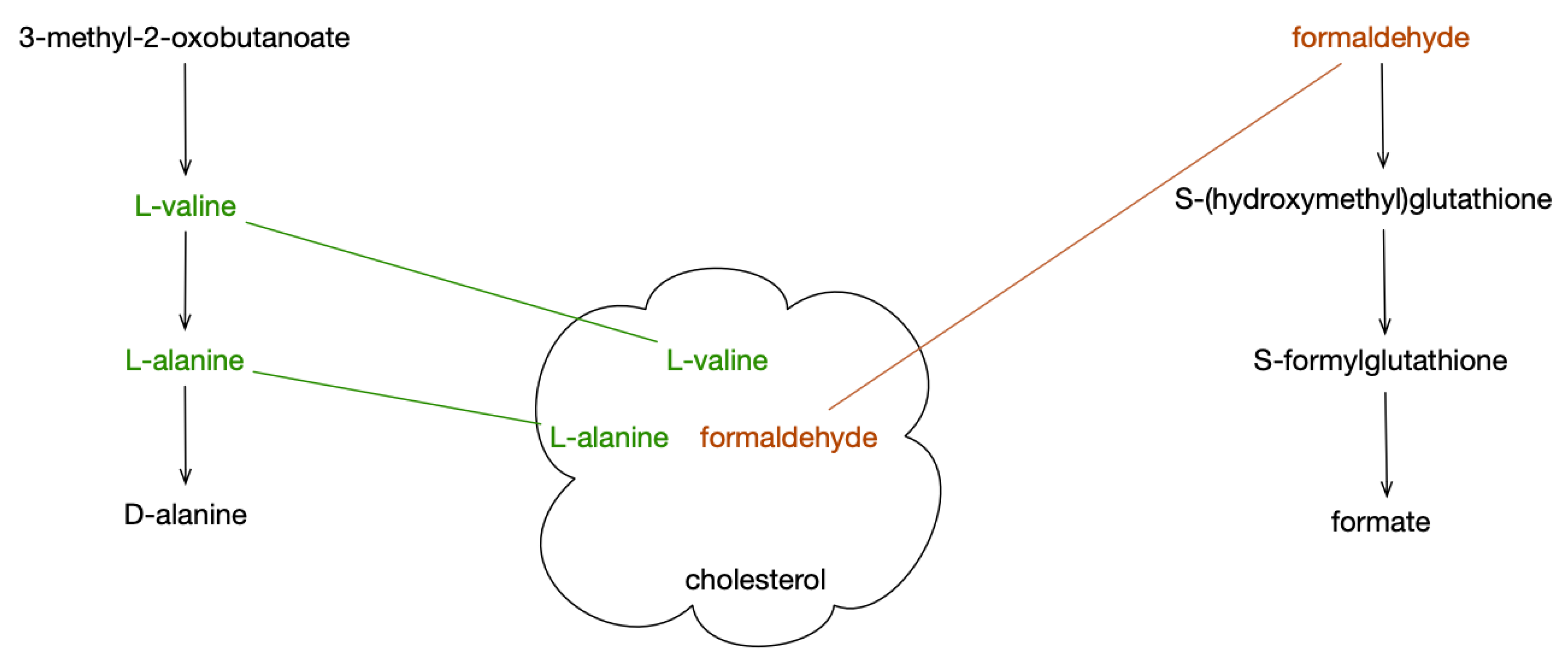
Pathway covering (
Figure 1) is a new method for analyzing metabolite sets against a set of pathways. It is based on the set-cover problem [
2] that has recently been applied to reducing redundancy among pathway databases [
3], but it has not, to our knowledge, been used to suggest pathways associated with changes in metabolite levels. We implemented pathway covering as part of the Pathway Tools [
4,
5] software and evaluated it using the HumanCyc Pathway/Genome Database (PGDB) and several datasets from Metabolomics Workbench (MWB) and another published study. HumanCyc and Pathway Tools are components of the BioCyc [
6] website and collection of more than 14,000 organism PGDBs.
The classic set-cover problem starts with a “universal” set of elements and a collection of sets of elements drawn from that universal set. The goal is identifying a smallest collection of these sets that covers the entire universal set of elements. One extension of this problem is to apply weights or costs to each of the sets that are combined to form a cover. This is called the weighted set-cover problem. We will refer to these weights as costs throughout this paper as our optimizer is designed to seek a minimum value solution.
In this work, the set to be covered consists of metabolites observed to have changed due to disease or experimental conditions, and the collection of sets meant to cover the input set of metabolites consists of the collection of sets of substrate compounds for each pathway in the organism. In this application, we assign costs to the pathways themselves, rather than their sets of substrate compounds.
The problem is finding a collection of sets (pathway substrates) that:
Assigning a cost to each pathway and then solving for a minimum-cost solution reduces the possible solutions to sets of pathways that are small, thereby avoiding “solutions” such as the set of all pathways that contain one or more substrates on the list. Different analysis goals will suggest different cost functions that will yield alternative solutions. Such goals might include the absolute smallest number of pathways, or pathways that are small or have a large a proportion of input substrates, or pathways with a particular function or class such as synthesis or degradation.
The problem of finding the smallest covering set is one of the well-known NP-hard problems [
2], and several approaches to solving or estimating solutions have been developed. These approaches may provide approximate or exact optima (in terms of minimizing cost) and will use different types of algorithms. Two common solution approaches are “greedy” algorithms [
7,
8] (approximate solutions), and algorithms that transform the set-cover problem into an equivalent NP-complete problem (exact solutions), such as integer linear programming (ILP), for which efficient tools for finding solutions have been developed. Although ILP is potentially exponential, our experience is that for the sets of metabolites discussed here, the solver returns a solution in no more than one second on modern hardware (which is faster than the process of mapping input names to database objects which precedes the solver).
In practice, the only input metabolites that can be included in a covering solution are those that are recognized and are substrates of at least one pathway in the database. Many compounds in organism metabolic databases, such as HumanCyc, are not assigned to any metabolic pathway. In addition, pathways include compounds not found in the input set. These compounds can be removed from consideration as they have no bearing on whether a pathway will cover compounds in the input set. By extension, any pathway with no compounds in the input set is eliminated from further consideration.
Therefore, the pathway covering problem is to cover the subset of input metabolites that are known to be substrates of at least one pathway, using the subsets of the substrate compounds of pathways that occur in the input set. The optimal solution, at this point, is the smallest set of pathways that cover the compounds in the input set.
This approach will always yield the smallest collection of pathways or, in some cases several collections of pathways of equal size. However, this solution may not be the most biologically meaningful. For example, using the constant cost function will prefer larger pathways that include more input compounds, since fewer pathways will be needed for the cover. These larger pathways are more likely to have branches and alternative routes such that a particular compound may not be essential to the pathway, whereas a smaller pathway is less likely to have alternative routes. There may also be prior justification to focus on pathways of a certain type (e.g., biosynthetic pathways).
We explored more than a dozen cost functions, but we will report results for the five functions (enumerated below) that we are making available in the released version of the tool.
2. Results
2.4. Study ST000061—Visceral and Subcutaneous Adipose Tissue
This study compared two types of adipose tissue (subcutaneous and visceral) from 50 colo-rectal cancer patients, though 59 samples are reported for both cell types. All significant changes were in the direction of higher metabolite levels in the visceral tissue. There were 39 named compounds (
Table 2) that showed a fold-change difference of 1.5 or greater and that also had a corrected
p-value less than 0.1. These 39 named compounds were resolved to 45 metabolites known to HumanCyc. Of these 45, 35 were substrates in one or more pathways. Because all the changes were in the same direction, the results from the pathway harmony cost function were omitted, as these were not meaningful in this case. The details of solutions for each cost function are listed in
Table A1 through
Table A4.
Table 3 summarizes the solution sets returned by each cost function. The second column shows the number of solutions returned for each cost function, while the third column shows the size range of the solution sets. Finally, column four indicates the largest number of compounds that were covered by a pathway in the solution set.
The constant cost function generated over 1 million solution sets, and apart from a representative solution of 20 pathways listed in
Table A1 in
Appendix A we did not explore these solutions further. Because the solver objective function is equal to the size of the solution for this cost function, all optimal solutions will be the same size. The smallest number of solutions was returned by covered compound sparseness, which also returned solutions that were smaller in size than the other cost functions. Hence we will prefer this cost function for future analyses.
Of the 35 metabolites resolved from the 39 names reported in the study, 12 cases had a metabolite that was consistently covered by a particular pathway, across all solutions returned by any cost function.
alpha-tocopherol covered by alpha-tocopherol degradation
arachidonic acid and ethanolamine covered by anandamide degradation
cholesterol covered by pregnenolone biosynthesis
L-isoleucine covered by isoleucine degradation
L-leucine covered by leucine degradation
L-tyrosine and L-phenylalanine covered by tyrosine biosynthesis
L-threonine covered by threonine degradation
L-valine covered by valine degradation
putrescine and L-ornithine covered by putrescine biosynthesis III
For eight compounds (alpha-tocopherol, arachidonic acid, ethanolamine, isoleucine, leucine, phenylalanine, threonine, and valine), the pathway selected was the only one in the database that specified the particular compound (as opposed to a class of compounds) as a substrate. Likewise, the presence of phenylalanine pulled in its degradation pathway which removed any advantage to including tyrosine in any other pathway. The absence of synthesis pathways is consistent with six of the seven listed amino acids being considered essential in humans. Likewise, alpha-tocopherol is a nutrient that humans cannot synthesize (vitamin E). Although both arachidonic acid and ethanolamine are specific substrates of multiple reactions in pathways, anandamide degradation is the only pathway in which they both occur.
The small solution set size returned by the compound sparseness cost function may reflect the function consistently covering seven compounds with one superpathway (“superpathway of conversion of glucose to acetyl CoA and entry into the TCA cycle”). The constant cost function also did this for at least some solutions. The remaining cost functions, size, and biosynthesis preferred, covered four of these seven with a smaller pathway (“Rapoport-Luebering glycolytic shunt”).
However, these four compounds were all introduced to the covering procedure by one input name (“glyceric acid”) which in HumanCyc is a class with four instance compounds. The glycolytic shunt is active in mammalian erythrocytes but is not reported to be active in adipose tissue.
Intuitively, if the input metabolite set represents a coordinated shift in metabolism as a result of the experiment, the size of its pathway covering set should be smaller than the pathway covering set for a set of metabolites of the same size, drawn at random from pathways in the organism. This notion was tested by generating 1000 sets of 34 compounds, drawn without replacement from all compounds that were substrates of at least one pathway. The covering algorithm was applied to each set, using the constant value cost function, and the size (number of pathways) in each solution was collected into a size distribution. Comparing the size S of the covering set returned for the study data (20) against the distribution, S was smaller than 994 coverings and equal in size to three coverings of random compound sets. This suggests that the pathway covering tool is finding a signal in the observed metabolite shifts that is absent in random collections of metabolites in pathways. As such it validates both the method and the dataset.
To compare pathway covering with enrichment, we performed a “compounds enriched for pathways” analysis of the 39 compounds used in the pathway covering analysis using HumanCyc as the database (see methods for details). A pathway enrichment analysis of the ST000061 data set (see Methods for details) returned ten individual pathways (and 13 pathway classes). We compared this result against the solutions returned by pathway covering using the covered compound sparseness function. The covering analysis returned four solutions, sharing 19 pathways with an additional five pathways appearing among alternative solutions (
Appendix A Table A4), which we included. For this data set, there was relatively little overlap between the individual pathways from the enrichment and covering analyses (
Table 4). Adding the five pathways that occurred in some covering solutions did not increase the overlap.
3. Discussion
Any method that interprets metabolite measurements in terms of changes in pathways will necessarily be dependent on the database of pathways used for interpretation. Furthermore, the requirements that a compound is in the database, is recognized by name, and exists in at least one reaction and one pathway is common to all pathway-based interpretations of metabolomics data. The necessity to identify reactions and pathways led us to avoid studies with a large lipidomics component. Lipids pose a particular challenge since there are relatively few pathways known that are specific to particular lipid species even though they are easy to detect and identify with current methods. The solution lies in continued growth and curation of pathway databases, along with the caution that results with this method will differ between databases with different sets of metabolites and pathways.
There are several other approaches to the interpretation of metabolite lists in terms of affected pathways. A review by Booth et al. [
15] divided the approaches at the time between visualization and enrichment (or over-representation)-based approaches. Although other, non-pathway-based approaches to interpreting metabolomics data (e.g., [
16]), have been developed, the majority of pathway-based tools have used enrichment, which is extensively reviewed by Marco-Ramell et al. [
17] and is also implemented in BioCyc [
18]. Enrichment methods ask the question: is the input metabolite set over-represented for metabolites within certain pathways relative to what would be expected by chance, based on the complete set of pathways and metabolites (present in pathways).
The review by Marco-Ramell et al. covered 13 different tools. These included cross database tools such as MetaboAnalyst and its tools MSEA [
19] and MetPA [
20], as well as MBRole [
21] and ConsensusPathDB [
22], which draw pathway data from multiple databases, most commonly KEGG and HumanCyc as well as more specialized databases such as OMIM. Marco-Ramel et al. found fairly small differences among the tools, with potentially the biggest issue being tools that used out-of-date versions of the databases.
For each compound in the input list, enrichment methods require a determination of statistical significance. If no pathway achieves a usable
p-value, either because of the pathway’s size or other properties of the database, nothing is reported. By contrast, the pathway covering algorithm assigns every compound to a pathway if such a pathway exists in the database, regardless of statistical significance. There will also be differences depending on the choice of statistical method (Hypergeometric/Fisher’s exact vs. Kolmogorov-Smirnov) and the correction for multiple comparisons (in most cases Benjamini-Hochberg [
13]).
When we compared the results of enrichment and pathway covering empirically, we found that in one case (ST000061) pathway covering returned more pathways, and in two cases enrichment returned substantially more pathways. Our interpretation is that pathway covering will return more results when enrichment p-values do not reach statistical significance (e.g., in experiments with relatively few observed metabolites), which can be an advantage if in fact a single metabolite is a valid signal for a pathway whose flux is indeed changing. In particular, enrichment methods may tend to miss large pathways since to exceed the p-value cutoff many metabolites for such pathways must be in the observed set. We expect pathway enrichment to return more results than pathway covering when a larger number of metabolites are observed, resulting in more pathways that exceed the p-value cutoff. One of our motivations for developing the pathway covering method was that enrichment sometimes returns many “overlapping” pathways that are all triggered by a small number of metabolites that are found in multiple pathways.
Visualization approaches that center on interpreting metabolomics data focus on visualizing metabolites (either presence on a list or level of change between samples) on a diagram, either as a set of pathways or metabolic overview diagram. Examples include multiple tools in the Pathways Tools suite [
18], also the PaintOmics [
23] and MetaMapp [
24] tools which integrate interpretation of spectra with metabolite visualization such as Pathos [
25].
Pathway covering, as implemented on the BioCyc website, provides visualization of metabolites in pathways but is really an alternative to enrichment methods. It differs from enrichment methods in that it can propose alternative solutions, both because of alternative cost functions and because of the existence of multiple solutions with the same objective function value. Enrichment approaches, for example, simply count compounds and pathways, whereas cost functions can focus on properties of pathways in addition to whether the pathway contain a particular compound as a substrate (as exemplified by our pathway harmony function).
Pathway covering proposes a small set of pathways, which potentially makes a solution easier for the user to evaluate than either a large set of enriched pathways, or a graphical display of compounds mapped onto a large and complex metabolic map diagram.
Author Contributions
Conceptualization, P.D.K. and M.L.; methodology, P.D.K. and P.E.M.; software, P.E.M.; formal analysis, M.L., P.E.M. and P.D.K.; investigation, P.E.M.; resources, P.D.K.; writing—original draft preparation, P.E.M.; writing—review and editing, P.D.K., M.L., P.E.O.; visualization, P.E.M.; supervision, P.D.K.; funding acquisition, P.D.K.
Funding
This research was funded supported by award number R03-CA211814 from the National Cancer Institute of the National Institutes of Health. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
Acknowledgments
We thank Richard Billington for implementing the user interface code.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| PGDB | Pathway Genome Database |
| MWB | Metabolomics Workbench |
| MDPI | Multidisciplinary Digital Publishing Institute |
References
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Sumner, K.S.N.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucl. Acids Res. 2015, 44, D463–D470. [Google Scholar] [CrossRef] [PubMed]
- Karp, R.M. Reducibilty Among Combinatorial Algorithms. In Complexity of Computer Computations; Springer: Berlin/Heidelberg, Germany, 1972; pp. 85–103. [Google Scholar]
- Stoney, R.; Schwartz, J.M.; Robertson, D.; Nenadic, G. Using set theory to reduce redundancy in pathway sets. BMC Bioinform. 2018, 19, 386. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Latendresse, M.; Paley, S.M.; Krummenacker, M.; Ong, Q.; Billington, R.; Kothari, A.; Weaver, D.; Lee, T.; Subhraveti, P.; et al. Pathway Tools version 19.0 update: Software for Pathway/Genome Informatics and Systems Biology. Brief. Bioinform. 2015. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.; Latendresse, M.; Paley, S.; Krummenacker, M.; Ong, Q.; Billington, R.; Kothari, A.; Weaver, D.; Lee, T.; Subhraveti, P.; et al. Pathway Tools version 19.0: Integrated Software for Pathway/Genome Informatics and Systems Biology. arXiv 2015, arXiv:1510.03964. [Google Scholar]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ausiello, G.; Bourgeois, N.; Giannakos, T.; Paschos, V. Greedy algorithms for on-line set-covering. Algorithmic Oper. Res. 2009, 4, 36–48. [Google Scholar]
- Chvatal, V. A greedy heuristic for the set-covering problem. Math. Oper. Res. 1979, 4, 233–235. [Google Scholar] [CrossRef]
- Metabolomics Workbench Project PR000058, Study ST000061. Available online: http://dx.doi.org/10.21228/M80018 (accessed on 6 February 2019).
- Metabolomics Workbench Project PR000535, Study ST000741. Available online: http://dx.doi.org/10.21228/M8HH47 (accessed on 6 February 2019).
- McDonnell, S.; Hwang, S.; Rolland, D.; Murga-Zamalloa, C.; Basrur, V.; Conlon, K.; Fermin, D.; Wolfe, T.; Raskind, A.; Ruan, C.; et al. Integrated phosphoproteomic and metabolomic profiling reveals NPM-ALK-mediated phosphorylation of PKM2 and metabolic reprogramming in anaplastic large call lymphoma. Blood 2013, 122, 958–968. [Google Scholar] [CrossRef]
- Supplemental Material from McDonnell13. Available online: http://www.bloodjournal.org/content/bloodjournal/suppl/2013/06/27/blood-2013-01-482026.DC1/blood-2013-01-482026-1.pdf (accessed on 24 February 2019).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Paley, S.; O’Maille, P.E.; Weaver, D.; Karp, P.D. Pathway collages: Personalized multi-pathway diagrams. BMC Bioinform. 2016, 17, 529–538. [Google Scholar] [CrossRef]
- Booth, S.; Weljie, A.; Turner, R. Computational tools for the secondary analysis of metabolomics experiments. Comput. Struct. Biotechnol. J. 2013, 4, e201301003. [Google Scholar] [CrossRef]
- Kuehne, A.; Mayr, U.; Sévin, D.; Claassen, M.; Zamboni, N. Metabolic network segmentation: A probabilistic graphical modeling approach to identify the sites and sequential order of metabolic regulation from non-targeted metabolomics data. PLoS Comput. Biol. 2017, 13, e1005577. [Google Scholar] [CrossRef] [PubMed]
- Marco-Ramell, A.; Alay, A.; Tulipani, S.; Urpi-Sarda, M. Evaluation and comparison of bioinformatic tools for the enrichment analysis of metabolomics data. BMC Bioinform. 2018, 19. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Holland, T.; Kothari, A.; Krummenacker, M.; Weaver, D.; Latendresse, M.; Paley, S. Computational Metabolomics Operations at BioCyc.org. Metabolite 2015, 5, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucl. Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef]
- Chagoyen, M. MBRole: Enrichment analysis of metabolomic data. Bioinformatics 2011, 27, 730–731. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Pentchev, K.; Galicka, H.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathKB: Toward a more complete picture of cell biology. Nucl. Acids Res. 2011, 39, D712–D717. [Google Scholar] [CrossRef] [PubMed]
- Hernaández-de Diego, R.; Tarazona, S.; Balzano-Nogueira, L.; Furió-Tarí, P.; Pappas, G.J., Jr.; Conesa, A. PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucl. Acids Res. 2018, 46, W503–W509. [Google Scholar] [CrossRef]
- Barupal, D.K.; Haldiya, P.K.; Wohlgemuth, G.; Kind, T.; Kothari, S.L.; Pinkerton, K.E.; Fiehn, O. MetaMapp: Mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinform. 2012, 13, 99. [Google Scholar] [CrossRef]
- Leader, D.; Burgess, K.; Creek, D.; Barrett, M. Pathos: A web facility that uses metabolic maps to display experimental changes in metabolites identified by mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 3422–3426. [Google Scholar] [CrossRef]
- Gamrath, G.; Koch, T.; Maher, S.; Rehfeldt, D. SCIP-Jack—A solver for STP and variants with parallelization extensions. Math. Program. Comput. 2017, 9, 231–296. [Google Scholar] [CrossRef]
- SCIP: Solving Constraint Integer Progams. Available online: http://scip.zib.de/ (accessed on 30 April 2019).
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).




{kind=link}
{kind=link}