Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community

 ,
,  , ,
, , 
 and
and 
Abstract
1. Introduction
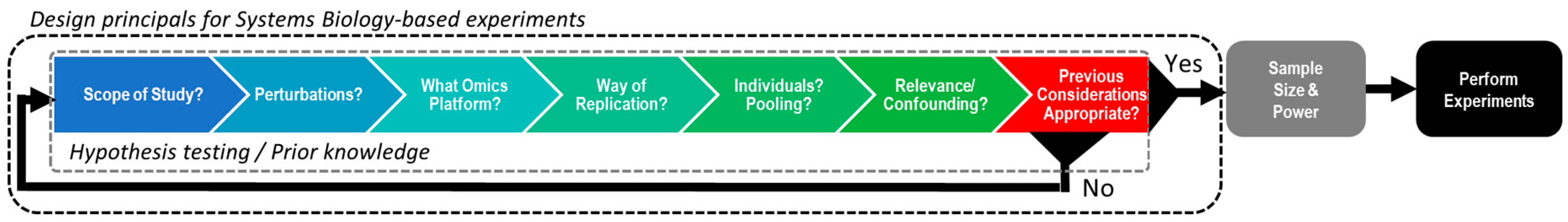
2. Designing Experiments Suitable for Multi-Omics Integration
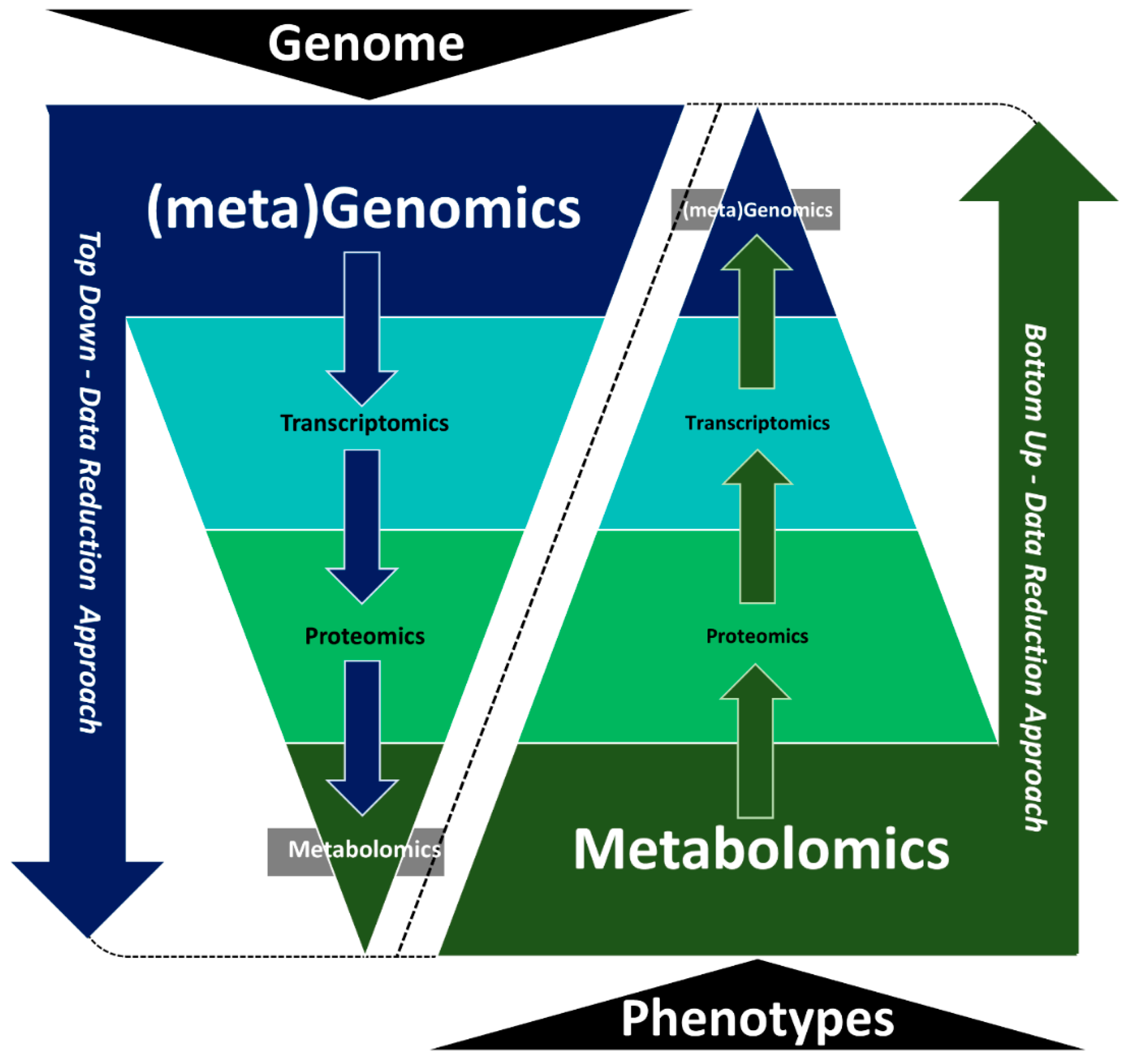
3. Multi-omics Data Integration
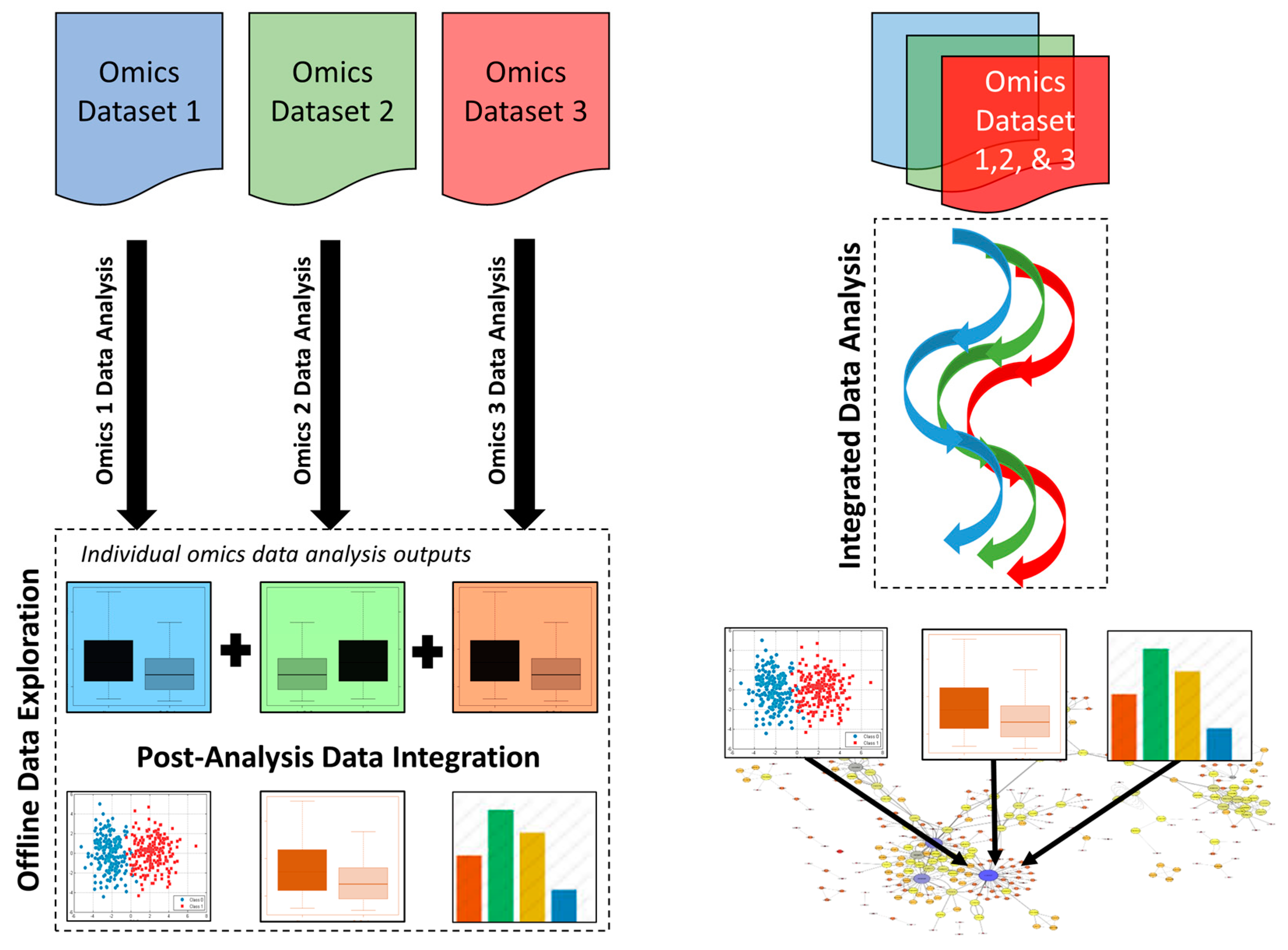
3.1. Post-Analysis Data Integration Approaches
3.2. Integrated Data Analysis Approaches
3.3. Systems Modeling
3.4. Software Tools, Databases and Approaches for Multi-Omics Integration
4. Challenges in Multi-Omics Integration
4.1. The Nature of the Omics Data Sets
4.2. Dispersed Data Sets and Non-Interoperable Tools
4.3. Inadequate Pathway and Data Visualization Tools
4.4. Failing to Demonstrate Utility
4.5. Limited Research Funding
5. Recommendations
- Adopt the sample collection, preparation and measurement standards used in metabolomics studies. This would ensure high-quality data collection in most multi-omics studies;
- Measure multi-omics data in a robust, quantitative manner to ensure reproducibility, enforce comparability and permit facile integration;
- Use reference standards, quality control (QC) samples, and universal standardized operating protocols (SOPs) to enable consistent multi-omics measurements across laboratories;
- Perform power analyses, where possible, prior to conducting large scale multi-omics studies; and,
- Create centralized data repositories, curated or reviewed software lists and improved software/database interoperability (adherence to FAIR data standards) to improve multi-omics integration;
- Improve or develop more comprehensive open source pathway databases and network visualization tools;
- Increase levels of funding and increase awareness of the need for metabolomics in multi-omics studies;
- Demonstrate clear utility of multi-omics studies to both the public and funding agencies; and,
- Undertake more community-driven activities to lead to the creation of multi-omics tools and resources better suited to the community’s needs.
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Breitling, R. What is systems biology? Front. Physiol. 2010, 1, 9. [Google Scholar] [CrossRef]
- Hillmer, R.A. Systems biology for biologists. PLoS Pathog. 2015, 11, e1004786. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.R.; Labow, M.; Reinhardt, M.; van Oostrum, J.; Peitsch, M.C. The application of systems biology to drug discovery. Curr. Opin. Chem. Biol. 2006, 10, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Cisek, K.; Krochmal, M.; Klein, J.; Mischak, H. The application of multi-omics and systems biology to identify therapeutic targets in chronic kidney disease. Nephrol. Dial. Transplant. 2016, 31, 2003–2011. [Google Scholar] [CrossRef]
- Hagemann, M.; Hesse, W.R. Systems and synthetic biology for the biotechnological application of cyanobacteria. Curr. Opin. Biotechnol. 2018, 49, 94–99. [Google Scholar] [CrossRef]
- Herrgard, M.J.; Swainston, N.; Dobson, P.; Dunn, W.B.; Arga, K.Y.; Arvas, M.; Bluthgen, N.; Borger, S.; Costenoble, R.; Heinemann, M.; et al. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat. Biotechnol. 2008, 26, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Xu, P. Production of chemicals using dynamic control of metabolic fluxes. Curr. Opin. Biotechnol. 2018, 53, 12–19. [Google Scholar] [CrossRef]
- Grav, L.M.; Sergeeva, D.; Lee, J.S.; de Mas, I.M.; Lewis, N.E.; Andersen, M.R.; Nielsen, L.K.; Lee, G.M.; Kildegaard, H.F. Minimizing clonal variation during mammalian cell line engineering for improved systems biology data generation. ACS Synth. Biol. 2018, 7, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, M.; Thorsson, V.; Li, B.; Rust, A.G.; Korb, M.; Kennedy, K.; Hai, T.; Bolouri, H.; Aderem, A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 2006, 441, 173. [Google Scholar] [CrossRef]
- Otero, J.M.; Nielsen, J. Industrial Systems Biology. Biotechnol. Bioeng. 2010, 105, 439–460. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J. Systems Biology of Metabolism. In Annual Review of Biochemistry; Kornberg, R.D., Ed.; Annual Reviews: Palo Alto, CA, USA, 2017; Volume 86, pp. 245–275. [Google Scholar]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Next-Generation DNA Sequencing Methods. Annu. Rev. Genom. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef] [PubMed]
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, 23. [Google Scholar] [CrossRef]
- Nassar, A.F.; Wu, T.; Nassar, S.F.; Wisnewski, A.V. UPLC–MS for metabolomics: A giant step forward in support of pharmaceutical research. Drug Discov. Today 2017, 22, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Beale, D.J.; Pinu, F.R.; Kouremenos, K.A.; Poojary, M.M.; Narayana, V.K.; Boughton, B.A.; Kanojia, K.; Dayalan, S.; Jones, O.A.H.; Dias, D.A. Review of recent developments in GC–MS approaches to metabolomics-based research. Metabolomics 2018, 14, 152. [Google Scholar] [CrossRef]
- Brunk, E.; George, K.W.; Alonso-Gutierrez, J.; Thompson, M.; Baidoo, E.; Wang, G.; Petzold, C.J.; McCloskey, D.; Monk, J.; Yang, L.; et al. Characterizing Strain Variation in Engineered E.coli Using a Multi-Omics-Based Workflow. Cell Syst. 2016, 2, 335–346. [Google Scholar] [CrossRef]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar] [CrossRef]
- Zampieri, M.; Sauer, U. Metabolomics-driven understanding of genotype-phenotype relations in model organisms. Curr. Opin. Syst. Biol. 2017, 6, 28–36. [Google Scholar] [CrossRef]
- Beale, D.J.; Karpe, A.V.; McLeod, J.D.; Gondalia, S.V.; Muster, T.H.; Othman, M.Z.; Palombo, E.A.; Joshi, D. An ‘omics’ approach towards the characterisation of laboratory scale anaerobic digesters treating municipal sewage sludge. Water Res. 2016, 88, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Mias, G.I.; Li-Pook-Than, J.; Jiang, L.; Lam, H.Y.; Chen, R.; Miriami, E.; Karczewski, K.J.; Hariharan, M.; Dewey, F.E.; et al. Personal Omics Profiling Reveals Dynamic Molecular and Medical Phenotypes. Cell 2012, 148, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Günther, O.P.; Shin, H.; Ng, R.T.; McMaster, W.R.; McManus, B.M.; Keown, P.A.; Tebbutt, S.J.; Lê Cao, K.-A. Novel Multivariate Methods for Integration of Genomics and Proteomics Data: Applications in a Kidney Transplant Rejection Study. OMICS J. Integr. Biol. 2014, 18, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.W.L.; Fermin, D.; Choi, K.P.; Ewing, R.; Choi, H. iOmicsPASS: A novel method for integration of multi-omics data over biological networks and discovery of predictive subnetworks. bioRxiv 2018, 374520. [Google Scholar] [CrossRef]
- Schloss, P.D. Identifying and overcoming threats to reproducibility, replicability, robustness, and generalizability in microbiome research. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.V.; Hu, Y.-J. Chapter Three—Integrative Analysis of Multi-omics Data for Discovery and Functional Studies of Complex Human Diseases. In Advances in Genetics; Friedmann, T., Dunlap, J.C., Goodwin, S.F., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 93, pp. 147–190. [Google Scholar]
- Strom, S.P. Fundamentals of RNA Analysis on Biobanked Specimens. In Biobanking: Methods and Protocols; Yong, W.H., Ed.; Springer: New York, NY, USA, 2019; pp. 345–357. [Google Scholar]
- O’Rourke, M.B.; Padula, M.P. Analysis of formalin-fixed, paraffin-embedded (FFPE) tissue via proteomic techniques and misconceptions of antigen retrieval. BioTechniques 2016, 60, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Coscia, F.; Lengyel, E.; Duraiswamy, J.; Ashcroft, B.; Bassani-Sternberg, M.; Wierer, M.; Johnson, A.; Wroblewski, K.; Montag, A.; Yamada, S.D.; et al. Multi-level Proteomics Identifies CT45 as a Chemosensitivity Mediator and Immunotherapy Target in Ovarian Cancer. Cell 2018, 175, 159–170.e116. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55. [Google Scholar] [CrossRef]
- Chetwynd, A.J.; Dunn, W.B.; Rodriguez-Blanco, G. Collection and Preparation of Clinical Samples for Metabolomics. In Metabolomics: From Fundamentals to Clinical Applications; Sussulini, A., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 19–44. [Google Scholar]
- Broadhurst, D.I.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Hou, S.; Wentzell, P.D. Regularized projection pursuit for data with a small sample-to-variable ratio. Metabolomics 2014, 10, 589–606. [Google Scholar] [CrossRef]
- Ren, S.; Hinzman, A.A.; Kang, E.L.; Szczesniak, R.D.; Lu, L.J. Computational and statistical analysis of metabolomics data. Metabolomics 2015, 11, 1492–1513. [Google Scholar] [CrossRef]
- Shi, Z.; Sellers, J.; Moult, J. Protein stability and in vivo concentration of missense mutations in phenylalanine hydroxylase. Proteins 2012, 80, 61–70. [Google Scholar] [CrossRef]
- Cattaneo, A.; Pariante, C.M. Integrating ‘omics’ approaches to prioritize new pathogenetic mechanisms for mental disorders. Neuropsychopharmacology 2018, 43, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Mallick, H.; Ma, S.; Franzosa, E.A.; Vatanen, T.; Morgan, X.C.; Huttenhower, C. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 2017, 18, 228. [Google Scholar] [CrossRef] [PubMed]
- Kimes, N.E.; Callaghan, A.V.; Aktas, D.F.; Smith, W.L.; Sunner, J.; Golding, B.T.; Drozdowska, M.; Hazen, T.C.; Suflita, J.M.; Morris, P.J. Metagenomic analysis and metabolite profiling of deep-sea sediments from the Gulf of Mexico following the Deepwater Horizon oil spill. Front. Microbiol. 2013, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Hultman, J.; Waldrop, M.P.; Mackelprang, R.; David, M.M.; McFarland, J.; Blazewicz, S.J.; Harden, J.; Turetsky, M.R.; McGuire, A.D.; Shah, M.B.; et al. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 2015, 521, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.C.; Tian, T.F.; Tseng, Y.J. 3Omics: A web-based systems biology tool for analysis, integration and visualization of human transcriptomic, proteomic and metabolomic data. BMC Syst. Biol. 2013, 7, 64. [Google Scholar] [CrossRef]
- Trygg, J. O2-PLS for qualitative and quantitative analysis in multivariate calibration. J. Chemom. 2002, 16, 283–293. [Google Scholar] [CrossRef]
- Trygg, J.; Wold, S. O2-PLS, a two-block (X–Y) latent variable regression (LVR) method with an integral OSC filter. J. Chemom. 2003, 17, 53–64. [Google Scholar] [CrossRef]
- Löfstedt, T.; Trygg, J. OnPLS—A novel multiblock method for the modelling of predictive and orthogonal variation. J. Chemom. 2011, 25, 441–455. [Google Scholar] [CrossRef]
- Kirwan, G.M.; Johansson, E.; Kleemann, R.; Verheij, E.R.; Wheelock, Å.M.; Goto, S.; Trygg, J.; Wheelock, C.E. Building multivariate systems biology models. Anal. Chem. 2012, 84, 7064–7071. [Google Scholar] [CrossRef]
- Rantalainen, M.; Cloarec, O.; Beckonert, O.; Wilson, I.D.; Jackson, D.; Tonge, R.; Rowlinson, R.; Rayner, S.; Nickson, J.; Wilkinson, R.W.; et al. Statistically integrated metabonomic−proteomic studies on a human prostate cancer xenograft model in mice. J. Proteome Res. 2006, 5, 2642–2655. [Google Scholar] [CrossRef] [PubMed]
- Reinke, S.N.; Galindo-Prieto, B.; Skotare, T.; Broadhurst, D.I.; Singhania, A.; Horowitz, D.; Djukanović, R.; Hinks, T.S.C.; Geladi, P.; Trygg, J.; et al. OnPLS-based multi-block data integration: A multivariate approach to interrogating biological interactions in asthma. Anal. Chem. 2018, 90, 13400–13408. [Google Scholar] [CrossRef] [PubMed]
- Bylesjö, M.; Eriksson, D.; Kusano, M.; Moritz, T.; Trygg, J. Data integration in plant biology: The O2PLS method for combined modeling of transcript and metabolite data. Plant J. 2007, 52, 1181–1191. [Google Scholar] [CrossRef]
- Srivastava, V.; Obudulu, O.; Bygdell, J.; Löfstedt, T.; Rydén, P.; Nilsson, R.; Ahnlund, M.; Johansson, A.; Jonsson, P.; Freyhult, E.; et al. OnPLS integration of transcriptomic, proteomic and metabolomic data shows multi-level oxidative stress responses in the cambium of transgenic hipI- superoxide dismutase Populus plants. BMC Genom. 2013, 14, 893. [Google Scholar] [CrossRef] [PubMed]
- Beale, D.J.; Crosswell, J.; Karpe, A.V.; Ahmed, W.; Williams, M.; Morrison, P.D.; Metcalfe, S.; Staley, C.; Sadowsky, M.J.; Palombo, E.A.; et al. A multi-omics based ecological analysis of coastal marine sediments from Gladstone, in Australia’s Central Queensland, and Heron Island, a nearby fringing platform reef. Sci. Total Environ. 2017, 609, 842–853. [Google Scholar] [CrossRef]
- Beale, D.J.; Crosswell, J.; Karpe, A.V.; Metcalfe, S.S.; Morrison, P.D.; Staley, C.; Ahmed, W.; Sadowsky, M.J.; Palombo, E.A.; Steven, A.D.L. Seasonal metabolic analysis of marine sediments collected from Moreton Bay in South East Queensland, Australia, using a multi-omics-based approach. Sci. Total Environ. 2018, 631–632, 1328–1341. [Google Scholar] [CrossRef]
- Kikuchi, J.; Ito, K.; Date, Y. Environmental metabolomics with data science for investigating ecosystem homeostasis. Prog. Nuclear Magn. Reson. Spectrosc. 2018, 104, 56–88. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Caraus, I.; Xia, J.; Li, C.; Wishart, D.S.; Bourque, G.; Li, S. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Wierling, C.; Herwig, R.; Lehrach, H. Resources, standards and tools for systems biology. Brief. Funct. Genom. 2007, 6, 240–251. [Google Scholar] [CrossRef]
- Shapiro, B.E.; Levchenko, A.; Meyerowitz, E.M.; Wold, B.J.; Mjolsness, E.D. Cellerator: Extending a computer algebra system to include biochemical arrows for signal transduction simulations. Bioinformatics 2003, 19, 677–678. [Google Scholar] [CrossRef]
- Wishart, D.S.; Yang, R.; Arndt, D.; Tang, P.; Cruz, J. Dynamic cellular automata: An alternative approach to cellular simulation. Silico Biol. 2005, 5, 139–161. [Google Scholar]
- Voss, K.; Heiner, M.; Koch, I. Steady state analysis of metabolic pathways using Petri nets. Silico Biol. 2003, 3, 367–387. [Google Scholar]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Varma, A.; Palsson, B.O. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl. Environ. Microbiol. 1994, 60, 3724. [Google Scholar]
- Schilling, C.H.; Edwards, J.S.; Letscher, D.; Palsson, B.Ø. Combining pathway analysis with flux balance analysis for the comprehensive study of metabolic systems. Biotechnol. Bioeng. 2000, 71, 286–306. [Google Scholar] [CrossRef]
- Lee, I.D.; Palsson, B.O. A Macintosh software package for simulation of human red blood cell metabolism. Comput. Methods Programs Biomed. 1992, 38, 195–226. [Google Scholar] [CrossRef]
- Varma, A.; Palsson, B.O. Metabolic flux balancing: Basic concepts, scientific and practical use. Bio/Technology 1994, 12, 994–998. [Google Scholar] [CrossRef]
- Tomita, M.; Hashimoto, K.; Takahashi, K.; Shimizu, T.S.; Matsuzaki, Y.; Miyoshi, F.; Saito, K.; Tanida, S.; Yugi, K.; Venter, J.C.; et al. E-CELL: Software environment for whole-cell simulation. Bioinformatics 1999, 15, 72–84. [Google Scholar] [CrossRef]
- Ishii, N.; Robert, M.; Nakayama, Y.; Kanai, A.; Tomita, M. Toward large-scale modeling of the microbial cell for computer simulation. J. Biotechnol. 2004, 113, 281–294. [Google Scholar] [CrossRef]
- Nakayama, Y.; Kinoshita, A.; Tomita, M. Dynamic simulation of red blood cell metabolism and its application to the analysis of a pathological condition. Theor. Biol. Med. Model. 2005, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Yachie-Kinoshita, A.; Nishino, T.; Shimo, H.; Suematsu, M.; Tomita, M. A metabolic model of human erythrocytes: Practical application of the E-Cell Simulation Environment. J. Biomed. Biotechnol. 2010, 2010, 642420. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Yachie-Kinoshita, A.; Hirayama, A.; Soga, T.; Suematsu, M.; Tomita, M. In silico modeling and metabolome analysis of long-stored erythrocytes to improve blood storage methods. J. Biotechnol. 2009, 144, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Yachie-Kinoshita, A.; Hirayama, A.; Soga, T.; Suematsu, M.; Tomita, M. Dynamic simulation and metabolome analysis of long-term erythrocyte storage in adenine-guanosine solution. PLoS ONE 2013, 8, e71060. [Google Scholar] [CrossRef] [PubMed]
- Mori, H. From the sequence to cell modeling: Comprehensive functional genomics in Escherichia coli. J. Biochem. Mol. Biol. 2004, 37, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Joyce, A.R.; Reed, J.L.; White, A.; Edwards, R.; Osterman, A.; Baba, T.; Mori, H.; Lesely, S.A.; Palsson, B.O.; Agarwalla, S. Experimental and computational assessment of conditionally essential genes in Escherichia coli. J. Bacteriol. 2006, 188, 8259–8271. [Google Scholar] [CrossRef]
- Tian, Z.; Faure, A.; Mori, H.; Matsuno, H. Identification of key regulators in glycogen utilization in E. coli based on the simulations from a hybrid functional Petri net model. BMC Syst. Biol. 2013, 7 (Suppl. 6), S1. [Google Scholar] [CrossRef]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A.; et al. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef]
- Cooling, M.T.; Hunter, P.; Crampin, E.J. Modelling biological modularity with CellML. IET Syst. Biol. 2008, 2, 73–79. [Google Scholar] [CrossRef]
- Nagasaki, M.; Saito, A.; Jeong, E.; Li, C.; Kojima, K.; Ikeda, E.; Miyano, S. Cell Illustrator 4.0: A computational platform for systems biology. Silico Biol. 2010, 10, 5–26. [Google Scholar]
- Sahoo, S.; Haraldsdottir, H.S.; Fleming, R.M.; Thiele, I. Modeling the effects of commonly used drugs on human metabolism. FEBS J. 2015, 282, 297–317. [Google Scholar] [CrossRef] [PubMed]
- Echeverri Olga, Y.; Salazar Diego, A.; Rodriguez-Lopez, A.; Janneth, G.; Almeciga-Diaz Carlos, J.; Barrera Luis, A. Understanding the metabolic consequences of human arylsulfatase a deficiency through a computational systems biology study. Cent. Nerv. Syst. Agents Med. Chem. 2017, 17, 72–77. [Google Scholar]
- Knecht, C.; Fretter, C.; Rosenstiel, P.; Krawczak, M.; Hutt, M.T. Distinct metabolic network states manifest in the gene expression profiles of pediatric inflammatory bowel disease patients and controls. Sci. Rep. 2016, 6, 32584. [Google Scholar] [CrossRef]
- Ma, D.; Yang, L.; Fleming, R.M.; Thiele, I.; Palsson, B.O.; Saunders, M.A. Reliable and efficient solution of genome-scale models of metabolism and macromolecular expression. Sci. Rep. 2017, 7, 40863. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.E.; Nagarajan, H.; Palsson, B.O. Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 2012, 10, 291. [Google Scholar] [CrossRef]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Drager, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Bauer, E.; Thiele, I. From metagenomic data to personalized in silico microbiotas: Predicting dietary supplements for Crohn’s disease. NPJ Syst. Biol. Appl. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Danielsdottir, A.D.; Krecke, M.; Merten, D.; Haraldsdottir, H.S.; et al. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res 2019, 47, D614–D624. [Google Scholar]
- Noecker, C.; Eng, A.; Srinivasan, S.; Theriot, C.M.; Young, V.B.; Jansson, J.K.; Fredricks, D.N.; Borenstein, E. Metabolic model-based integration of microbiome taxonomic and metabolomic profiles elucidates mechanistic links between ecological and metabolic variation. mSystems 2016, 1. [Google Scholar] [CrossRef]
- Erickson, A.R.; Cantarel, B.L.; Lamendella, R.; Darzi, Y.; Mongodin, E.F.; Pan, C.; Shah, M.; Halfvarson, J.; Tysk, C.; Henrissat, B.; et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS ONE 2012, 7, e49138. [Google Scholar] [CrossRef]
- Srinivasan, S.; Morgan, M.T.; Fiedler, T.L.; Djukovic, D.; Hoffman, N.G.; Raftery, D.; Marrazzo, J.M.; Fredricks, D.N. Metabolic signatures of bacterial vaginosis. mBio 2015, 6. [Google Scholar] [CrossRef]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef]
- Jansson, J.; Willing, B.; Lucio, M.; Fekete, A.; Dicksved, J.; Halfvarson, J.; Tysk, C.; Schmitt-Kopplin, P. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS ONE 2009, 4, e6386. [Google Scholar] [CrossRef] [PubMed]
- Jozefczuk, S.; Klie, S.; Catchpole, G.; Szymanski, J.; Cuadros-Inostroza, A.; Steinhauser, D.; Selbig, J.; Willmitzer, L. Metabolomic and transcriptomic stress response of Escherichia coli. Mol. Syst. Biol. 2010, 6, 364. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.; Mains, A.; Virk, B.; Rodriguez, N.; Murdoch, S.; Pearce, J.; Bergmann, S.; Le Novère, N.; Casanueva, O. Multi-Omics and Genome-Scale Modeling Reveal a Metabolic Shift During C. elegans Aging. Front. Mol. Biosci. 2019, 6, 364. [Google Scholar] [CrossRef]
- Fondi, M.; Liò, P. Multi -omics and metabolic modelling pipelines: Challenges and tools for systems microbiology. Microbiol. Res. 2015, 171, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Vaz, F.M.; Ferdinandusse, S.; van Kuilenburg, A.B.P.; Kemp, S.; van Karnebeek, C.D.; Waterham, H.R.; Houtkooper, R.H. Translational Metabolism: A multidisciplinary approach towards precision diagnosis of inborn errors of metabolism in the omics era. J. Inherit. Metab. Dis. 2019, 42, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Biswapriya, B.M.; Carl, L.; Michael, O.; Laura, A.C. Integrated omics: Tools, advances and future approaches. J. Mol. Endocrinol. 2019, 62, R21–R45. [Google Scholar]
- Haas, R.; Zelezniak, A.; Iacovacci, J.; Kamrad, S.; Townsend, S.; Ralser, M. Designing and interpreting ‘multi-omic’ experiments that may change our understanding of biology. Curr. Opin. Syst. Biol. 2017, 6, 37–45. [Google Scholar] [CrossRef]
- Bult, C.J.; Eppig, J.T.; Kadin, J.A.; Richardson, J.E.; Blake, J.A.; Mouse Genome Database Group. The Mouse Genome Database (MGD): Mouse biology and model systems. Nucleic Acids Res. 2008, 36, D724–D728. [Google Scholar] [CrossRef]
- Marygold, S.J.; Crosby, M.A.; Goodman, J.L.; FlyBase, C. Using FlyBase, a database of Drosophila genes and genomes. Methods Mol. Biol. (Clifton, N.J.) 2016, 1478, 1–31. [Google Scholar]
- Howe, K.L.; Bolt, B.J.; Cain, S.; Chan, J.; Chen, W.J.; Davis, P.; Done, J.; Down, T.; Gao, S.; Grove, C.; et al. WormBase 2016: Expanding to enable helminth genomic research. Nucleic Acids Res. 2016, 44, D774–D780. [Google Scholar] [CrossRef]
- Sajed, T.; Marcu, A.; Ramirez, M.; Pon, A.; Guo, A.C.; Knox, C.; Wilson, M.; Grant, J.R.; Djoumbou, Y.; Wishart, D.S. ECMDB 2.0: A richer resource for understanding the biochemistry of E. coli. Nucleic Acids Res 2016, 44, D495–D501. [Google Scholar] [CrossRef]
- Karp, P.D.; Ong, W.K.; Paley, S.; Billington, R.; Caspi, R.; Fulcher, C.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; et al. The EcoCyc Database. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef]
- Ramirez-Gaona, M.; Marcu, A.; Pon, A.; Guo, A.C.; Sajed, T.; Wishart, N.A.; Karu, N.; Djoumbou Feunang, Y.; Arndt, D.; Wishart, D.S. YMDB 2.0: A significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 2017, 45, D440–D445. [Google Scholar] [CrossRef] [PubMed]
- Schläpfer, P.; Zhang, P.; Wang, C.; Kim, T.; Banf, M.; Chae, L.; Dreher, K.; Chavali, A.K.; Nilo-Poyanco, R.; Bernard, T.; et al. Genome-wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol. 2017, 173, 2041. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, K.A.; Starr, B.; Wong, E.D.; Dalusag, K.S.; Hellerstedt, S.T.; Lang, O.W.; Nash, R.S.; Skrzypek, M.S.; Engel, S.R.; Cherry, J.M. Outreach and online training services at the Saccharomyces Genome Database. Database 2017, 2017, bax002. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef] [PubMed]
- Kale, N.S.; Haug, K.; Conesa, P.; Jayseelan, K.; Moreno, P.; Rocca-Serra, P.; Nainala, V.C.; Spicer, R.A.; Williams, M.; Li, X.; et al. Metabolights: An open-access database repository for metabolomics data. Curr. Protoc. Bioinform. 2016, 53, 14.13.1–14.13.18. [Google Scholar]
- Fahy, E.; Alvarez-Jarreta, J.; Brasher, C.J.; Nguyen, A.; Hawksworth, J.I.; Rodrigues, P.; Meckelmann, S.; Allen, S.M.; O’Donnell, V.B. LipidFinder on LIPID MAPS: Peak filtering, MS searching and statistical analysis for lipidomics. Bioinformatics 2018, 35, 685–687. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 2016, 44, D447–D456, Discussion 101–103, 119–128, 244–152. [Google Scholar] [CrossRef]
- Kanehisa, M. The KEGG database. Novartis Found. Symp. 2002, 247, 91–101. [Google Scholar]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Altman, T.; Dale, J.M.; Dreher, K.; Fulcher, C.A.; Gilham, F.; Kaipa, P.; Karthikeyan, A.S.; Kothari, A.; Krummenacker, M.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2010, 38, D473–D479. [Google Scholar] [CrossRef]
- Beale, D.J.; Karpe, A.V.; Ahmed, W. Beyond metabolomics: A review of multi-omics-based approaches. In Microbial Metabolomics: Applications in Clinical, Environmental, and Industrial Microbiology; Beale, D.J., Kouremenos, K.A., Palombo, E.A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 289–312. [Google Scholar]
- Lourenço, A.; Ferreira, A.; Veiga, N.; Machado, I.; Pereira, M.O.; Azevedo, N.F. BiofOmics: A web platform for the systematic and standardized collection of high-throughput biofilm data. PLoS ONE 2012, 7, e39960. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Hemert, J.V.; Dickerson, J.A. OmicsAnalyzer: A Cytoscape plug-in suite for modeling omics data. Bioinformatics 2010, 26, 2995–2996. [Google Scholar] [CrossRef][Green Version]
- Enjalbert, B.; Jourdan, F.; Portais, J.-C. Intuitive visualization and analysis of multi-omics data and application to Escherichia coli carbon metabolism. PLoS ONE 2011, 6, e21318. [Google Scholar] [CrossRef]
- King, Z.A.; Dräger, A.; Ebrahim, A.; Sonnenschein, N.; Lewis, N.E.; Palsson, B.O. Escher: A web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput. Biol. 2015, 11, e1004321. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.T.; Reiss, D.J.; Bonneau, R.; Baliga, N.S. The Gaggle: An open-source software system for integrating bioinformatics software and data sources. BMC Bioinform. 2006, 7, 176. [Google Scholar] [CrossRef]
- Machado, D.; Herrgård, M. Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput. Biol. 2014, 10, e1003580. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Fjell, C.D.; Mayer, M.L.; Pena, O.M.; Wishart, D.S.; Hancock, R.E. INMEX--a web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 2013, 41, W63–W70. [Google Scholar] [CrossRef]
- Kamburov, A.; Cavill, R.; Ebbels, T.M.; Herwig, R.; Keun, H.C. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 2011, 27, 2917–2918. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2013, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Tokimatsu, T.; Sakurai, N.; Suzuki, H.; Ohta, H.; Nishitani, K.; Koyama, T.; Umezawa, T.; Misawa, N.; Saito, K.; Shibata, D. KaPPA-view: A web-based analysis tool for integration of transcript and metabolite data on plant metabolic pathway maps. Plant Physiol. 2005, 138, 1289–1300. [Google Scholar] [CrossRef]
- Lin, K.; Kools, H.; de Groot, P.J.; Gavai, A.K.; Basnet, R.K.; Cheng, F.; Wu, J.; Wang, X.; Lommen, A.; Hooiveld, G.J.; et al. MADMAX - Management and analysis database for multiple ~omics experiments. J. Integr. Bioinform. 2011, 8, 160. [Google Scholar] [CrossRef]
- Usadel, B.; Nagel, A.; Thimm, O.; Redestig, H.; Blaesing, O.E.; Palacios-Rojas, N.; Selbig, J.; Hannemann, J.; Piques, M.C.; Steinhauser, D.; et al. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of coresponding genes, and comparison with known responses. Plant Physiol. 2005, 138, 1195–1204. [Google Scholar] [CrossRef]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. Cell Mol. Biol. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Kaever, A.; Landesfeind, M.; Feussner, K.; Mosblech, A.; Heilmann, I.; Morgenstern, B.; Feussner, I.; Meinicke, P. MarVis-Pathway: Integrative and exploratory pathway analysis of non-targeted metabolomics data. Metabolomics 2015, 11, 764–777. [Google Scholar] [CrossRef]
- Wagele, B.; Witting, M.; Schmitt-Kopplin, P.; Suhre, K. MassTRIX reloaded: Combined analysis and visualization of transcriptome and metabolome data. PLoS ONE 2012, 7, e39860. [Google Scholar] [CrossRef]
- Karnovsky, A.; Weymouth, T.; Hull, T.; Tarcea, V.G.; Scardoni, G.; Laudanna, C.; Sartor, M.A.; Stringer, K.A.; Jagadish, H.V.; Burant, C.; et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 2012, 28, 373–380. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLOS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Bouhaddani, S.e.; Uh, H.-W.; Jongbloed, G.; Hayward, C.; Klarić, L.; Kiełbasa, S.M.; Houwing-Duistermaat, J. Integrating omics datasets with the OmicsPLS package. BMC Bioinform. 2018, 19, 371. [Google Scholar] [CrossRef]
- Wheeler, H.E.; Aquino-Michaels, K.; Gamazon, E.R.; Trubetskoy, V.V.; Dolan, M.E.; Huang, R.S.; Cox, N.J.; Im, H.K. Poly-omic prediction of complex traits: OmicKriging. Genetic Epidemiol. 2014, 38, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Droste, P.; Miebach, S.; Niedenführ, S.; Wiechert, W.; Nöh, K. Visualizing multi-omics data in metabolic networks with the software Omix—A case study. Biosystems 2011, 105, 154–161. [Google Scholar] [CrossRef]
- Garcia-Alcalde, F.; Garcia-Lopez, F.; Dopazo, J.; Conesa, A. Paintomics: A web based tool for the joint visualization of transcriptomics and metabolomics data. Bioinformatics 2011, 27, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.R.; Evelo, C.T. PathVisio 3: An extendable pathway analysis toolbox. PLoS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef]
- Neuweger, H.; Persicke, M.; Albaum, S.P.; Bekel, T.; Dondrup, M.; Huser, A.T.; Winnebald, J.; Schneider, J.; Kalinowski, J.; Goesmann, A. Visualizing post genomics data-sets on customized pathway maps by ProMeTra-aeration-dependent gene expression and metabolism of Corynebacterium glutamicum as an example. BMC Syst. Biol. 2009, 3, 82. [Google Scholar] [CrossRef]
- Eriksson, L.; Byrne, T.; Johansson, E.; Trygg, J.; Vikström, C. Multi- and Megavariate Data Analysis Basic Principles and Applications, Volume 1, 3rd ed.; MKS Umetrics AB: Umea, Sweden, 2013. [Google Scholar]
- Junker, B.H.; Klukas, C.; Schreiber, F. VANTED: A system for advanced data analysis and visualization in the context of biological networks. BMC Bioinform. 2006, 7, 1–13. [Google Scholar]
- Grimplet, J.; Cramer, G.R.; Dickerson, J.A.; Mathiason, K.; Van Hemert, J.; Fennell, A.Y. VitisNet: “Omics” integration through grapevine molecular networks. PLoS ONE 2009, 4, e8365. [Google Scholar] [CrossRef] [PubMed]
- Aderem, A. Systems biology: Its practice and challenges. Cell 2005, 121, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Dihazi, H.; Asif, A.R.; Beißbarth, T.; Bohrer, R.; Feussner, K.; Feussner, I.; Jahn, O.; Lenz, C.; Majcherczyk, A.; Schmidt, B.; et al. Integrative omics - from data to biology. Expert Rev. Proteom. 2018, 15, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, M.; da Veiga Leprevost, F.; Squizzato, S.; Park, Y.M.; Haug, K.; Carroll, A.J.; Spalding, D.; Paschall, J.; Wang, M.; et al. Discovering and linking public omics data sets using the Omics Discovery Index. Nat. Biotechnol. 2017, 35, 406–409. [Google Scholar] [CrossRef]
- Kuo, W.P.; Jenssen, T.K.; Butte, A.J.; Ohno-Machado, L.; Kohane, I.S. Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics 2002, 18, 405–412. [Google Scholar] [CrossRef]
- Guo, Y.; Sheng, Q.; Li, J.; Ye, F.; Samuels, D.C.; Shyr, Y. Large scale comparison of gene expression levels by microarrays and RNAseq using TCGA data. PLoS ONE 2013, 8, e71462. [Google Scholar] [CrossRef]
- Sinha, R.; Abnet, C.C.; White, O.; Knight, R.; Huttenhower, C. The microbiome quality control project: Baseline study design and future directions. Genome Biol. 2015, 16, 276. [Google Scholar] [CrossRef] [PubMed]
- Tabb, D.L.; Vega-Montoto, L.; Rudnick, P.A.; Variyath, A.M.; Ham, A.J.; Bunk, D.M.; Kilpatrick, L.E.; Billheimer, D.D.; Blackman, R.K.; Cardasis, H.L.; et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2010, 9, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.T.; Landry, J.-R. RNA-Seq—Quantitative measurement of expression through massively parallel RNA-sequencing. Methods 2009, 48, 249–257. [Google Scholar] [CrossRef]
- Nayfach, S.; Pollard, K.S. Toward accurate and quantitative comparative metagenomics. Cell 2016, 166, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Zeevi, D.; Korem, T.; Zmora, N.; Israeli, D.; Rothschild, D.; Weinberger, A.; Ben-Yacov, O.; Lador, D.; Avnit-Sagi, T.; Lotan-Pompan, M.; et al. Personalized nutrition by prediction of glycemic responses. Cell 2015, 163, 1079–1094. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabasi, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar] [CrossRef]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Boccaletti, S.; Bianconi, G.; Criado, R.; del Genio, C.I.; Gómez-Gardeñes, J.; Romance, M.; Sendiña-Nadal, I.; Wang, Z.; Zanin, M. The structure and dynamics of multilayer networks. Phys. Rep. 2014, 544, 1–122. [Google Scholar] [CrossRef]
- Kivelä, M.; Arenas, A.; Barthelemy, M.; Gleeson, J.P.; Moreno, Y.; Porter, M.A. Multilayer networks. J. Complex Netw. 2014, 2, 203–271. [Google Scholar] [CrossRef]
- Price, N.D.; Magis, A.T.; Earls, J.C.; Glusman, G.; Levy, R.; Lausted, C.; McDonald, D.T.; Kusebauch, U.; Moss, C.L.; Zhou, Y.; et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat. Biotechnol. 2017, 35, 747–756. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ (Clin. Res. Ed.) 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed]
- Bashiardes, S.; Godneva, A.; Elinav, E.; Segal, E. Towards utilization of the human genome and microbiome for personalized nutrition. Curr. Opin. Biotechnol. 2018, 51, 57–63. [Google Scholar] [CrossRef]
- Ogura, T.; Date, Y.; Tsuboi, Y.; Kikuchi, J. Metabolic dynamics analysis by massive data integration: Application to tsunami-affected field soils in Japan. ACS Chem. Biol. 2015, 10, 1908–1915. [Google Scholar] [CrossRef]
- Ara, T.; Enomoto, M.; Arita, M.; Ikeda, C.; Kera, K.; Yamada, M.; Nishioka, T.; Ikeda, T.; Nihei, Y.; Shibata, D.; et al. Metabolonote: A Wiki-Based Database for Managing Hierarchical Metadata of Metabolome Analyses. Front. Bioeng. Biotechnol. 2015, 3, 38. [Google Scholar] [CrossRef]
- Ćwiek, H.; Krajewski, P.; Klukas, C.; Chen, D.; Lange, M.; Weise, S.; Scholz, U.; van Dijk, A.D.J.; Nap, J.P.; Fiorani, F.; et al. Towards recommendations for metadata and data handling in plant phenotyping. J. Exp. Bot. 2015, 66, 5417–5427. [Google Scholar]
- Meyer, R.S. Encouraging metadata curation in the Diversity Seek initiative. Nature Plants 2015, 1, 15099. [Google Scholar] [CrossRef]
- Diamandis, E.P. Cancer biomarkers: Can we turn recent failures into success? J. Natl. Cancer Inst. 2010, 102, 1462–1467. [Google Scholar] [CrossRef]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome profiling in human diseases: New advances and perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef]
- Trivedi, D.K.; Hollywood, K.A.; Goodacre, R. Metabolomics for the masses: The future of metabolomics in a personalized world. New Horizons Transl. Med. 2017, 3, 294–305. [Google Scholar] [CrossRef]
- Phillips, K.A.; Deverka, P.A.; Hooker, G.W.; Douglas, M.P. Genetic Test Availability And Spending: Where Are We Now? Where Are We Going? Health Aff. (Proj. Hope) 2018, 37, 710–716. [Google Scholar] [CrossRef]
- Yurkovich, J.T.; Palsson, B.O. Quantitative -omic data empowers bottom-up systems biology. Curr. Opin. Biotechnol. 2018, 51, 130–136. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Potential Limitations | Strategies to Overcome Limitation |
|---|---|
Limited biomass of sample
|
|
| Heterogeneity of cell type/ composition (e.g., microbiome community, whole organism, tissue or single cell). Proportions of multiple cell types in a sample can change substantially and shift omics profile [27]. |
|
| Differences in specific biomolecules in sample types (e.g., urine may have many metabolites but very few proteins, DNA and RNA in comparison to blood, stool or tissue samples). |
|
| Technical artifacts, including batch effects. |
|
Multiple testing
|
|
| Background contamination (e.g., in a microbiome study stool samples will have host DNA, RNA, protein, and metabolites) |
|
| Differences in analytical platforms and integrating data sets of multi-omics that measure fundamentally different biomolecules |
|
| Software Tool | Omics Integrated | Domain | Functionality | Type of license | Reference |
|---|---|---|---|---|---|
| 3Omics |
| Medical (human) |
| Open | [41] |
| BiofOmics |
| Biofilm |
| Open | [113] |
| BioCyc/MetaCyc |
| Unspecified |
| Open | [111] |
| Cell Illustrator 5.0 |
| Unspecified |
| Licensed | [74] |
| CellML (Open source XML language) |
| Unspecified |
| Open | [73] |
| COBRA |
| Unspecified |
| Open | [12,79] |
| Cytoscape with MODAM, and; Cytoscape with OmicsAnalyzer |
| Unspecified |
| Open | [114,115] |
| E-Cell |
| Unspecified (Cells) |
| Open | [63] |
| Escher |
| Unspecified |
| Open (MIT license) | [116] |
| Gaggle | Variety of omics platform bioinformatics solutions | Unspecified |
| Open | [117] |
| GIM3E (Gene Inactivation Moderated by Metabolism, Metabolomics and Expression) |
| Unspecified |
| Open; Phython based and requires COBRApy 0.2.x. | [118] |
| INMEX (Integrative meta-analysis of expression data) |
| Medical and Clinical |
| Open | [119] |
| IMPaLA (Integrated Molecular Pathway Level Analysis) |
| Medical and clinical |
| Academic only | [120] |
| Ingenuity Pathway Analysis |
| Medical (human) and clinical. |
| Commercial | [121] |
| IOMA (Integrative Omics-Metabolic Analysis) |
| Unspecified |
| Open | [20] |
| KaPPA-View |
| Plants |
| Open | [122] |
| MADMAX (Management and analysis database for multiple omics experiments) |
| Plants, Medical and Clinical |
| Open | [123] |
| MapMan |
| Plants (developed for use with Arabidopsis. Includes more species) |
| Open | [124,125] |
| MarVis-Pathway (Marker Visualization Pathway) |
| Unspecified |
| Academic only | [126] |
| MassTrix |
| Unspecified |
| Open | [127] |
| MetaboAnalyst |
| Plants, Microbial, Microbiome, Medical and Clinical |
| Open | [53] |
| MetaboLights |
| Unspecified |
| Open | [106] |
| MetScape 2 |
| Medical and Clinical |
| Open | [128] |
| mixOmics (R package) |
| Unspecified |
| Open | [129] |
| OmicsPLS |
| Unspecified |
| Open | [130] |
| Omickriging (R package) |
| Unspecified |
| Open | [131] |
| Omix visualization tool |
| Unspecified |
| Annual license fee | [132] |
| PaintOmics |
| 100 top species of different biological kingdoms |
| Open | [133] |
| PathVisio 3 |
| Unspecified |
| Open (Apache) | [134] |
| ProMeTra |
| Medical and Clinical |
| Open | [135] |
| Reactome |
| Unspecified |
| Open | [110] |
| Recon3D |
| Human |
| Open | [80] |
| SimCell |
| Unspecified |
| Open | [56] |
| SIMCA |
| Unspecified |
| Commercial | [136] |
| VANTED (Visualization and Analysis of Networks with related Experimental Data) |
| Unspecified |
| Open | [137] |
| VitisNet |
| Grapes |
| Open | [138] |
| xCellerator |
| Unspecified |
| Open | [55] |
| Database | Omics | Domain | Functionality | Type of license | Reference |
|---|---|---|---|---|---|
| ChEBI |
| Unspecified |
| Open | [105] |
| E. coli metabolome database (ECMDB) |
| Microbial |
| Open | [97] |
| FlyBase |
| Drosophila |
| License | [95] |
| GenBank (database) |
| Numerous (over 100,000 organisms) |
| Open | [103] |
| Human Metabolome Database (HMDB) |
| Human |
| Open | [102] |
| KEGG |
| PlantsAnimalsMicrobes |
| Open and licensed. | [109] |
| Lipid Maps |
| Unspecified |
| Open | [107] |
| Plant Metabolic Network (PMN) |
| Plants |
| Open | [100] |
| PRIDE |
| Unspecified |
| Open | [108] |
| ProMeTra |
| Medical and Clinical |
| Open | [135] |
| Reactome |
| Unspecified |
| Open | [110] |
| Saccharomyces genome database (SGD) |
| Microbe (yeast) |
| Open | [101] |
| UniProt |
| Unspecified |
| Open | [104] |
| Wormbase |
| Helminths |
| Open | [96] |
| Yeast metabolome data (YMDB) |
| Microbe (yeast) |
| Open | [99] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community. Metabolites 2019, 9, 76. https://doi.org/10.3390/metabo9040076
Pinu FR, Beale DJ, Paten AM, Kouremenos K, Swarup S, Schirra HJ, Wishart D. Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community. Metabolites. 2019; 9(4):76. https://doi.org/10.3390/metabo9040076
Chicago/Turabian StylePinu, Farhana R., David J. Beale, Amy M. Paten, Konstantinos Kouremenos, Sanjay Swarup, Horst J. Schirra, and David Wishart. 2019. "Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community" Metabolites 9, no. 4: 76. https://doi.org/10.3390/metabo9040076
APA StylePinu, F. R., Beale, D. J., Paten, A. M., Kouremenos, K., Swarup, S., Schirra, H. J., & Wishart, D. (2019). Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community. Metabolites, 9(4), 76. https://doi.org/10.3390/metabo9040076