Untargeted Metabolomics-Based Screening Method for Inborn Errors of Metabolism using Semi-Automatic Sample Preparation with an UHPLC- Orbitrap-MS Platform

 ,
,
Abstract
:1. Introduction
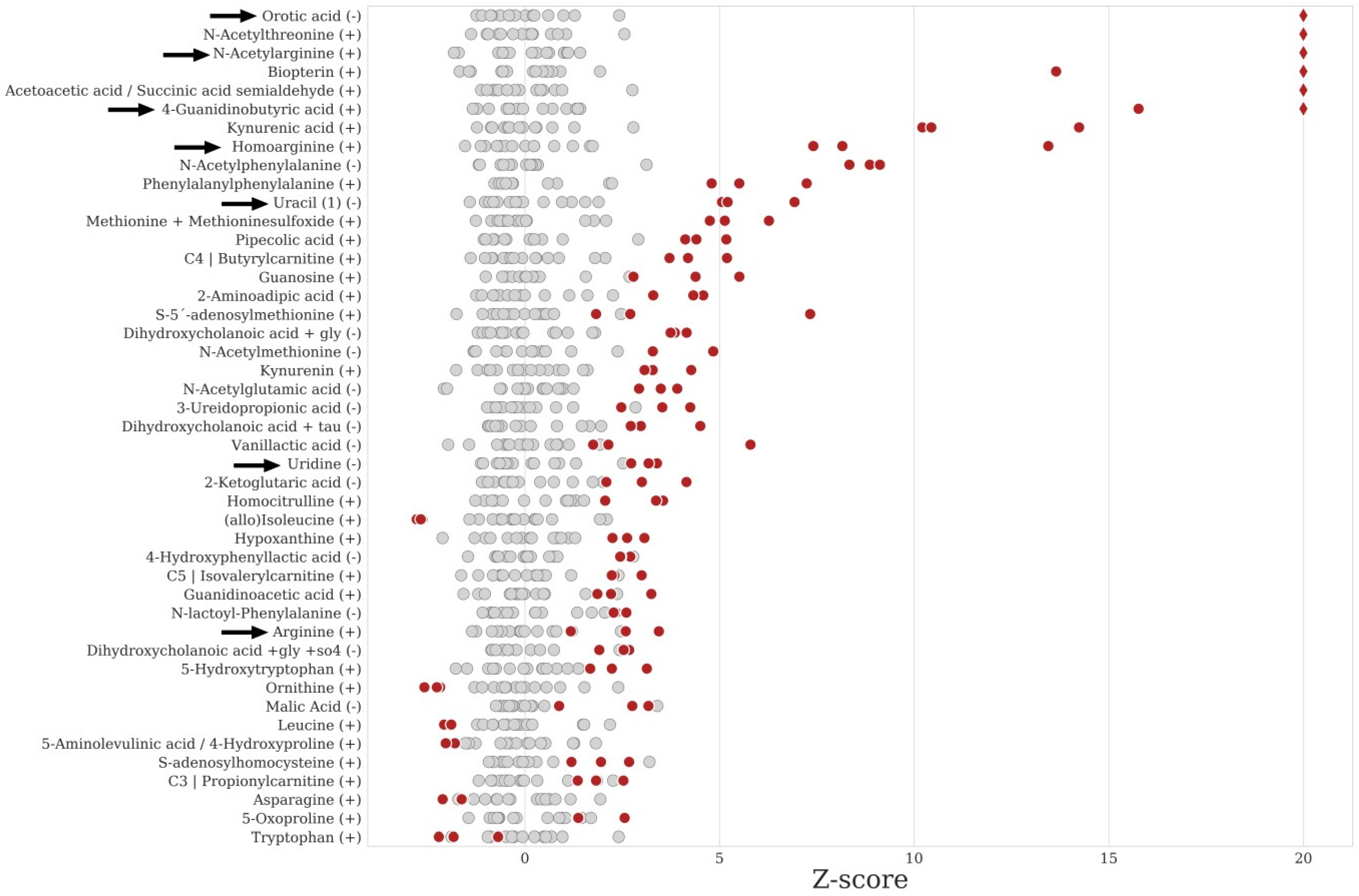
2. Results
2.1. Analytical Performance
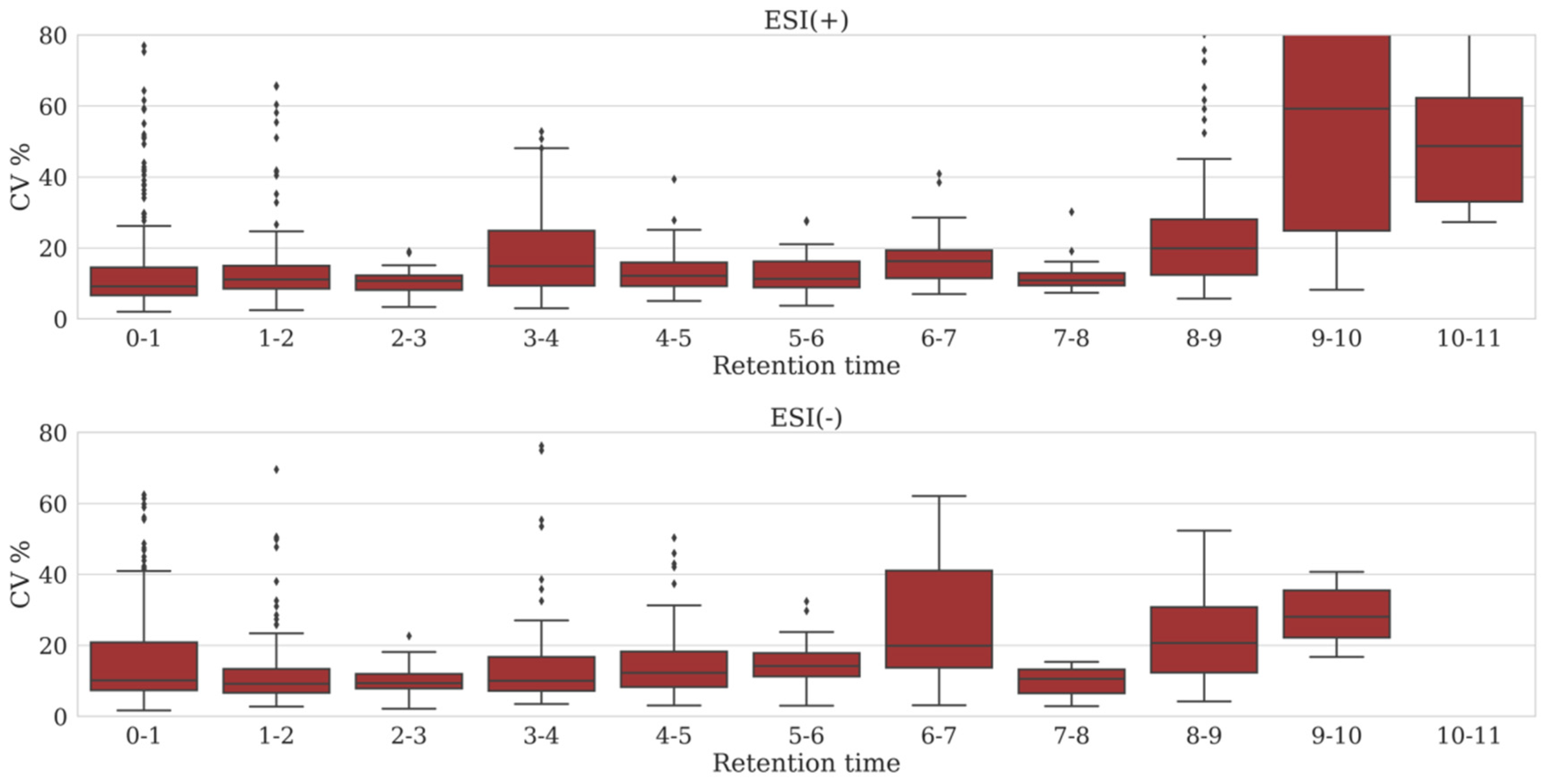
2.2. Retention Time Stability
2.3. Variation in Peak Area
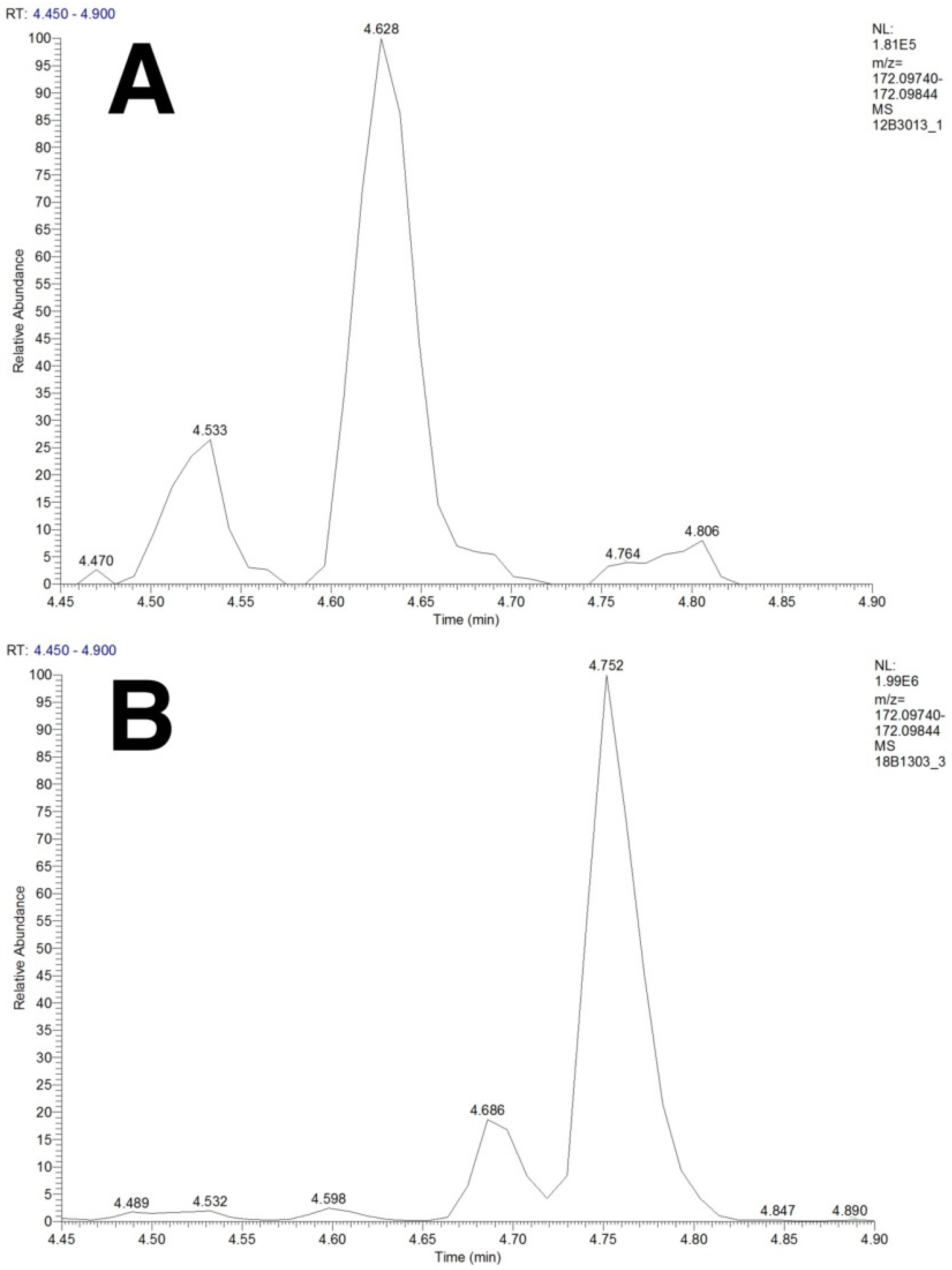
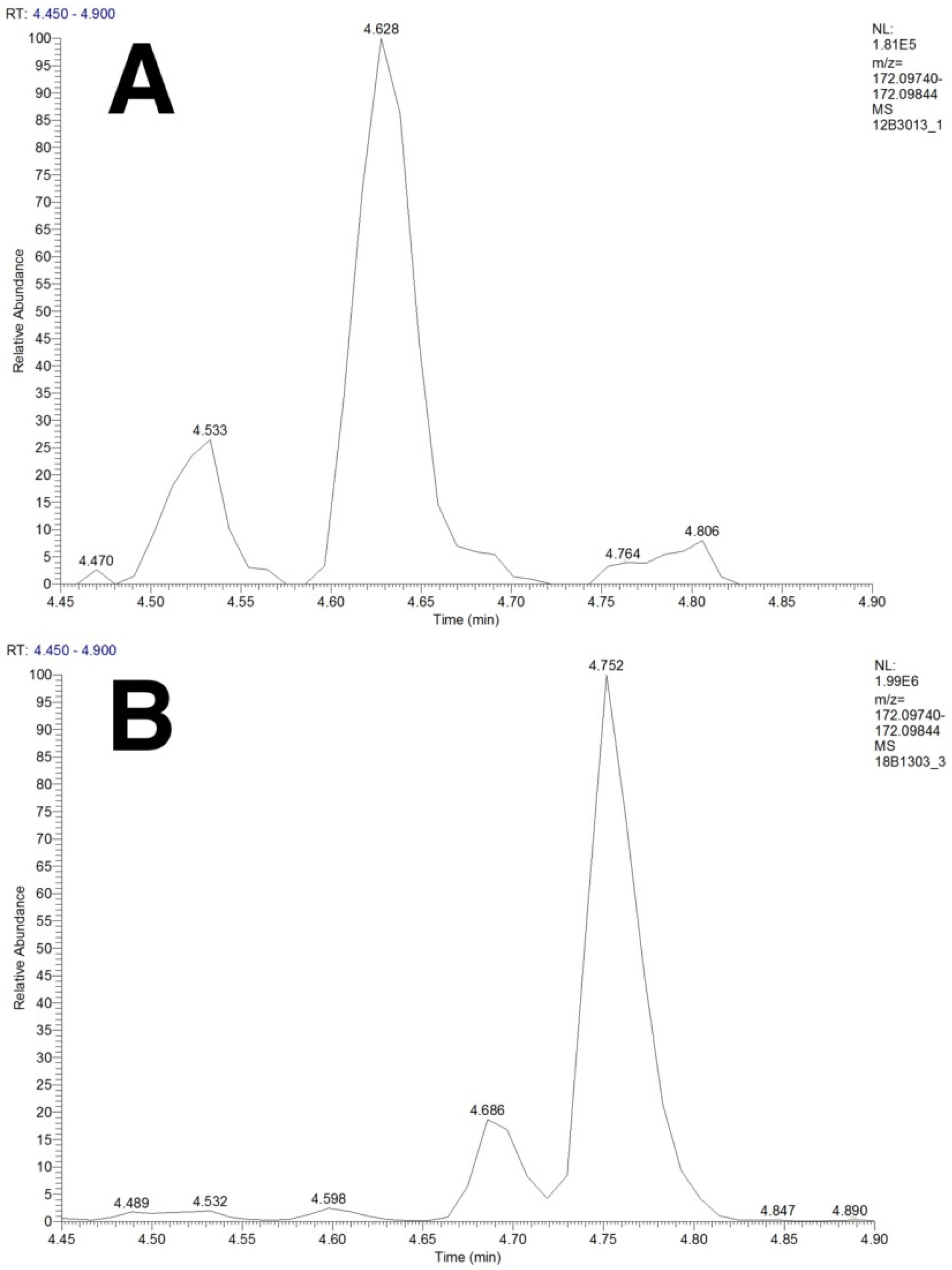
2.4. Separation of Isobaric and Isomeric Species
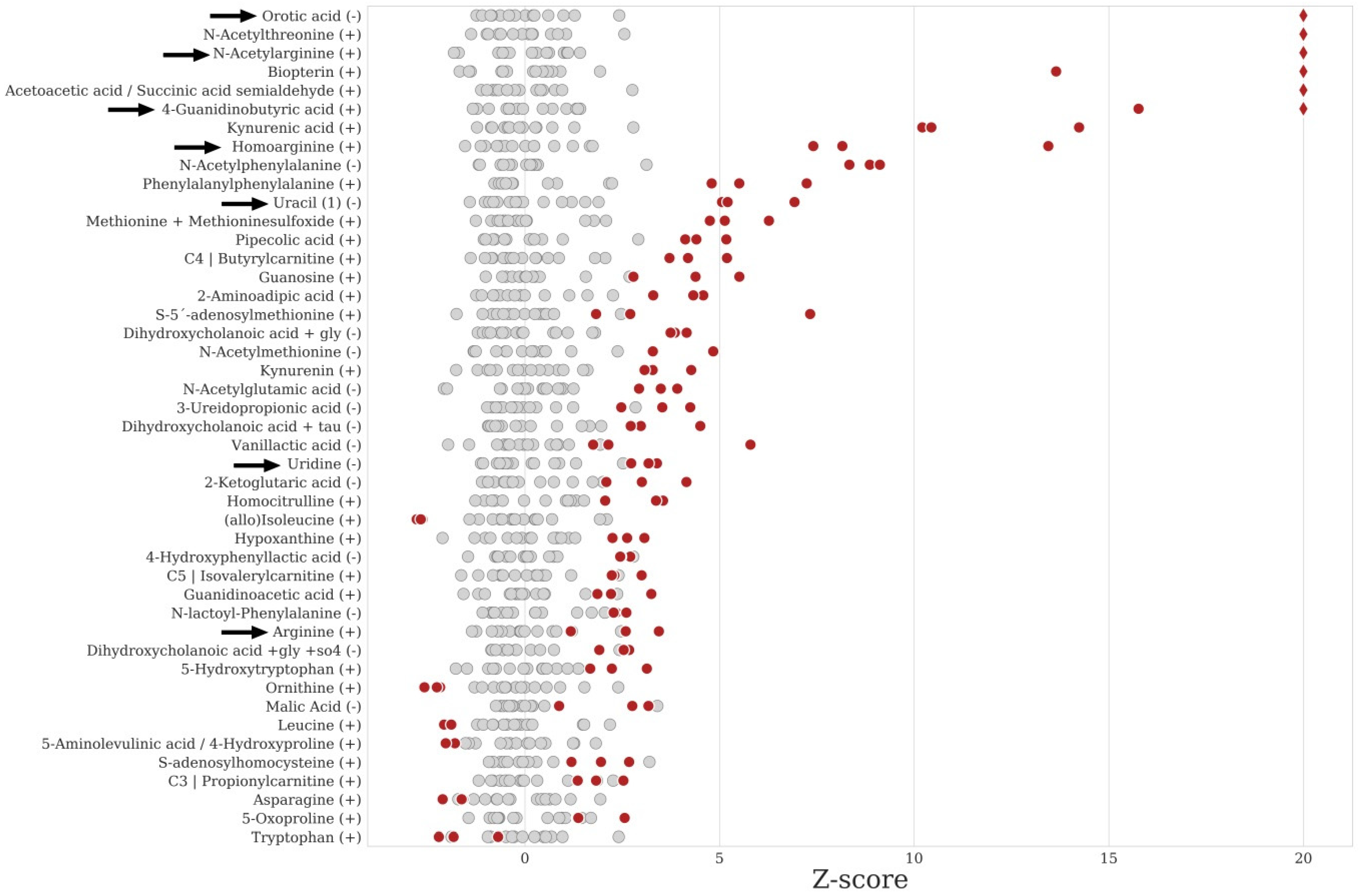
2.5. Data Analysis and IEM Detection
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Sample Selection
4.3. Semi-Automated Sample Preparation Procedure
4.4. UHPLC-Orbitrap-MS(/MS) Analysis
- Inclusion list set at on (do not pick others)
- Inclusion list set at on (do pick others), exclusion list set at on
- Inclusion list set at off, exclusion list set at on
4.5. Quality Control
4.5.1. Retention Time Stability
4.5.2. Within Batch Peak Area Variation
4.5.3. Between-Batch Peak Area Variation
4.5.4. Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharer, J.D. An Overview of Biochemical Genetics. Curr. Protoc. Hum. Genet. 2016, 89, 17.1.1–17.1.16. [Google Scholar] [PubMed]
- Jacob, M.; Malkawi, A.; Albast, N.; Al Bougha, S.; Lopata, A.; Dasouki, M.; Abdel Rahman, A.M. A targeted metabolomics approach for clinical diagnosis of inborn errors of metabolism. Anal. Chim. Acta 2018, 1025, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.J.; Eggington, M.; Kahler, S.G. Comprehensive screening of urine samples for inborn errors of metabolism by electrospray tandem mass spectrometry. Clin. Chem. 2002, 48, 1970–1980. [Google Scholar] [PubMed]
- Sandlers, Y. The future perspective: Metabolomics in laboratory medicine for inborn errors of metabolism. Transl. Res. 2017, 189, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Mussap, M.; Zaffanello, M.; Fanos, V. Metabolomics: A challenge for detecting and monitoring inborn errors of metabolism. Biochim. Clin. 2019, 43, 14–23. [Google Scholar] [CrossRef]
- Tebani, A.; Abily-Donval, L.; Afonso, C.; Marret, S.; Bekri, S. Clinical metabolomics: The new metabolic window for inborn errors of metabolism investigations in the post-genomic era. Int. J. Mol. Sci. 2016, 17, 1167. [Google Scholar] [CrossRef] [PubMed]
- Wevers, R.A.; Blau, N. Think big—Think omics. J. Inherit. Metab. Dis. 2018, 41, 281–283. [Google Scholar] [CrossRef]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.D.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E.; Engelke, U.F.H.; de Boer, S.; Hoegen, B.; Kwast, H.J.T.; van de Vorst, M.; Huigen, M.C.D.G.; Keularts, I.M.L.W.; et al. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef]
- Körver-Keularts, I.M.L.W.; Wang, P.; Waterval, H.W.A.H.; Kluijtmans, L.A.J.; Wevers, R.A.; Langhans, C.D.; Scott, C.; Habets, D.D.J.; Bierau, J. Fast and accurate quantitative organic acid analysis with LC-QTOF/MS facilitates screening of patients for inborn errors of metabolism. J. Inherit. Metab. Dis. 2018, 41, 415–424. [Google Scholar] [CrossRef]
- Haijes, H.; Willemsen, M.; van der Ham, M.; Gerrits, J.; Pras-Raves, M.; Prinsen, H.; van Hasselt, P.; de Sain-van der Velden, M.; Verhoeven-Duif, N.; Jans, J. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites 2019, 9, 12. [Google Scholar] [CrossRef]
- Lv, H.; Palacios, G.; Hartil, K.; Kurland, I.J. Advantages of tandem LC-MS for the rapid assessment of tissue-specific metabolic complexity using a pentafluorophenylpropyl stationary phase. J. Proteome Res. 2011, 10, 2104–2112. [Google Scholar] [CrossRef]
- Boudah, S.; Olivier, M.F.; Aros-Calt, S.; Oliveira, L.; Fenaille, F.; Tabet, J.C.; Junot, C. Annotation of the human serum metabolome by coupling three liquid chromatography methods to high-resolution mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 966, 34–47. [Google Scholar] [CrossRef]
- Yoshida, H.; Mizukoshi, T.; Hirayama, K.; Miyano, H. Comprehensive analytical method for the determination of hydrophilic metabolites by high-performance liquid chromatography and mass spectrometry. J. Agric. Food Chem. 2007, 55, 551–560. [Google Scholar] [CrossRef]
- Needham, S.R.; Brown, P.R.; Duff, K.; Bell, D. Optimized stationary phases for the high-performance liquid chromatography-electrospray ionization mass spectrometric analysis of basic pharmaceuticals. J. Chromatogr. A 2000, 869, 159–170. [Google Scholar] [CrossRef]
- Bell, D.S.; Jones, A.D. Solute attributes and molecular interactions contributing to U-shape”retention on a fluorinated high-performance liquid chromatography stationary phase. J. Chromatogr. A 2005, 1073, 99–109. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- Crombez, E.A.; Cederbaum, S.D. Hyperargininemia due to liver arginase deficiency. Mol. Genet. Metab. 2005, 84, 243–251. [Google Scholar] [CrossRef]
- Mizutani, N.; Hayakawa, C.; Ohya, Y.; Watanabe, K.; Watanabe, Y.; Mori, A. Guanidino compounds in hyperargininemia. Tohoku J. Exp. Med. 1987, 153, 197–205. [Google Scholar] [CrossRef]
- Marescau, B.; De Deyn, P.P.; Lowenthal, A.; Qureshi, I.A.; Antonozzi, I.; Bachmann, C.; Cederbaum, S.D.; Cerone, R.; Chamoles, N.; Colombo, J.P.; et al. Guanidino Compound Analysis as a Complementary Diagnostic Parameter for Hyperargininemia: Follow-Up of Guanidino Compound Levels during Therapy. Pediatr. Res. 1990, 27, 297–303. [Google Scholar] [CrossRef]
- Marescau, B.; Deshmukh, D.R.; Kockx, M.; Possemiers, I.; Qureshi, I.A.; Wiechert, P.; De Deyn, P.P. Guanidino compounds in serum, urine, liver, kidney, and brain of man and some ureotelic animals. Metabolism 1992, 41, 526–532. [Google Scholar] [CrossRef]
- Marescaua, B.; Qureshi, I.A.; De Deyn, P.; Letarte, J.; Ryba, R.; Lowenthal, A. Guanidino compounds in plasma, urine and cerebrospinal fluid of hyperargininemic patients during therapy. Clin. Chim. Acta 1985, 146, 21–27. [Google Scholar] [CrossRef]
- Kennedy, A.D.; Wittmann, B.M.; Evans, A.M.; Miller, L.A.D.; Toal, D.R.; Lonergan, S.; Elsea, S.H.; Pappan, K.L. Metabolomics in the clinic: A review of the shared and unique features of untargeted metabolomics for clinical research and clinical testing. J. Mass Spectrom. 2018, 53, 1143–1154. [Google Scholar] [CrossRef]
- Carmical, J.; Brown, S. The impact of phospholipids and phospholipid removal on bioanalytical method performance. Biomed. Chromatogr. 2016, 30, 710–720. [Google Scholar] [CrossRef]
- Václavík, J.; Coene, K.L.M.; Vrobel, I.; Najdekr, L.; Friedecký, D.; Karlíková, R.; Mádrová, L.; Petsalo, A.; Engelke, U.F.H.; van Wegberg, A.; et al. Structural elucidation of novel biomarkers of known metabolic disorders based on multistage fragmentation mass spectra. J. Inherit. Metab. Dis. 2018, 41, 407–414. [Google Scholar] [CrossRef]
- Van Karnebeek, C.D.M.; Bonafé, L.; Wen, X.-Y.; Tarailo-Graovac, M.; Balzano, S.; Royer-Bertrand, B.; Ashikov, A.; Garavelli, L.; Mammi, I.; Turolla, L.; et al. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nat. Genet. 2016, 48, 777. [Google Scholar] [CrossRef]
- Graham, E.; Lee, J.; Price, M.; Tarailo-Graovac, M.; Matthews, A.; Engelke, U.; Tang, J.; Kluijtmans, L.A.J.; Wevers, R.A.; Wasserman, W.W.; et al. Integration of genomics and metabolomics for prioritization of rare disease variants: A 2018 literature review. J. Inherit. Metab. Dis. 2018, 41, 435–445. [Google Scholar] [CrossRef]
- MzCloud Advanced Mass Spectral Database. Available online: www.mzcloud.org (accessed on 19 September 2019).
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, 521–526. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, 603–610. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0-The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, 801–807. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Inborn Error of Metabolism a | Z-Score b | p-Value c | ID d |
|---|---|---|---|
| 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (N = 1) | |||
| 2-Methyl-3-hydroxybutyric acid [↑] (+/−) | NF | NF | - |
| Tiglylglycine [↑] (−) | 7.0 * | 2.99 × 10−3 | 2b |
| 3-Methylcrotonyl-CoA-carboxylase deficiency (N = 1) | |||
| 3-Hydroxyisovaleric acid [↑] (+/−) | NF | NF | - |
| 3-Methylcrotonylglycine [↑] (−) | 33.7 * | 1.50 × 10−2 | 2b |
| 3-Hydroxyisovalerylcarnitine [↑] (+) | 1811.7 * | 4.35 × 10−3 | 2b |
| Adenylosuccinate lyase deficiency (N = 1) | |||
| SAICAR [↑] (+/−) | NF | NF | - |
| Succinyladenosine [↑] (−) | 57.2 * | 2.80 × 10−3 | 2b |
| Alkaptonuria (N = 1) | |||
| Homogentisic acid [↑] (+) | 0.6 | 3.78 × 10−1 | 2b |
| Alpha-Methylacyl-CoA racemase deficiency (N = 2) | |||
| Pristanoyl-carnitine [↑] (+/−) | NF | NF | - |
| Phytanoyl-carnitine [↑] (+/−) | NF | NF | - |
| Dihydroxycholestanoic acid + Gly [↑] (−) | INF/INF | <1.1 × 10−16/<1.1 × 10−16 | 2b |
| Dihydroxycholestanoic acid + Tau [↑] (−) | 121.5 */34.1 * | 1.95 × 10−3/1.51 × 10−2 | 2b |
| Trihydroxycholestanoic acid + Gly [↑] (−) | 462.6 */386.6 * | 1.70 × 10−3/1.45 × 10−2 | 2b |
| Trihydroxycholestanoic acid + Tau [↑] (−) | 500.4 */326.5 * | 9.07 × 10−4/1.70 × 10−2 | 2b |
| Aminoacylase I deficiency (N = 1) | |||
| N-Acetylalanine [↑] (−) | 8.7 * | 1.74 × 10−2 | 2b |
| N-Acetylarginine [↑] (−) | 0.4 | 1.69 × 10−1 | 2b |
| N-Acetylglutamic acid [↑] (−) | 362.9 * | 1.19 × 10−2 | 2b |
| N-Acetylglycine [↑] (−) | 8.9 * | 1.06 × 10−2 | 2b |
| N-Acetylleucine [↑] (−) | 23.0 * | 1.31 × 10−2 | 2b |
| N-Acetylmethionine [↑] (−) | 644.7 * | 8.44 × 10−3 | 2b |
| N-Acetylserine [↑] (−) | NF | NF | - |
| N-Acetylthreonine [↑] (+) | 1259.4 * | 1.31 × 10−2 | 2b |
| Arginase deficiency (N = 1) | |||
| 2-Oxoarginine [↑] (−) | NF e | NF | - |
| 4-Guanidinobutyric acid [↑] (−) | 25.7 * | 1.18 × 10−2 | 2b |
| Arginine [↑] (+) | 2.4 | 5.14 × 10−2 | 1 |
| Glutamine + Glutamic acid [↑] (−) | −0.1 | 7.51 × 10−1 | 1 |
| Guanidinoacetic acid [↑] (−) | 2.7 * | 6.08 × 10−6 | 1 |
| Homoarginine [↑] (+) | 9.7 * | 3.45 × 10−2 | 2b |
| N-Acetylarginine [↑] (−) | 137.2 * | 3.20 × 10−4 | 2b |
| Orotic acid [↑] (−) | 32.4 * | 5.85 × 10−4 | 2b |
| Uracil [↑] (−) | 5.7 * | 4.02 × 10−3 | 1 |
| Uridine [↑] (−) | 3.1 * | 1.60 × 10−6 | 1 |
| Argininic acid [↑] (+/−) | NF e | NF | - |
| Argininosuccinic aciduria (N = 3) | |||
| Arginine [↓] (+) | −0.9 *–−0.3 | 2.01 × 10−1–2.98 × 10−2 | 1 |
| Argininosuccinic acid [↑] (−) | 4797 *–13,725 * | 2.96 × 10−2–5.40 × 10−3 | 2b |
| Citrulline [↑] (−) | 10 *–32 * | 4.87 × 10−3–2.83 × 10−4 | 1 |
| Cytidine [↑] (−) | 3580 */2.1 */NF | 9.78 × 10−3/7.33 × 10−6/NF | 1 |
| Glutamine + Glutamic acid [↑] (+) | 2.2 */−1.8 */NF | 6.99 × 10−3/7.51 × 10−6/NF | 1 |
| Homocitrulline [↑] (+) | 1.7 *–4.5 | 6.69 × 10−4–2.81 × 10−5 | 2b |
| N-Acetylcitrulline [↑] (−) | 6.2 *–42.7 * | 8.23 × 10−3–9.02 × 10−4 | 2b |
| Orotic acid [↑] (−) | 0.1/−0.5/NF | 6.61 × 10−1/1.08 × 10−1/NF | 2b |
| Uracil [↑] (−) | −0.7 */−1.9 * /NF | 1.89 × 10−2/5.20 × 10−6/NF | 1 |
| Uridine [↑] (−) | −0.4–3.4 * | 1.70 × 10−1–1.06 × 10−3 | 1 |
| Beta-ketothiolase deficiency (N = 2) | |||
| 2-Methyl-3-hydroxybutyric acid [↑] (−) | 5.8 */15.7 * | 2.66 × 10−2/1.03 × 10−3 | 4 |
| 2-Methylacetoacetic acid [↑] (−) | 0.5/NF | 5.84 × 10−1/NF | 4 |
| Tiglylcarnitine [↑] (+) | 0.3/NF | 0.726/NF | 2b |
| Tiglylglycine [↑] (−) | 190 */217 * | 1.23 × 10−2/7.96 × 10−5 | 2b |
| Beta-mannosidosis (N = 1) | |||
| GlcNAc-Man [↑] (−) | 383.9 * | 5,73 × 10−3 | 2b |
| Carbamoyl phosphate synthetase I deficiency (N = 2) | |||
| Glutamine + Glutamic acid [↑] (+) | 4.7 */0.59 | 6.43 × 10−5/1.4 × 10−1 | 1 |
| Citrullinemia type I (N = 1) | |||
| Arginine [↓] (+) | 0.5 | 4.06 × 10−1 | 1 |
| Citrulline [↑] (+) | 128.7 * | 3.64 × 10−3 | 1 |
| Citrulline lactam [↑] (+) | 202,9 | 9.13 × 10−2 | 4 |
| Glutamine + Glutamic acid [↑] (+) | −2.4 * | 3.28 × 10−2 | 1 |
| N-Acetylcitrulline [↑] (−) | 335.2 * | 3.77 × 10−3 | 2b |
| Orotic acid [↑] (−) | 0.3 | 3.64 × 10−1 | 2b |
| Uracil [↑] (−) | 0.1 | 8.50 × 10−1 | 1 |
| Uridine [↑] (−) | 1.4 * | 6.41 × 10−3 | 1 |
| Glutamate formiminotransferase deficiency (N = 1) | |||
| Formiminoglutamic acid [↑] (+) | 2031.0 * | 7.13 × 10−3 | 2b |
| Hydantion-5-propionic acid [↑] (−) | 12.0 * | 2.24 × 10−5 | 2b |
| Glutaric aciduria I (N = 2) | |||
| 3-Hydroxyglutaric acid [↑] (−) | 2.1 */NF | 5.49 × 10−5/NF | 4 |
| Glutarylcarnitine [↑] (+) | 317.4 */338.6 * | 5.57 × 10−3/4.72 × 10−3 | 1 |
| Glutaric acid [↑] (−) | 0.7/2.5 * | 3.05 × 10−5/2.16 × 10−7 | 1 |
| Glutarylglycine [↑] (−) | 228.8 */513.4 * | 2.90 × 10−3/9.19 × 10−4 | 4 |
| Glutaric aciduria II (N = 2) | |||
| 2-Hydroxyglutaric acid [↑] (−) | 39.1 */73.7 * | 6.27 × 10−3/2.23 × 10−3 | 4 |
| 3-Hydroxyglutaric acid [↑] (−) | 2.9 */NF | 2,15 × 10−5/NF | 4 |
| Ethylmalonic acid [↑] (−) | 1.2/53.6 * | 1.61 × 10−1/5.02 × 10−3 | 1 |
| Glutaric acid [↑] (−) | 1.8 */2.1 * | 9.31 × 10−6/3.03 × 10−5 | 1 |
| Hexanoylglycine [↑] (−) | 391.2 */1797.4 * | 6.37 × 10−3/1.05 × 10−3 | 2b |
| Isobutyrylglycine [↑] (−) | 16.4 */NF | 4,56 × 10−3/NF | 2b |
| Isovaleryglycine [↑] (−) | 51.5 */1.1 * | 7.78 × 10−3/3.37 × 10−2 | 2b |
| (Iso)butyrylcarnitine [↑] (+) | 30.2 */110.2 * | 5.65 × 10−4/8.95 × 10−4 | 1 |
| Isovalerylcarnitine [↑] (+) | 189.6 */34.1 * | 9.08 × 10−4/3.06 × 10−3 | 1 |
| Glutarylcarnitine [↑] (+) | 28.7 */85.9 * | 2.37 × 10−3/9.32 × 10−4 | 2b |
| Hexanoylcarnitine [↑] (+) | 124.5 */184.6 * | 3.24 × 10−3/9.02 × 10−3 | 1 |
| Octanoylcarnitine [↑] (+) | 101.8 */123.1 * | 2.71 × 10−3/6.21 × 10−4 | 1 |
| Decanoylcarnitine [↑] (+) | 72.0 */70.4 * | 1.73 × 10−3/1.47 × 10−3 | 1 |
| Dodecanoylcarnitine [↑] (+) | 0.9/80.0 * | 7.21 × 10−2/1.01 × 10−2 | 1 |
| Tetradecanoylcarnitine [↑] (+) | 125.7/101.7 * | 1.51 × 10−1/5.78 × 10−3 | 1 |
| Tetradecenoylcarnitine [↑] (+) | 100.8 */61.9 * | 4.70 × 10−2/2.82 × 10−4 | 1 |
| Hexadecanoylcarnitine [↑] (+) | 6.1/9.9 * | 1.99 × 10−1/1.97 × 10−2 | 1 |
| Hexadecenoylcarnitine [↑] (+) | 189.3/130.2 * | 1.16 × 10−1/5.25 × 10−3 | 1 |
| Octadecanoylcarnitine [↑] (+) | 4.2/4.5 | 1.80 × 10−1/1.43 × 10−1 | 1 |
| Oleoylcarnitine [↑] (+) | 7.5/11.8 * | 1.87 × 10−1/4.68 × 10−2 | 1 |
| Linoleoylcarnitine [↑] (+) | 13.9/21.5 * | 1.54 × 10−1/1.09 × 10−3 | 1 |
| Homocystinuria (CBS deficiency) (N = 3) | |||
| Cysteinyl-homocysteine [↑] (+/−) | NF | NF | - |
| Homocysteine [↑] (+) | 0.5–1.2 * | 9.93 × 10−2–4.93 × 10−3 | 1 |
| Homocystine [↑] (+/−) | NF | NF | - |
| Methionine + Methionine sulfoxide [↑] (+) | 6.5 *–55.1 * | 5.01 × 10−3–2.71 × 10−4 | 1 |
| Homocysteic acid [↑] (+/−) | NF | NF | - |
| Isovaleric acidemia (N = 1) | |||
| 3-Hydroxyisovaleric acid [↑] (−) | −1.8 * | 8.52 × 10−5 | 1 |
| 4-Hydroxyisovaleric acid [↑] (−) | NF | NF | - |
| Isovalerylcarnitine [↑] (+) | 69.2 * | 1.40 × 10−4 | 1 |
| Isovaleryglycine [↑] (−) | 159.9 * | 8.82 × 10−4 | 1 |
| Long-chain-3-hydroxyacyl CoA dehydrogenase deficiency (N = 2) | |||
| 3-Hydroxyadipic acid [↑] (−) | NF | NF | - |
| 3-Hydroxydecanedioic acid [↑] (−) | NF | NF | - |
| 3-Hydroxytetradecenoylcarnitine [↑] (+) | 11.7 */19.6 * | 8.84 × 10−3/2.56 × 10−3 | 2b |
| 3-Hydroxytetradecanoylcarnitine [↑] (+) | 37.9 */16.9 * | 1.90 × 10−2/3.54 × 10−2 | 2b |
| 3-Hydroxyhexadecenoylcarnitine [↑] (+) | 142.4 */61.6 * | 2.39 × 10−2/2.94 × 10−2 | 2b |
| 3-Hydroxyhexadecanoylcarnitine [↑] (+) | 402.1/67.3 | 8.01 × 10−2/1.62 × 10−1 | 2b |
| 3-Hydroxyoleoylcarnitine [↑] (+) | 1141.8/149.3 | 7.30 × 10−2/1.56 × 10−1 | 2b |
| 3-Hydroxyoctadecanoylcarnitine [↑] (+) | 754.6/35.2 | 9.32 × 10−2/1.93 × 10−1 | 2b |
| Sebacic acid [↑] (−) | 6.1 */2.3 * | 1.92 × 10−2/5.01 × 10−3 | 4 |
| Suberic acid [↑] (−) | 2.2/1.6 * | 2.06 × 10−1/3.44 × 10−2 | 4 |
| Lysinuric protein intolerance (N = 2) | |||
| Arginine [↓] (+) | −1.5 */2.4 * | 6.40 × 10−3/3.04 × 10−6 | 1 |
| Glutamine + Glutamic acid [↑] (+) | 3.2 */6.4 * | 3.46 × 10−4/1.83 × 10−13 | 1 |
| Lysine [↓] (+) | −1.6 */−1.9 * | 2.50 × 10−5/1.38 × 10−5 | 1 |
| Ornithine [↓] (+) | −2.1 */1.1 | 5.87 × 10−1/3.41 × 10−6 | 1 |
| Orotic acid [↑] (−) | 4.0 */NF | 3.27 × 10−2/NF | 4 |
| Malonyl-CoA decarboxylase deficiency (N = 1) | |||
| Malonylcarnitine [↑] (+) | 132.7 * | 9.84 × 10−3 | 2b |
| Malonic acid [↑] (−) | 0.1 | 8.39 × 10−1 | 1 |
| Maple syrup urine disease (N = 2) | |||
| (allo)Isoleucine [↑] (+) | 3.1 */17.7 * | 3.91 × 10−3/4.95 × 10−3 | 1 |
| 2-Hydroxy-3-methylbutyric acid [↑] (−) | NF | NF | - |
| 2-Hydroxy-3-methylvaleric acid [↑] (−) | NF | NF | - |
| 2-Hydroxy-4-methylvaleric acid [↑] (−) | NF | NF | - |
| 2-Keto-3-methylbutyric acid [↑] (−) | NF | NF | - |
| 2-Keto-3-methylvaleric acid [↑] (−) | −1.9 */25.5 * | 2.95 × 10−6/4.52 × 10−2 | 4 |
| 2-Keto-4-methylvaleric acid [↑] (−) | −2.0 */1.4 | 1.45 × 10−6/1.48 × 10−1 | 4 |
| Leucine [↑] (+) | 3.1 */22.1 * | 6.85 × 10−3/4.25 × 10−3 | 1 |
| Valine [↑] (+) | 1.2/5.2 * | 5.61 × 10−2/4.63 × 10−3 | 1 |
| Medium chain acyl-CoA dehydrogenase deficiency (N = 4) | |||
| 5-Hydroxyhexanoic acid [↑] (−) | NF | NF | - |
| 7-Hydroxyoctanoic acid [↑] (−) | NF | NF | - |
| Decenoylcarnitine [↑] (+) | 8.8 *–45.1 * | 4.24 × 10−3–6.42 × 10−5 | 1 |
| Hexanoylcarnitine [↑] (+) | 11.2 *–54.3 * | 2.12 × 10−3–1.11 × 10−5 | 1 |
| Octanoylcarnitine [↑] (+) | 25.5 *–60.7 * | 4.63 × 10−3–1.87 × 10−3 | 1 |
| Decenedioic acid [↑] (−) | NF | NF | - |
| Hexanoic acid/Trans-cyclohexan × 10-1,2-diol [↑] (−) | −0.4–1.9 | 6.69 × 10−1–2.51 × 10−1 | 1 |
| Hexanoylglycine [↑] (−) | 29.3 *–125.4 * | 1.23 × 10−2–6.83 × 10−3 | 2b |
| Octanoic acid [↑] (−) | 26.5 *–61.5 * | 9.37 × 10−3–2.57 × 10−3 | 4 |
| Octanoylglycine [↑] (−) | 169.5 *–1281.0 | 9.76 × 10−3–2.02 × 10−3 | 2b |
| Phenylpropionylglycine [↑] (−) | 220.0 *–1629.8 * | 9.89 × 10−3–2.44 × 10−3 | 2b |
| Sebacic acid [↑] (−) | 0.0–−0.3 | 9.94 × 10−1–3.90 × 10−1 | 4 |
| Suberic acid [↑] (−) | −0.1–0.6 * | 8.34 × 10−1–3.44 × 10−2 | 4 |
| Suberylglycine [↑] (−) | 61.3 *–350.5 * | 1.90 × 10−2–3.83 × 10−3 | 2b |
| Undecanoylcarnitine [↑] (+) | −0.6–0.1 | 7.88 × 10−1–9.32 × 10−2 | 4 |
| Heptanoylcarnitine [↑] (+) | 3.1 *–10.7 * | 2.13 × 10−2–6.56 × 10−4 | 4 |
| Nonanoylcarnitine [↑] (+) | 53.7 *–110.5 * | 4.37 × 10−3–8.91 × 10−4 | 4 |
| Methylmalonyl-CoA mutase deficiency (N = 1) | |||
| Propionylcarnitine [↑] (+) | 102.2 * | 1.91 × 10−3 | 1 |
| Methylmalonylcarnitine [↑] (+) | 2201.5 * | 1.51 × 10−2 | 1 |
| Methylcitric acid (1) [↑] (−) | 1204.5 * | 1.12 × 10−3 | 1 |
| Methylcitric acid (2) [↑] (−) | 4.5 * | 3.41 × 10−5 | 1 |
| Methylmalonic acid [↑] (−) | 221.5 * | 2.81 × 10−3 | 1 |
| Mevalonic aciduria (N = 1) | |||
| Mevalonic acid [↑] (−) | 0.9 | 9.00 × 10−2 | 4 |
| Mevalonolactone [↑] (−) | NF | NF | - |
| Organic cation transporter 2 deficiency (N = 1) | |||
| Carnitine [↓] (+) | −2.2 * | 2.39 × 10−3 | 1 |
| Ornithine aminotransferase (N = 1) | |||
| 3-Amino-2-piperidone [↑] (+) | 29.5 * | 7.96 × 10−4 | 4 |
| Guanidinoacetic acid [↓] (+) | −1.2 * | 2.60 × 10−4 | 1 |
| Ornithine [↑] (+) | 31.7 * | 2.51 × 10−3 | 1 |
| Ornithine transcarbamylase deficiency (N = 2) | |||
| Citrulline [↓] (+) | 1.8 */1.8 * | 2.09 × 10−3/5.18 × 10−6 | 1 |
| Glutamine + Glutamic acid [↑] (+) | 1.5 * | 7.07 × 10−5 | 1 |
| Orotic acid [↑] (−) | 0.2/NF | 0.394/NF | 4 |
| Uridine [↑] (−) | 7.3 */4.8 * | 2.36 × 10−6/7.07 × 10−3 | 1 |
| Phenylketonuria (N = 3) | |||
| 2-Hydroxyphenylacetic acid [↑] (−) | NF | NF | 1 |
| Glutamylphenylalanine [↑] (+) | 64.3 */31.5 */NF | 1.30 × 10−3/1.44 × 10−2/NF | 1 |
| N-Acetylphenylalanine [↑] (−) | 42.7 *–136.2 * | 3.33 × 10−2–1.35 × 10−3 | 1 |
| N-Lactoyl-phenylalanine [↑] (−) | 39.9 */41.6 * | 2.77 × 10−2–9.96 × 10−3 | 1 |
| Phenylacetic acid [↑] (−) | 1.7 *–6.0 * | 1.01 × 10−2–3.03 × 10−5 | 1 |
| Phenylalanine [↑] (+) | 29.0 *–88.1 * | 9.42 × 10−3–<1.1 × 10−16 | 1 |
| Phenylalanylphenylalanine [↑] (+) | 10.3 */33.3 */NF | 1.10 × 10−2/1.21 × 10−2 /NF | 1 |
| Phenyllactic acid [↑] (−) | 71.8 */125.4 */NF | 3.33 × 10−3/1.04 × 10−2/NF | 1 |
| Phenylpyruvic acid [↑] (−) | −0.8 */−0.1/NF | 8.01 × 10−3/7.57 × 10−1/NF | 1 |
| alpha-N-Phenylacetylglutamine [↑] (−) | 1.4 *–4.2 * | 3.64 × 10−4–3.20 × 10−6 | 1 |
| Propionic acidemia (N = 1) | |||
| 2-Methyl-3-hydroxybutyric acid [↑] (−) | NF | NF | - |
| 3-Hydroxypropionic acid [↑] (−) | NF | NF | - |
| 3-Hydroxyvaleric acid [↑] (−) | NF | NF | - |
| 3-Ketovaleric acid [↑] (−) | NF | NF | - |
| Propionylcarnitine [↑] (+) | 137.4 * | 1.13 × 10−4 | 1 |
| Glycine [↑] (+) | 14.5 * | 5.46 × 10−3 | 1 |
| Methylcitric acid (1) [↑] (−) | 1001.9 * | 6.26 × 10−3 | 1 |
| Methylcitric acid (2) [↑] (−) | 576.1 * | 2.84 × 10−3 | 1 |
| Propionylglycine [↑] (−) | 950.7 * | 1.27 × 10−3 | 2b |
| Tiglylglycine [↑] (−) | NF e | NF | - |
| Thymidine phosphorylase deficiency (N = 1) | |||
| Deoxyuridine [↑] (−) | 370.9 * | 6.97 × 10−4 | 1 |
| Thymidine [↑] (−) | 64.6 * | 2.20 × 10−3 | 1 |
| Thymine [↑] (−) | NF | NF | - |
| Uracil [↑] (−) | −0.9 | 1.12 × 10−1 | 1 |
| Tyrosinemia I (N = 2) | |||
| 4-Hydroxyphenylacetic acid [↑] (−) | 140.8 */NF | 6.53 × 10−3/NF | 1 |
| 4-Hydroxyphenyllactic acid [↑] (−) | 389.5 */1013.8 * | 6.02 × 10−3/3.39 × 10−3 | 1 |
| 4-Hydroxyphenylpyruvic acid [↑] (−) | 4.2 */0.6 | 7.29 × 10−7/5.69 × 10−1 | 1 |
| N-Acetyltyrosine [↑] (−) | 32.4 */96.0 * | 9.55 × 10−3/5.73 × 10−3 | 1 |
| Phenylpyruvic acid [↑] (+) | 113.8 */NF | 4.09 × 10−3/NF | 1 |
| Succinylacetone [↑] (−) | NF | NF | - |
| Tyrosine [↑] (+) | 18.8 */36.9 * | 4.82 × 10−3/1.88 × 10−4 | 1 |
| Very long chain acyl-CoA dehydrogenase deficiency (N = 1) | |||
| Tetradecanoylcarnitine [↑] (+) | 36.6 * | 6.31 × 10−3 | 2b |
| Tetradecenoylcarnitine [↑] (+) | 85.5 * | 1.97 × 10−2 | 2b |
| Tetradecadienoylcartinine [↑] (+) | 42.9 * | 1.38 × 10−2 | 2b |
| Hexadecanoylcarnitine [↑] (+) | 6.8 * | 9.69 × 10−3 | 1 |
| Octadecanoylcarnitine [↑] (+) | 2.7 * | 1.06 × 10−2 | 1 |
| Oleoylcarnitine [↑] (+) | 5.9 * | 4.26 × 10−5 | 2b |
| Carnitine palmitoyltransferase II (N = 2) | |||
| Hexadecanoylcarnitine [↑] (+) | 9.6 */21.6 * | 9.01 × 10−3/2.28 × 10−2 | 1 |
| Octadecanoylcarnitine [↑] (+) | 12.6 */33.4 * | 1.22 × 10−2/2.70 × 10−2 | 1 |
| Oleoylcarnitine [↑] (+) | 7.2 */21.4 * | 4.10 × 10−3/4.06 × 10−2 | 2b |
| Sebacic acid [↑] (−) | 0.2/−0.3 | 7.59 × 10−1/4.69 × 10−1 | 4 |
| Suberic acid [↑] (−) | 0.7/−0.1 | 3.78 × 10−1/7.82 × 10−1 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonte, R.; Bongaerts, M.; Demirdas, S.; Langendonk, J.G.; Huidekoper, H.H.; Williams, M.; Onkenhout, W.; Jacobs, E.H.; Blom, H.J.; Ruijter, G.J.G. Untargeted Metabolomics-Based Screening Method for Inborn Errors of Metabolism using Semi-Automatic Sample Preparation with an UHPLC- Orbitrap-MS Platform. Metabolites 2019, 9, 289. https://doi.org/10.3390/metabo9120289
Bonte R, Bongaerts M, Demirdas S, Langendonk JG, Huidekoper HH, Williams M, Onkenhout W, Jacobs EH, Blom HJ, Ruijter GJG. Untargeted Metabolomics-Based Screening Method for Inborn Errors of Metabolism using Semi-Automatic Sample Preparation with an UHPLC- Orbitrap-MS Platform. Metabolites. 2019; 9(12):289. https://doi.org/10.3390/metabo9120289
Chicago/Turabian StyleBonte, Ramon, Michiel Bongaerts, Serwet Demirdas, Janneke G. Langendonk, Hidde H. Huidekoper, Monique Williams, Willem Onkenhout, Edwin H. Jacobs, Henk J. Blom, and George J. G. Ruijter. 2019. "Untargeted Metabolomics-Based Screening Method for Inborn Errors of Metabolism using Semi-Automatic Sample Preparation with an UHPLC- Orbitrap-MS Platform" Metabolites 9, no. 12: 289. https://doi.org/10.3390/metabo9120289
APA StyleBonte, R., Bongaerts, M., Demirdas, S., Langendonk, J. G., Huidekoper, H. H., Williams, M., Onkenhout, W., Jacobs, E. H., Blom, H. J., & Ruijter, G. J. G. (2019). Untargeted Metabolomics-Based Screening Method for Inborn Errors of Metabolism using Semi-Automatic Sample Preparation with an UHPLC- Orbitrap-MS Platform. Metabolites, 9(12), 289. https://doi.org/10.3390/metabo9120289