Metabolomic Analyses Reveal Extensive Progenitor Cell Deficiencies in a Mouse Model of Duchenne Muscular Dystrophy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
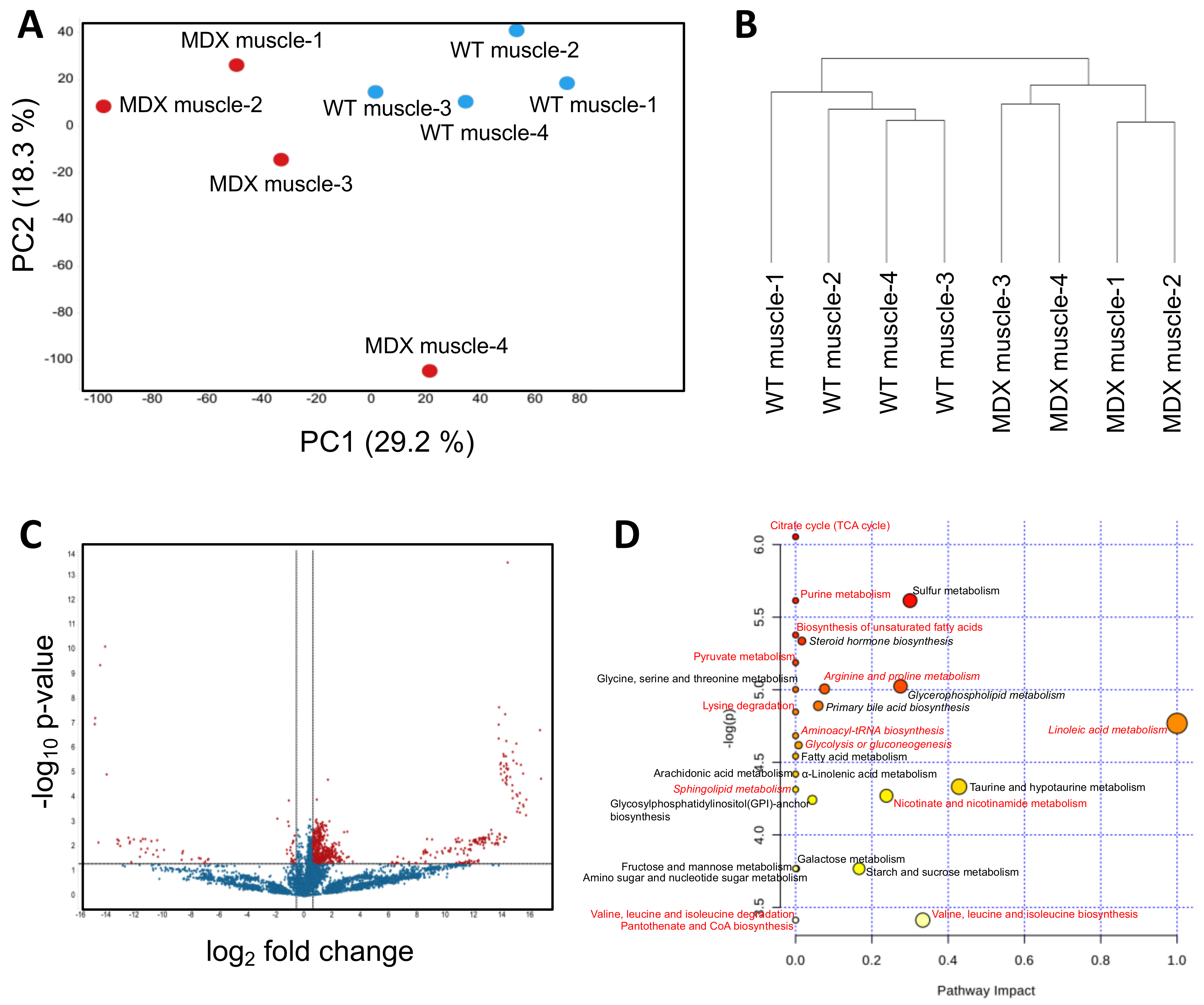
2.1. Satellite Cells (SC) Isolated from Dystrophic Mice Exhibit Metabolite Imbalances
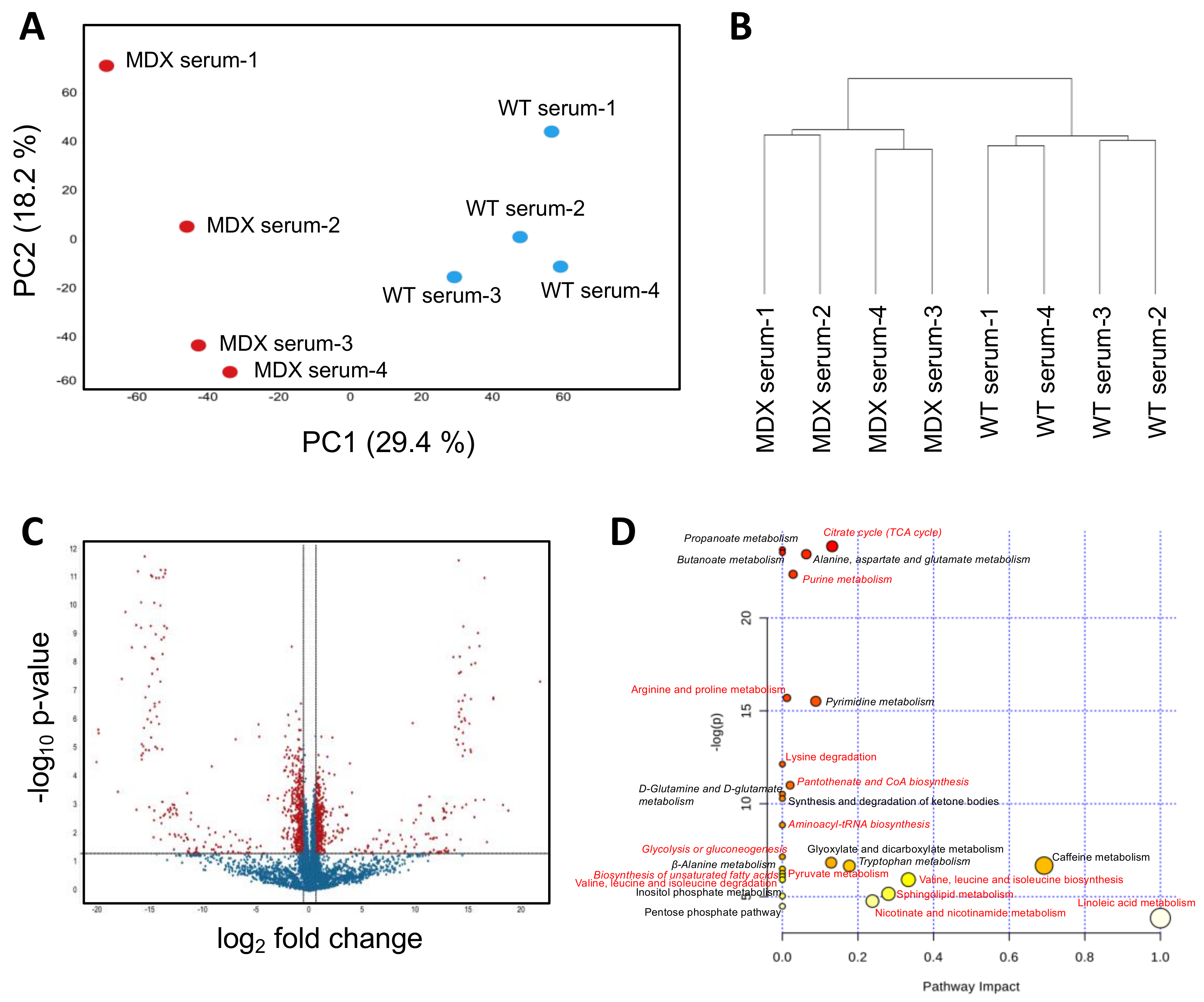
2.2. Dystrophic Mice Have Serum Metabolite Alterations
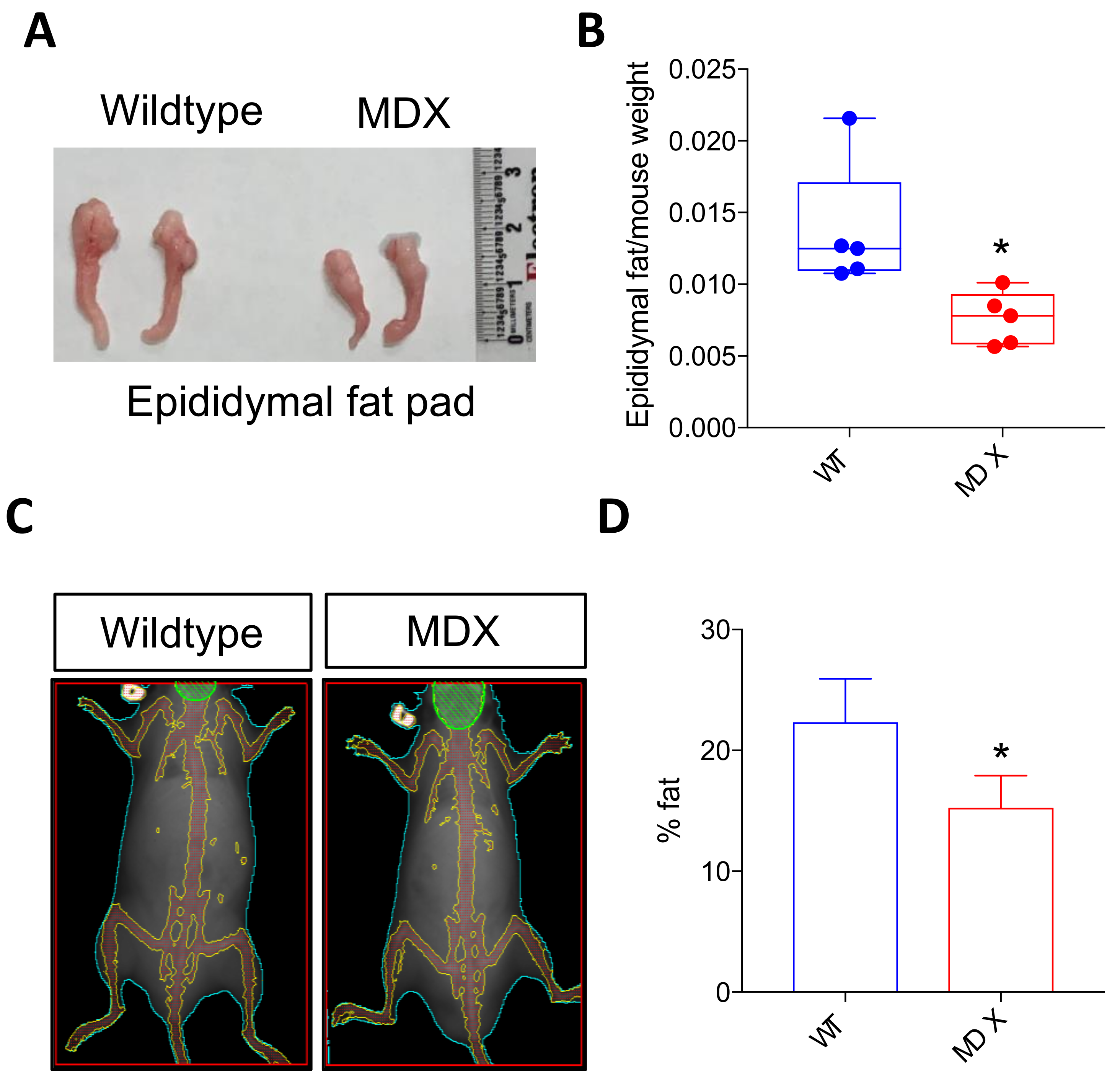
2.3. Dystrophic Mice Exhibit Adipose Tissue Abnormalities
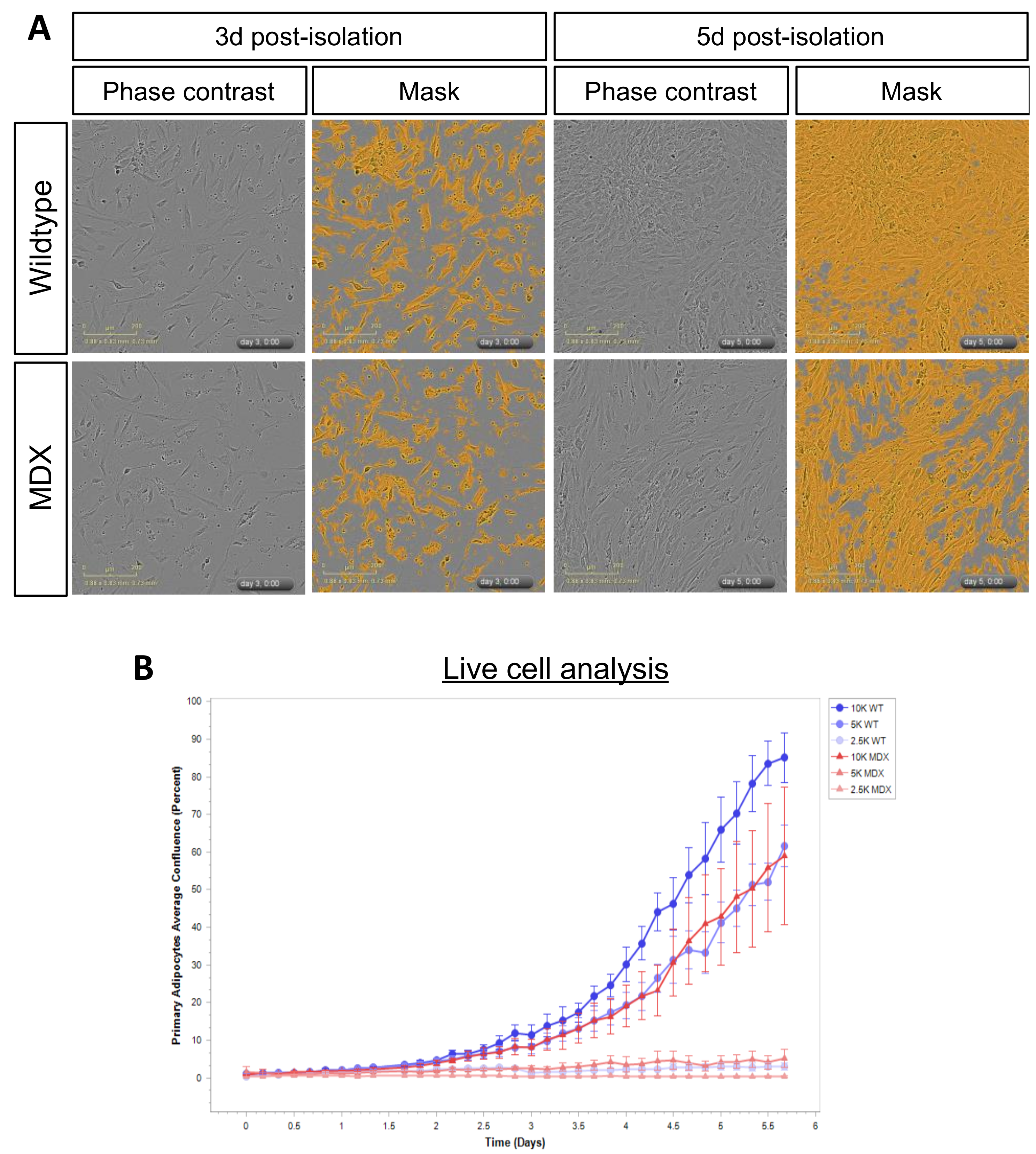
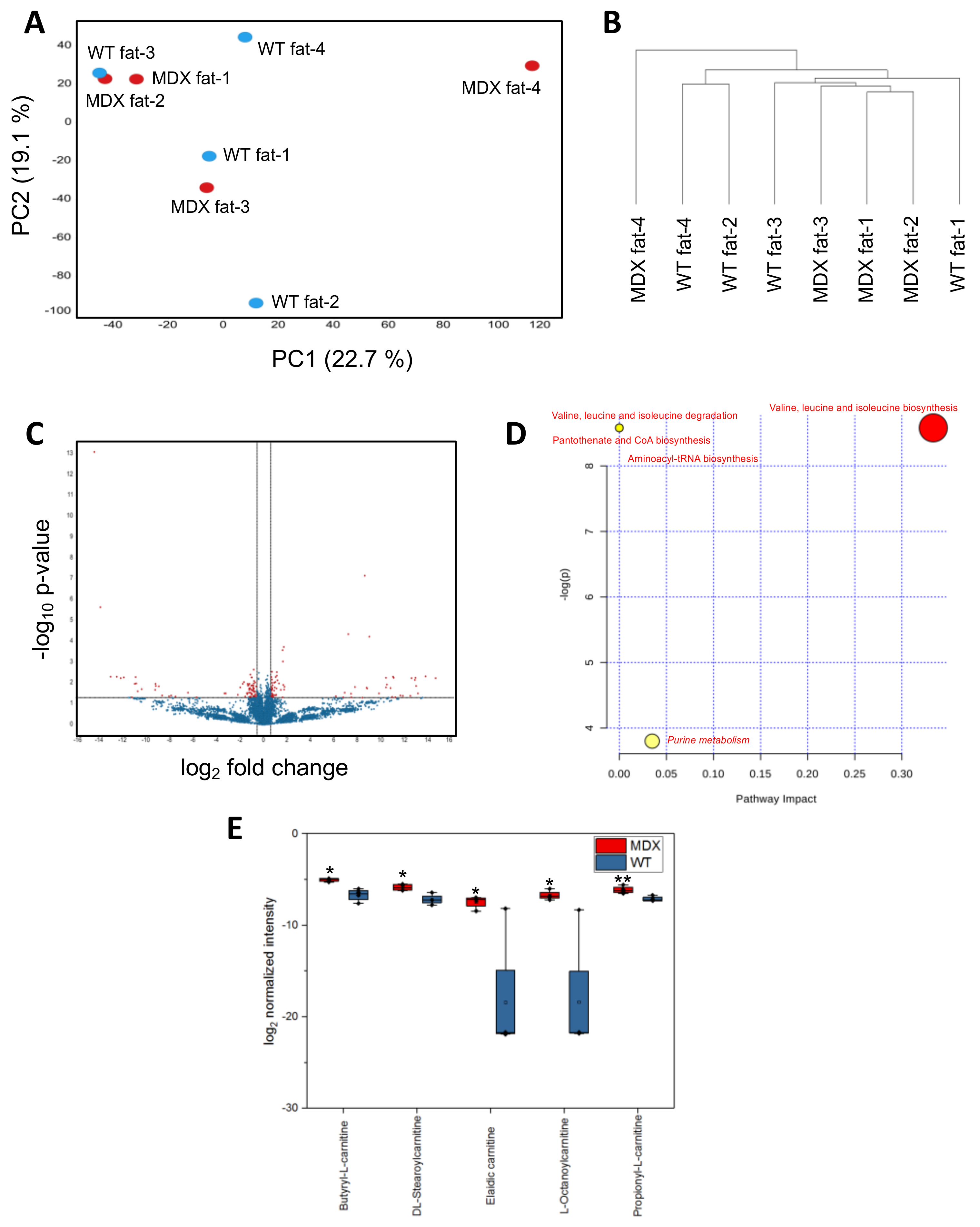
2.4. MDX Adipose Progenitor Cells (APCs) Have Altered Metabolite Profiles
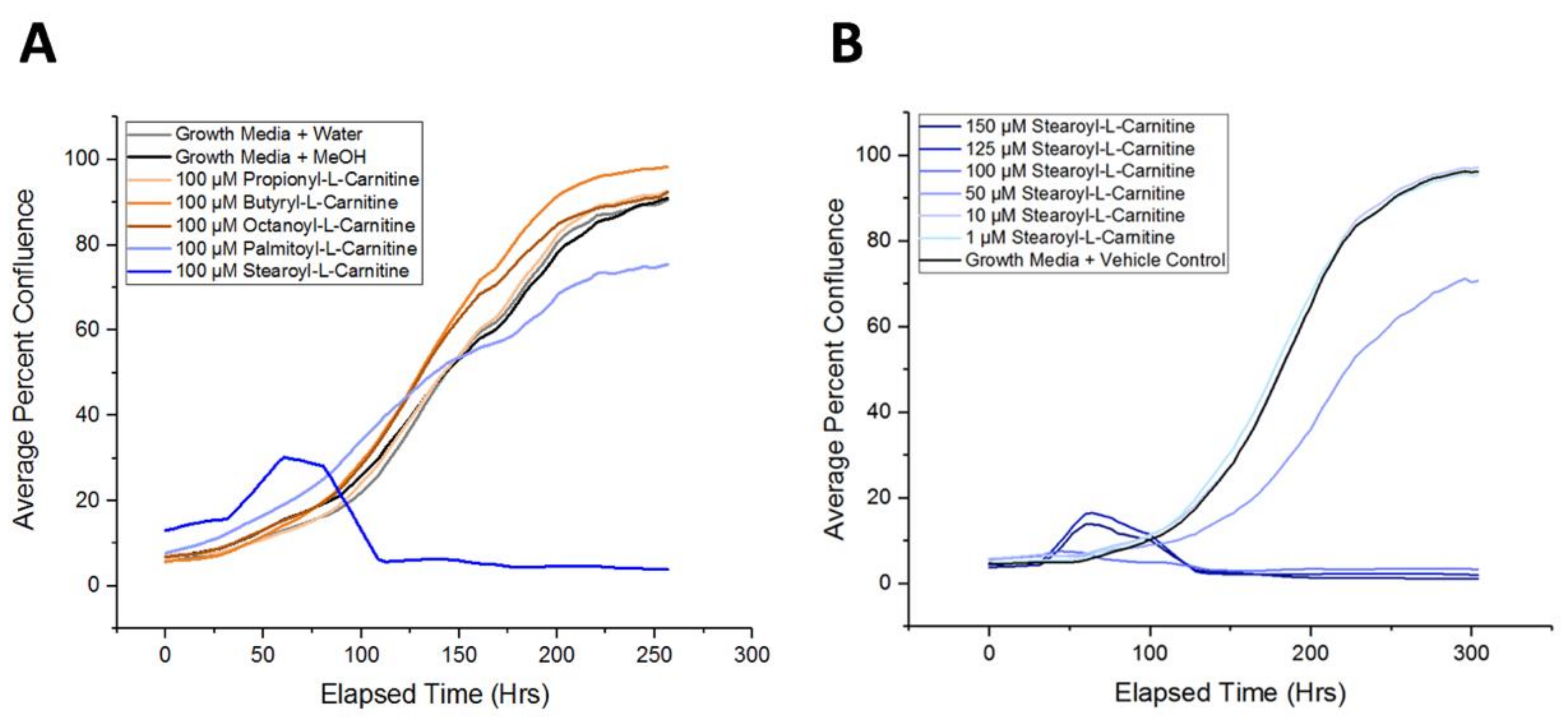
2.5. Long-Chain Acylcarnitines Inhibit APC Expansion
3. Discussion
4. Conclusions
5. Methods
5.1. Satellite Cell Isolation
5.2. Adipose Progenitor Cell Isolation
5.3. Nontargeted Metabolomics
5.4. Statistical Analysis and Metabolic Pathway Analysis
5.5. Animals and Imaging
5.6. Cell Culture and Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nudel, U.; Zuk, D.; Einat, P.; Zeelon, E.; Levy, Z.; Neuman, S.; Yaffe, D. Duchenne muscular dystrophy gene product is not identical in muscle and brain. Nature 1989, 337, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Leikina, E.S.; Plotnikov, N.N.; Prokopenko, L.I. Basic methods of prevention of helminthiases, their improvement and development. Med. Parazitol. (Mosk) 1974, 43, 259–265. [Google Scholar] [PubMed]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, J.G.; Hahn, H.S.; Wong, B.L.; Lorenz, J.N.; Wenisch, A.S.; Levin, L.S. Evolution of the mdx mouse cardiomyopathy: Physiological and morphological findings. Neuromuscul. Disord. 2004, 14, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.S.; Metzger, J.; Reyes, M.; Townsend, D.; Faulkner, J.A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007, 21, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.M.; Teng, H.; Nichol, M.C.; Cunningham, J.C.; Wilkinson, R.S.; Sanes, J.R. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell 1997, 90, 729–738. [Google Scholar] [CrossRef]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef]
- Abdullah, M.; Kornegay, J.N.; Honcoop, A.; Parry, T.L.; Balog-Alvarez, C.J.; O’Neal, S.K.; Bain, J.R.; Muehlbauer, M.J.; Newgard, C.B.; et al. Nontargeted Metabolomics Analysis of Golden Retriever Muscular Dystrophy-Affected Muscles Reveals Alterations in Arginine and Proline Metabolism, and Elevations in Glutamic and Oleic Acid In Vivo. Metabolites 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite Cells in Muscular Dystrophy-Lost in Polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Even, P.C.; Decrouy, A.; Chinet, A. Defective regulation of energy metabolism in mdx-mouse skeletal muscles. Biochem. J. 1994, 304, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Kemp, G.J.; Taylor, D.J.; Dunn, J.F.; Frostick, S.P.; Radda, G.K. Cellular energetics of dystrophic muscle. J. Neurol. Sci. 1993, 116, 201–206. [Google Scholar] [CrossRef]
- Younkin, D.P.; Berman, P.; Sladky, J.; Chee, C.; Bank, W.; Chance, B. 31P NMR studies in Duchenne muscular dystrophy: age-related metabolic changes. Neurology 1987, 37, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.K.; Yadav, R.; Mukherjee, S.; Pal, L.; Sinha, N. Abnormal lipid metabolism in skeletal muscle tissue of patients with muscular dystrophy: In vitro, high-resolution NMR spectroscopy based observation in early phase of the disease. Magn. Reson. Imaging 2017, 38, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, M.; Khairallah, R.; Young, M.E.; Dyck, J.R.; Petrof, B.J.; Des Rosiers, C. Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. J. Mol. Cell Cardiol. 2007, 43, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Tracey, I.; Dunn, J.F.; Radda, G.K. Brain metabolism is abnormal in the mdx model of Duchenne muscular dystrophy. Brain 1996, 119, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Spitali, P.; Hettne, K.; Tsonaka, R.; Sabir, E.; Seyer, A.; Hemerik, J.B.A.; Goeman, J.J.; Picillo, E.; Ergoli, M.; Politano, L.; et al. Cross-sectional serum metabolomic study of multiple forms of muscular dystrophy. J. Cell Mol. Med. 2018, 22, 2442–2448. [Google Scholar] [CrossRef] [PubMed]
- Yablonka-Reuveni, Z.; Anderson, J.E. Satellite cells from dystrophic (mdx) mice display accelerated differentiation in primary cultures and in isolated myofibers. Dev. Dyn. 2006, 235, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Rudnicki, M.A. Targeting muscle stem cell intrinsic defects to treat Duchenne muscular dystrophy. NPJ Regen. Med. 2016, 1, 16006. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Sincennes, M.C.; Chevalier, F.P.; Brun, C.E.; Lacaria, M.; Segalés, J.; Muñoz-Cánoves, P.; Ming, H.; Rudnicki, M.A. The Dystrophin Glycoprotein Complex Regulates the Epigenetic Activation of Muscle Stem Cell Commitment. Cell Stem. Cell 2018, 22, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Wissing, E.R.; Boyer, J.G.; Kwong, J.Q.; Sargent, M.A.; Karch, J.; McNally, E.M.; Otsu, K.; Molkentin, J.D. P38α MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Hum. Mol. Genet. 2014, 23, 5452–5463. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, F.; Guyot, S.; Logerot, M.; Beney, L.; Gervais, P.; Demarquoy, J. Exploration of lipid metabolism in relation with plasma membrane properties of Duchenne muscular dystrophy cells: influence of L-carnitine. PLoS ONE 2012, 7, e49346. [Google Scholar] [CrossRef] [PubMed]
- Aartsma–Rus, A.; Spitali, P. Circulating Biomarkers for Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2015, 2, S49–S58. [Google Scholar] [CrossRef] [PubMed]
- Boca, S.M.; Nishida, M.; Harris, M.; Rao, S.; Cheema, A.K.; Gill, K.; Seol, H.; Morgenroth, L.P.; Henricson, E.; McDonald, C.; et al. Discovery of Metabolic Biomarkers for Duchenne Muscular Dystrophy within a Natural History Study. PLoS ONE 2016, 11, e0153461. [Google Scholar] [CrossRef] [PubMed]
- Martigne, L.; Salleron, J.; Mayer, M.; Cuisset, J.M.; Carpentier, A.; Neve, V.; Tiffreau, V.; Guimber, D.; Gottrand, F. Natural evolution of weight status in Duchenne muscular dystrophy: A retrospective audit. Br. J. Nutr. 2011, 105, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Satomura, S.; Yokota, I.; Tatara, K.; Naito, E.; Ito, M.; Kuroda, Y. Paradoxical weight loss with extra energy expenditure at brown adipose tissue in adolescent patients with Duchenne muscular dystrophy. Metabolism 2001, 50, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Connolly, A.M.; Keeling, R.M.; Mehta, S.; Pestronk, A.; Sanes, J.R. Three mouse models of muscular dystrophy: the natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin alpha2-deficient mice. Neuromuscul. Disord. 2001, 11, 703–712. [Google Scholar] [CrossRef]
- Chi, M.M.; Hintz, C.S.; McKee, D.; Felder, S.; Grant, N.; Kaiser, K.K.; Lowry, O.H. Effect of Duchenne muscular dystrophy on enzymes of energy metabolism in individual muscle fibers. Metabolism 1987, 36, 761–767. [Google Scholar] [CrossRef]
- Nishio, H.; Wada, H.; Matsuo, T.; Horikawa, H.; Takahashi, K.; Nakajima, T.; Matsuo, M.; Nakamura, H. Glucose, free fatty acid and ketone body metabolism in Duchenne muscular dystrophy. Brain Dev. 1990, 12, 390–402. [Google Scholar] [CrossRef]
- Carroll, J.E.; Villadiego, A.; Brooke, M.H. Increased long chain acyl CoA in Duchenne muscular dystrophy. Neurology 1983, 33, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Atri, S.; Sharma, M.C.; Sarkar, C.; Jagannathan, N.R. Skeletal muscle metabolism in Duchenne muscular dystrophy (DMD): An in-vitro proton NMR spectroscopy study. Magn. Reson. Imaging 2003, 21, 145–153. [Google Scholar] [CrossRef]
- Okada, K.; Manabe, S.; Sakamoto, S.; Ohnaka, M.; Niiyama, Y. Protein and energy metabolism in patients with progressive muscular dystrophy. J. Nutr. Sci. Vitaminol. (Tokyo) 1992, 38, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Aguer, C.; McCoin, C.S.; Knotts, T.A.; Thrush, A.B.; Ono-Moore, K.; McPherson, R.; Dent, R.; Hwang, D.H.; Adams, S.H.; Harper, M.E. Acylcarnitines: Potential implications for skeletal muscle insulin resistance. FASEB J. 2015, 29, 336–345. [Google Scholar] [CrossRef] [PubMed]
- McCoin, C.S.; Knotts, T.A.; Ono-Moore, K.D.; Oort, P.J.; Adams, S.H. Long-chain acylcarnitines activate cell stress and myokine release in C2C12 myotubes: calcium-dependent and -independent effects. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E990–E1000. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, D.; Yin, H.; Tong, H.; Li, S.; Yan, Y. Fatty acids promote bovine skeletal muscle satellite cell differentiation by regulating ELOVL3 expression. Cell Tissue Res. 2018, 373, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Wen, Y.; Kuroda, K.; Hannon, K.; Rudnicki, M.A.; Kuang, S. Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Dis. Model. Mech. 2014, 7, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Carmen, L.; Maria, V.; Morales-Medina, J.C.; Vallelunga, A.; Palmieri, B.; Iannitti, T. Role of proteoglycans and glycosaminoglycans in Duchenne muscular dystrophy. Glycobiology 2018. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.S.; Doles, J.D. Single cell transcriptome analysis of muscle satellite cells reveals widespread transcriptional heterogeneity. Gene 2017, 636, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Golde, W.T.; Gollobin, P.; Rodriguez, L.L. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim. (N. Y.) 2005, 34, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Dutta, T.; Chai, H.S.; Ward, L.E.; Ghosh, A.; Persson, X.M.; Ford, G.C.; Kudva, Y.C.; Sun, Z.; Asmann, Y.W.; Kocher, J.P.; et al. Concordance of changes in metabolic pathways based on plasma metabolomics and skeletal muscle transcriptomics in type 1 diabetes. Diabetes 2012, 61, 1004–1016. [Google Scholar] [CrossRef] [PubMed]
- Dutta, T.; Kudva, Y.C.; Persson, X.M.; Schenck, L.A.; Ford, G.C.; Singh, R.J.; Carter, R.; Nair, K.S. Impact of Long-Term Poor and Good Glycemic Control on Metabolomics Alterations in Type 1 Diabetic People. J. Clin. Endocrinol. Metab. 2016, 101, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Dutta, T.; Persson, X.M.; Mielke, M.M.; Petersen, R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS ONE 2013, 8, e63644. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]
- Chapman, V.M.; Miller, D.R.; Armstrong, D.; Caskey, C.T. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. USA 1989, 86, 1292–1296. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, J.; Cho, D.S.; Doles, J.D. Metabolomic Analyses Reveal Extensive Progenitor Cell Deficiencies in a Mouse Model of Duchenne Muscular Dystrophy. Metabolites 2018, 8, 61. https://doi.org/10.3390/metabo8040061
Joseph J, Cho DS, Doles JD. Metabolomic Analyses Reveal Extensive Progenitor Cell Deficiencies in a Mouse Model of Duchenne Muscular Dystrophy. Metabolites. 2018; 8(4):61. https://doi.org/10.3390/metabo8040061
Chicago/Turabian StyleJoseph, Josiane, Dong Seong Cho, and Jason D. Doles. 2018. "Metabolomic Analyses Reveal Extensive Progenitor Cell Deficiencies in a Mouse Model of Duchenne Muscular Dystrophy" Metabolites 8, no. 4: 61. https://doi.org/10.3390/metabo8040061
APA StyleJoseph, J., Cho, D. S., & Doles, J. D. (2018). Metabolomic Analyses Reveal Extensive Progenitor Cell Deficiencies in a Mouse Model of Duchenne Muscular Dystrophy. Metabolites, 8(4), 61. https://doi.org/10.3390/metabo8040061