Validation and Automation of a High-Throughput Multitargeted Method for Semiquantification of Endogenous Metabolites from Different Biological Matrices Using Tandem Mass Spectrometry

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Metabolite Extraction Protocol and Instrumentation
2.3. Method Validation
2.3.1. Selectivity and Specificity
2.3.2. Linearity, Accuracy, and Precision
2.3.3. Recovery and Matrix Effect
2.3.4. Stability of the Metabolites
2.3.5. Carryover
2.3.6. QC Samples
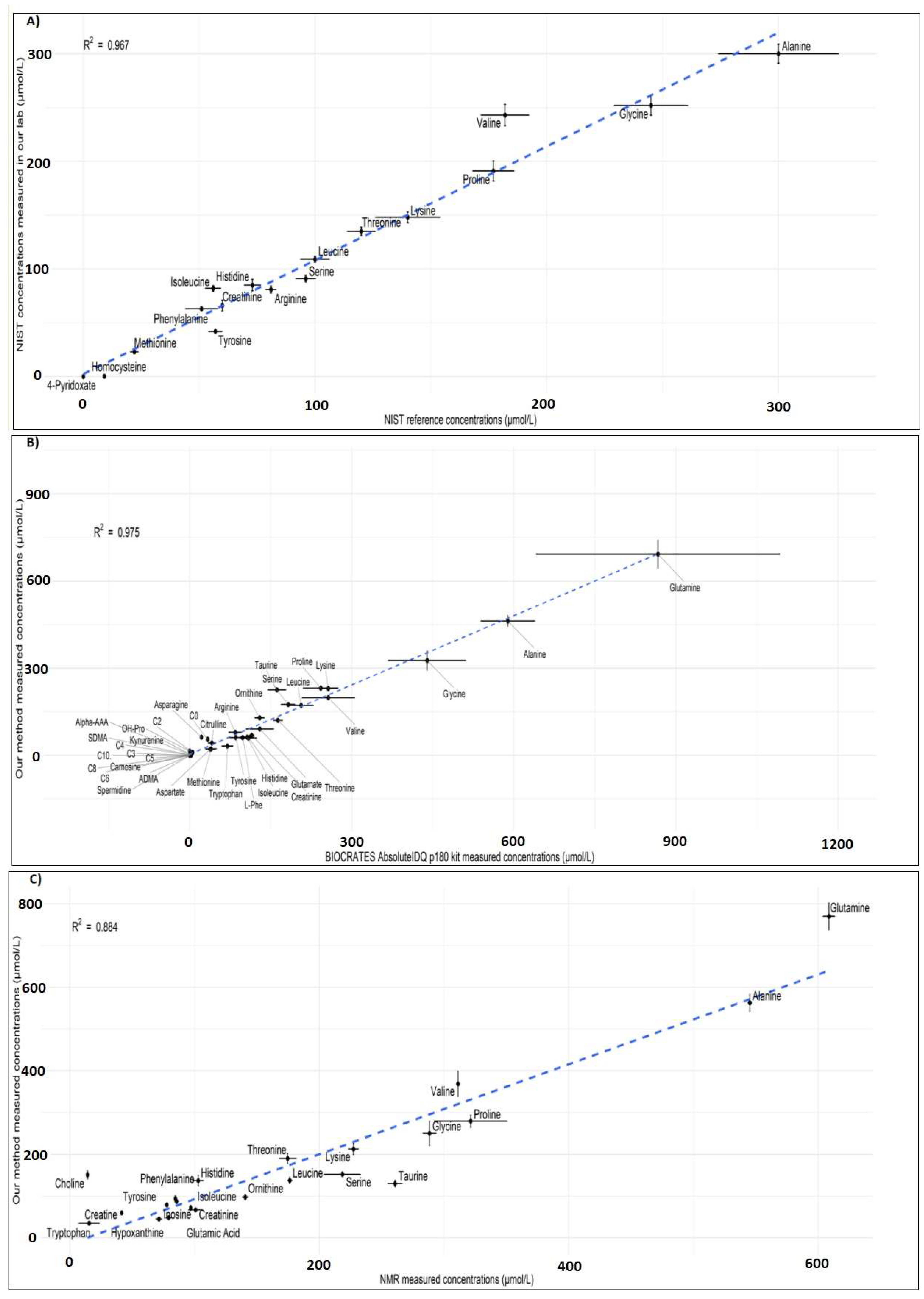
2.3.7. Comparison with Reference Material
2.3.8. Cross-Platform Comparison
2.4. Statistical Analyses
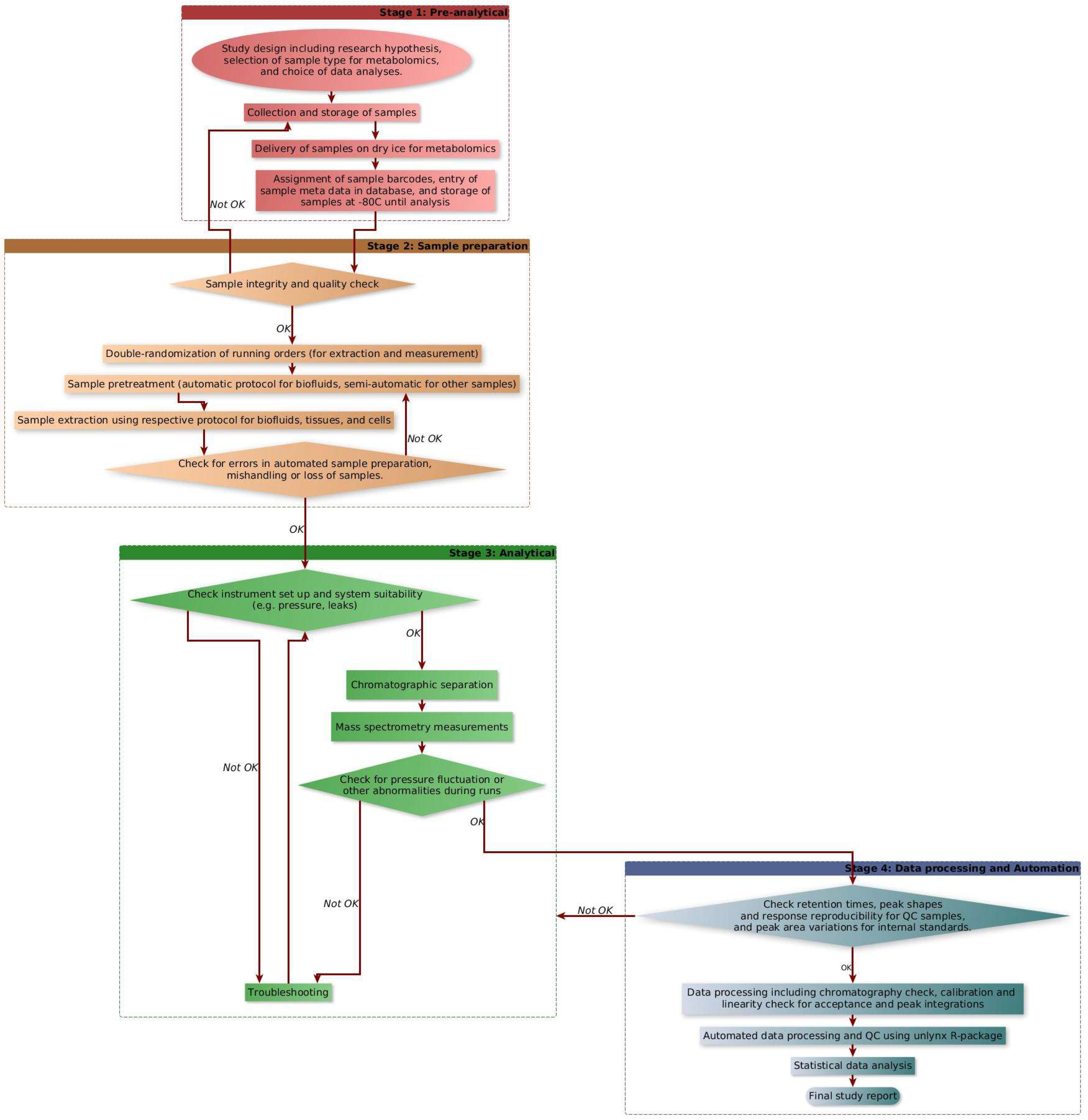
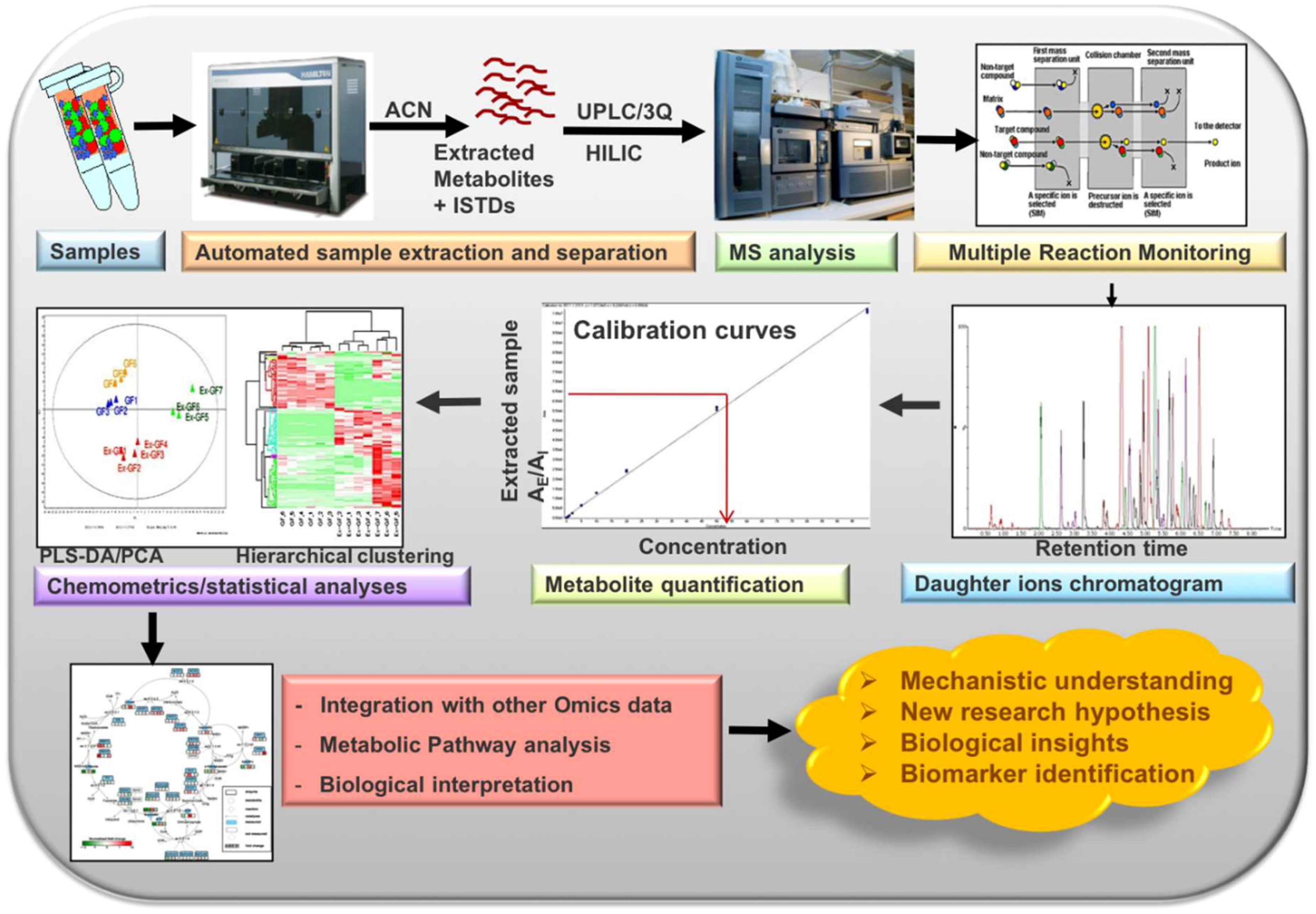
2.5. Automated Data Processing
- (i)
- Molecular weight normalization, in which the ppb values are normalized by the molecular weight of each compound, thereby converting the data from ppb units to µmoles.
- (ii)
- Process efficiency correction for the semiquantification of metabolites without internal standards.
- (iii)
- Normalization using dilution factor for specific sample type if dilution was needed.
- (iv)
- Cell number normalization (for cell samples) to convert the concentration values per million cells.
- (v)
- Calculation of mean, standard deviation, and relative standard deviation (RSD) of molecular concentrations (resulting from the previous steps) for each phenotypic group.
- (vi)
- Outlier detection in each phenotypic group; if the concentration value of a compound in a sample is more than one or two standard deviations (SD) away from the mean of the phenotypic group, then it is marked as an outlier in the Excel data set in two different colors.
- (vii)
- QC check by comparing the RSD of QC samples in the current dataset against the internal database of QC sample RSDs (based on interday RSDs recorded over one year).
3. Results and Discussions
3.1. Extraction Method Optimization
3.2. Method Validation
3.2.1. Selectivity and Specificity
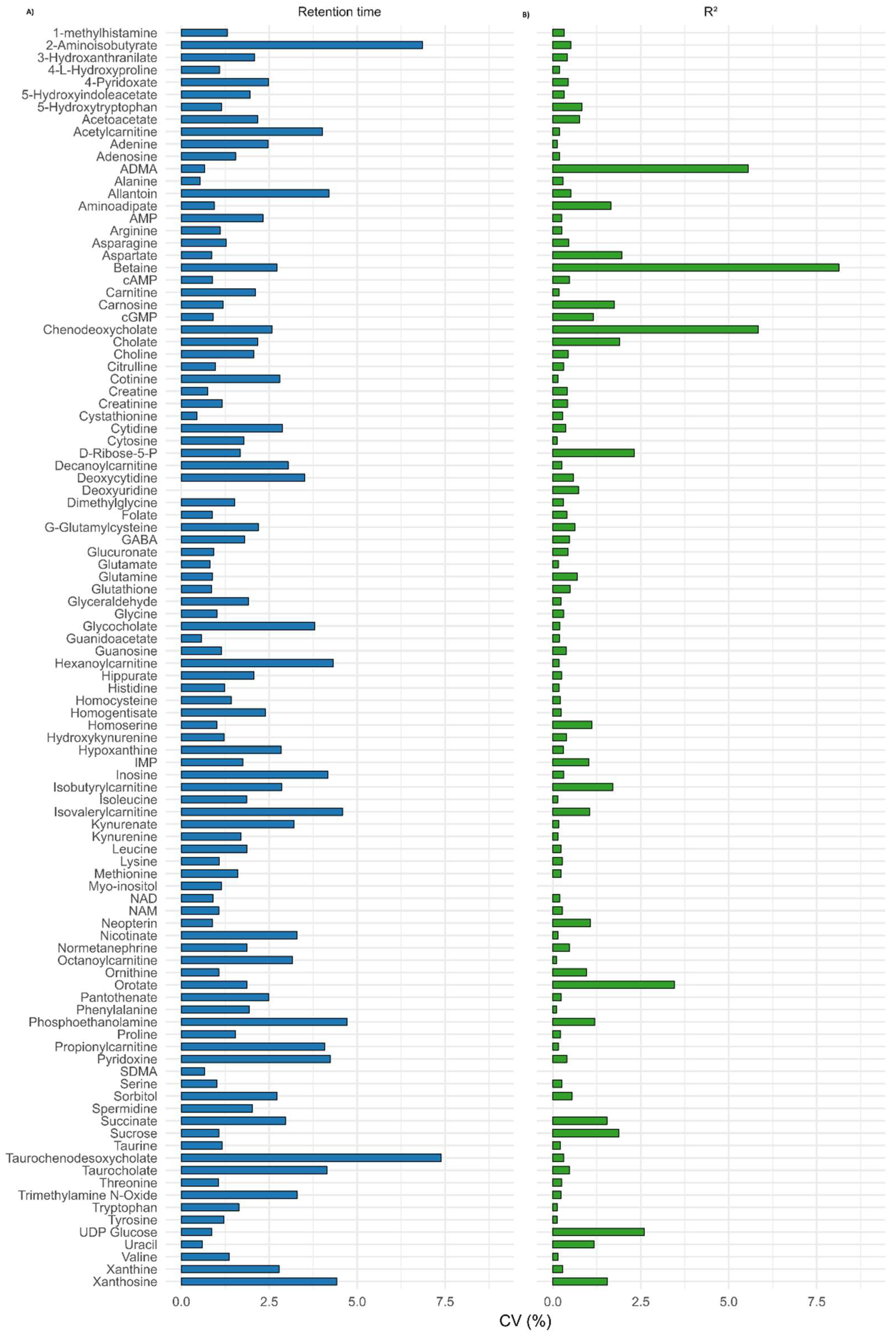
3.2.2. Linearity, Accuracy, and Precision
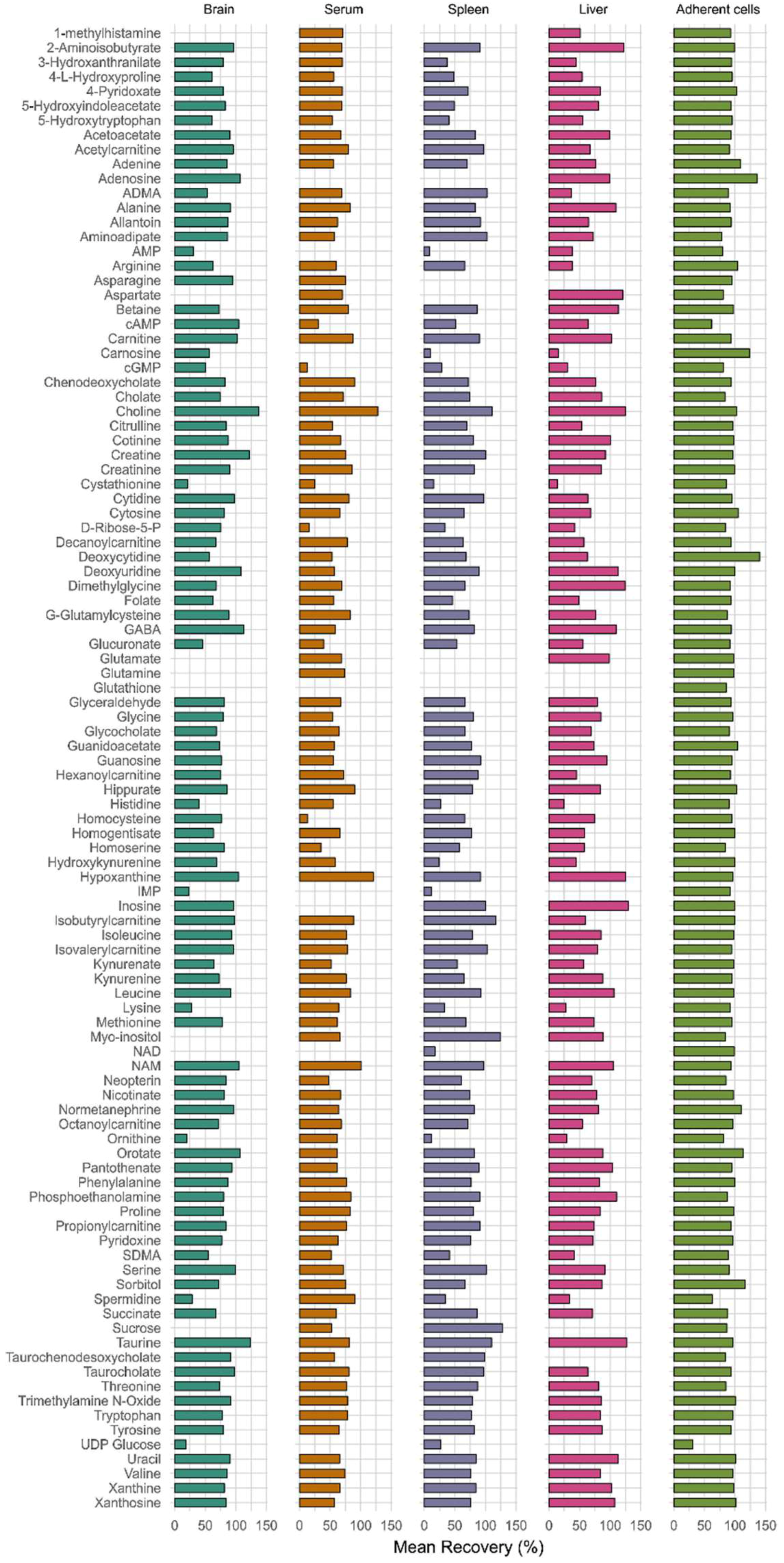
3.2.3. Recovery and Matrix Effect
3.2.4. Stability
3.2.5. Carryover
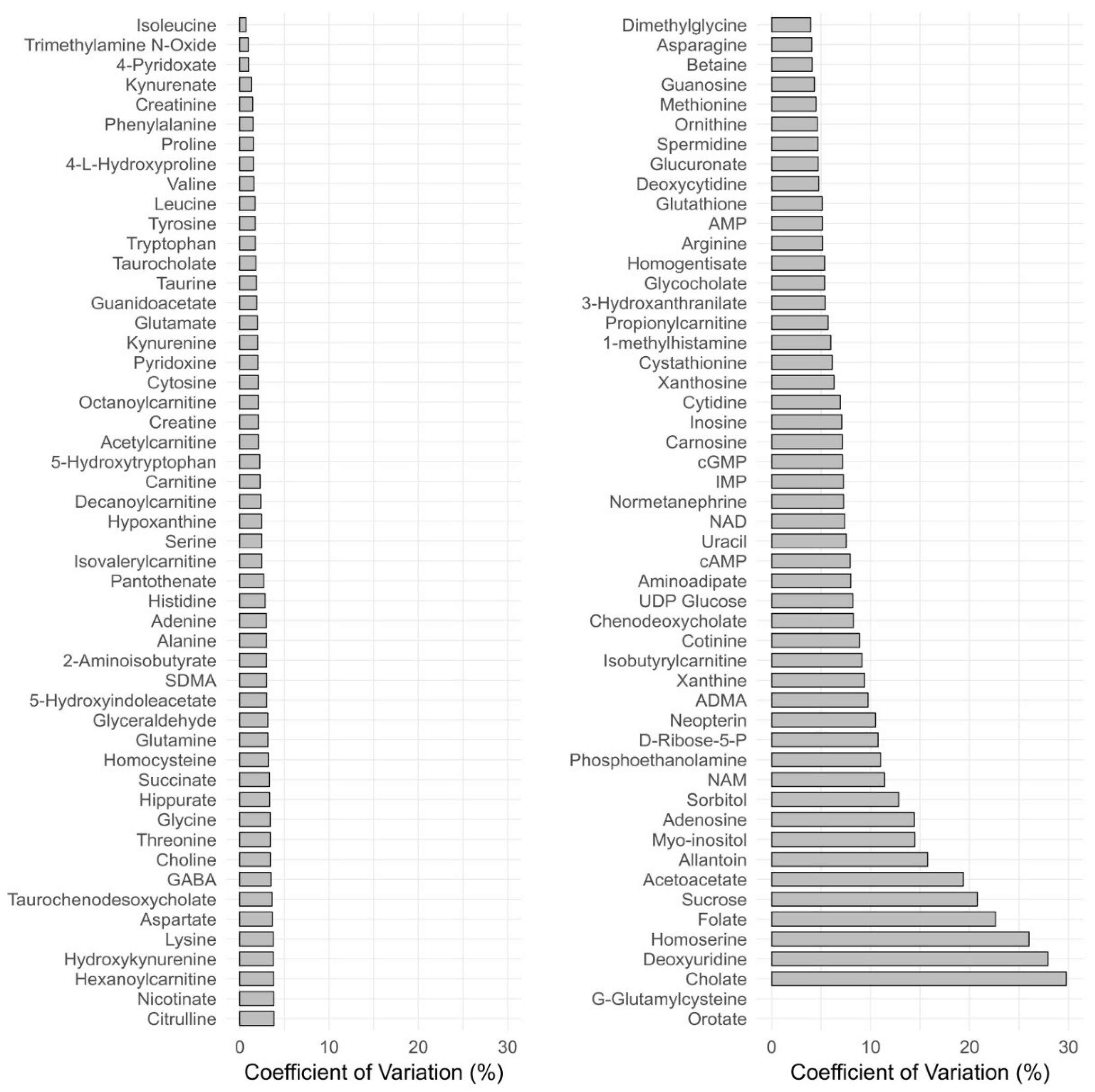
3.2.6. Reproducibility
3.2.7. Quality Management
3.2.8. Robustness and Cross-Platform Comparison
3.3. Automated Data Processing
3.4. Applicability of the Method
4. Conclusions
- (i)
- Optimization: well-characterized protocols for various biological matrices from different organisms enabled to study wide variety of research projects.
- (ii)
- Accuracy/Precision: the targeted and semiquantitative analysis using 102 external 11-point calibration curves, including 12 labeled internal standards in every analysis, made it possible to compare the data within and between the studies.
- (iii)
- Quality management: standard operating protocols, good laboratory practices, strict quality-management system, and proper documentation using electronic laboratory notebook enabled to check/retrieve very old data.
- (iv)
- High-throughput: automated sample preparation and short analysis time (17.5 min), enabled high-throughput capabilities, which is the most desired feature for large-scale analyses.
- (v)
- Stability: long-term stability studies in stock and intermediate solutions, wet extract, and freeze-thaw stability studies, critical for projects based on clinical/biobank samples.
- (vi)
- Automation: downstream data processing steps in an automated manner reduces the interbatch variation and human errors, valuable for analyzing population cohorts and in epidemiological studies.
- (vii)
- Reproducibility: low %CV of concentrations, retention times, correlation coefficient of calibration curves for an extended period of time, very important in metabolomics studies when comparing the data produced at different points of time.
- (viii)
- Reliability: as shown by the excellent correlation between metabolite concentrations measured using our method and the NIST SRM plasma reference values, our method serves a standardised and reliable platform for metabolomics studies.
- (ix)
- Robustness: Our results demonstrate an excellent cross-platform comparability with two completely different analytical platforms, a highly desirable criterion in multicenter studies when comparing the data across different laboratories using different instrumentation, protocols and analytical platforms.
- (x)
- Data sharing: the huge QC sample database of healthy adults (N = 539) collected for six years and shared with the scientific community, provide normal reference values such as those provided in the HMDB database.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gowda, G.A.N.; Djukovic, D. Overview of mass spectrometry-based metabolomics: Opportunities and challenges. Methods Mol. Biol. 2014, 1198, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Dudley, E.; Yousef, M.; Wang, Y.; Griffiths, W.J. Targeted metabolomics and mass spectrometry. Adv. Protein Chem. Struct. Biol. 2010, 80, 45–83. [Google Scholar] [CrossRef] [PubMed]
- Raterink, R.-J.; Lindenburg, P.W.; Vreeken, R.J.; Ramautar, R.; Hankemeier, T. Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics. TrAC Trends Anal. Chem. 2014, 61, 157–167. [Google Scholar] [CrossRef]
- Gong, Z.-G.; Hu, J.; Wu, X.; Xu, Y.-J. The recent developments in sample preparation for mass spectrometry-based metabolomics. Crit. Rev. Anal. Chem. 2017, 47, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Abdel Rahman, A.M.; Pawling, J.; Ryczko, M.; Caudy, A.A.; Dennis, J.W. Targeted metabolomics in cultured cells and tissues by mass spectrometry: Method development and validation. Anal. Chim. Acta 2014, 845, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Villas-Bôas, S.G.; Mas, S.; Åkesson, M.; Smedsgaard, J.; Nielsen, J. Mass spectrometry in metabolome analysis. Mass Spectrom. Rev. 2005, 24, 613–646. [Google Scholar] [CrossRef] [PubMed]
- Virgiliou, C.; Sampsonidis, I.; Gika, H.G.; Raikos, N.; Theodoridis, G.A. Development and validation of a hilic-ms/ms multitargeted method for metabolomics applications. Electrophoresis 2015, 36, 2215–2225. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Naviaux, J.C.; Bright, A.T.; Wang, L.; Naviaux, R.K. A robust, single-injection method for targeted, broad-spectrum plasma metabolomics. Metabolomics 2017, 13, 122. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Qi, C.-B.; Zhu, Q.-F.; Yuan, B.-F.; Feng, Y.-Q. Determination of thiol metabolites in human urine by stable isotope labeling in combination with pseudo-targeted mass spectrometry analysis. Sci. Rep. 2016, 6, 21433. [Google Scholar] [CrossRef] [PubMed]
- Rosebrock, A.P. Targeted full-scan lc-ms metabolomics: Simultaneous quantitation of knowns and feature discovery provide the best of both worlds. Bioanalysis 2017, 9, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Li, G.; Seymour, A.B. High-throughput and multiplexed lc/ms/mrm method for targeted metabolomics. Anal. Chem. 2010, 82, 5527–5533. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Breitkopf, S.B.; Yang, X.; Asara, J.M. A positive/negative ion–switching, targeted mass spectrometry–based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012, 7, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Zhang, P.; Zhu, J.; Raftery, D. Globally Optimized Targeted Mass Spectrometry: Reliable Metabolomics Analysis with Broad Coverage. Anal. Chem. 2015, 87, 12355–12362. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.D.; Souza, A.L.; Gerszten, R.E.; Clish, C.B. Targeted metabolomics. Curr. Protoc. Mol. Biol. 2012, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Chen, Y.-T.; Chen, C.-L.; Li, L. Development of a universal metabolome-standard method for long-term lc-ms metabolome profiling and its application for bladder cancer urine-metabolite-biomarker discovery. Anal. Chem. 2014, 86, 6540–6547. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsström, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Roman-Garcia, P.; Quiros-Gonzalez, I.; Mottram, L.; Lieben, L.; Sharan, K.; Wangwiwatsin, A.; Tubio, J.; Lewis, K.; Wilkinson, D.; Santhanam, B.; et al. Vitamin b12–dependent taurine synthesis regulates growth and bone mass. J. Clin. Investig. 2014, 124, 2988–3002. [Google Scholar] [CrossRef] [PubMed]
- Nikkanen, J.; Forsström, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamäki, J.; Roivainen, A.; Marjamäki, P.; et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools And Remodel One-Carbon Metabolism. Cell Metab. 2016, 23, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Ahola, S.; Auranen, M.; Isohanni, P.; Niemisalo, S.; Urho, N.; Buzkova, J.; Velagapudi, V.; Lundbom, N.; Hakkarainen, A.; Muurinen, T.; et al. Modified Atkins diet induces subacute selective ragged red fiber lysis in mitochondrial myopathy patients. EMBO Mol. Med. 2016, 11, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, J.; Tomašić, N.; Kotarsky, H.; Hansson, E.; Velagapudi, V.; Kallijärvi, J.; Fellman, V. Effect of high-carbohydrate diet on plasma metabolomics in mice with mitochondrial respiratory chain complex III deficiency due to a Bcs1l mutation. Int. J. Mol. Sci. 2016, 17, 1824. [Google Scholar] [CrossRef] [PubMed]
- Szibor, M.; Dhandapani, P.K.; Dufour, E.; Holmström, K.M.; Zhuang, Y.; Salwig, I.; Wittig, I.; Heidler, J.; Gizatullina, Z.; Gainutdinov, T.; et al. Broad AOX expression in a genetically tractable mouse model does not disturb normal physiology. Dis. Model. Mech. 2017, 2, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Schatton, D.; Pla-Martin, D.; Marx, M.C.; Hansen, H.; Mourier, A.; Nemazanyy, I.; Pessia, A.; Zentis, P.; Corona, T.; Kondylis, V.; et al. CLUH regulates mitochondrial metabolism by controlling translation and decay of target mRNAs. J. Cell Biol. 2017, 216, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Purhonen, J.; Rajendran, J.; Mörgelin, M.; Uusi-Rauva, K.; Katayama, S.; Krjutskov, K.; Einarsdottir, E.; Velagapudi, V.; Kere, J.; Jauhiainen, M.; et al. Ketogenic diet attenuates hepatopathy in mouse model of respiratory chain complex III deficiency caused by a Bcs1l mutation. Sci. Rep. 2017, 7, 957. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Wang, L.; Jackson, C.; Pessia, A.; Riikka, K.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Folate Cycle and the Integrated Stress Response in Mitochondrial Disease. Cell Metab. 2017, 26, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.A.; Quintaneiro, L.M.; Norvaisas, P.; Lui, P.P.; Wilson, M.P.; Leung, K.Y.; Herrera-Dominguez, L.; Sudiwala, S.; Pessia, A.; Clayton, P.T.; et al. Host-Microbe Co-metabolism Dictates Cancer Drug Efficacy in C. elegans. Cell 2017, 169, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Puhka, M.; Takatalo, M.; Nordberg, M.E.; Valkonen, S.; Nandania, J.; Aatonen, M.; Yliperttula, M.; Laitinen, S.; Velgapudi, V.; Mirtti, T.; et al. Metabolomic profiling of extracellular vesicles and alternative normalisation methodsreveal enriched metabolites and strategies to study prostate cancer related changes. Theranostics 2017, 7, 3824–3841. [Google Scholar] [CrossRef] [PubMed]
- Schrade, A.; Kyrönlahti, A.; Akinrinade, O.; Pihlajoki, M.; Häkkinen, M.; Fischer, S.; Alastalo, T.-P.; Velagapudi, V.; Toppari, J.; Wilson, D.B.; et al. GATA4 is a key regulator of steroidogenesis and glycolysis in mouse Leydig cells. Endocrinology 2015, 156, 1860–1872. [Google Scholar] [CrossRef] [PubMed]
- Schrade, A.; Kyrönlahti, A.; Akinrinade, O.; Pihlajoki, M.; Fischer, S.; Rodriguez, V.M.; Velagapudi, V.; Toppari, J.; Wilson, D.B.; Heikinheimo, M. GATA4 regulates blood-testis barrier function and lactate metabolism in mouse Sertoli cells. Endocrinology 2016, 157, 2416–2431. [Google Scholar] [CrossRef] [PubMed]
- Ali-Sisto, T.; Tolmunen, T.; Toffol, E.; Viinamäki, H.; Mäntyselkä, P.; Valkonen-Korhonen, M.; Honkalampi, K.; Ruusunen, A.; Velagapudi, V.; Lehto, S.M. Purine metabolism is dysregulated in patients with major depressive disorder. Psychoneuroendocrinology 2016, 70, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kolho, K.-L.; Pessia, A.; Jaakkola, T.; de Vos, W.M.; Velagapudi, V. Fecal and serum metabolomics in pediatric inflammatory bowel disease. J. Crohn’s Colitis 2017, 11, 321–334. [Google Scholar] [CrossRef]
- Fu, Y.; Gaelings, L.; Söderholm, S.; Belanov, S.; Nandania, J.; Nyman, T.A.; Matikainen, S.; Anders, S.; Velagapudi, V.; Kainov, D.E. NJ872 inhibits influenza A virus replication without altering cellular antiviral responses. Antiviral Res. 2016, 133, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Gaelings, L.; Söderholm, S.; Bugai, A.; Fu, Y.; Nandania, J.; Schepens, B.; Lorey, M.B.; Tynell, J.; Ginste, L.V.; Goffic, R.L.; et al. Regulation of Kynurenine Biosynthesis during Influenza Virus Infection. FEBS J. 2017, 284, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Kuivanen, S.; Bespalov, M.M.; Nandania, J.; Ianevski, A.; Velagapudi, V.; De Brabander, J.K.; Kainov, D.E.; Vapalahti, O. Obatoclax, saliphenylhalamide and gemcitabine inhibit Zika virus infection in vitro and differentially affect cellular signaling, transcription and metabolism. Antiviral Res. 2017, 139, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Bulanova, D.; Ianevski, A.; Bugai, A.; Akimov, Y.; Kuivanen, S.; Paavilainen, H.; Kakkola, L.; Nandania, J.; Turunen, L.; Ohman, T.; et al. Antiviral Properties of Chemical Inhibitors of Cellular Anti-Apoptotic Bcl-2 Proteins. Viruses 2017, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Zinkevičienė, A.; Kainov, D.; Girkontaitė, I.; Lastauskienė, E.; Kvedarienė, V.; Fu, Y.; Anders, S.; Velagapudi, V. Activation of Tryptophan and Phenylalanine Catabolism in the Remission Phase of Allergic Contact Dermatitis: A Pilot Study. Int. Arch. Allergy Immunol. 2016, 170, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.M.; Fogarty, C.L.; Katayama, S.; Vihinen, H.; Keskitalo, S.; Ilander, M.; Krjutškov, K.; Mustjoki, S.; Lehto, M.; Hautala, T.; et al. Combined immunodeficiency with hypoglycemia caused by mutations in hypoxia up-regulated 1. J. Allergy Clin. Immunol. 2017, 139, 1391–1393. [Google Scholar] [CrossRef] [PubMed]
- Rey, G.; Valekunja, U.K.; Feeney, K.A.; Wulund, L.; Milev, N.B.; Stangherlin, A.; Bollepalli, L.; Velagapudi, V.; O’Neill, J.S.; Reddy, A.B. The Pentose Phosphate Pathway Regulates the Circadian Clock. Cell Metab. 2016, 24, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Mäntyselkä, P.; Ali-Sisto, T.; Kautiainen, H.; Savolainen, J.; Niskanen, L.; Viinamäki, H.; Velagapudi, V.; Lehto, S.M. The association between musculoskeletal pain and circulating ornithine: A population-based study. Pain Med. 2017, 18, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on Bioanalytical Method Validation; European Medicines Agency: London, UK, 2011. [Google Scholar]
- Yamil, S.-M.; Mark, S.L.; Lisa, E.K.; Maureen, L.S.; Kelly, H.T.; Paul, A.R.; William, G.M.; Daniel, W.B.; Tracey, B.S.; Dmitrii, V.T.; et al. Metabolite Profiling of a NIST Standard Reference Material for Human Plasma (SRM 1950): GC/MS, LC/MS, NMR and Clinical Laboratory Analyses, Libraries and Web-based resources. Anal. Chem. 2013, 24, 11725–11731. [Google Scholar] [CrossRef]
- Siskos, A.P.; Jain, P.; Römisch-Margl, W.; Bennett, M.; Achaintre, D.; Asad, Y.; Marney, L.; Richardson, L.; Koulman, A.; Griffin, J.L.; et al. Interlaboratory Reproducibility of a Targeted Metabolomics Platform for Analysis of Human Serum and Plasma. Anal. Chem. 2017, 89, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Mihaleva, V.V.; Korhonen, S.P.; van Duynhoven, J.; Niemitz, M.; Vervoort, J.; Jacobs, D.M. Automated quantum mechanical total line shape fitting model for quantitative NMR-based profiling of human serum metabolites. Anal. Bioanal. Chem. 2014, 406, 3091–3102. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.D. Mcmc methods for multi-response generalized linear mixed models: The mcmcglmm R package. J. Stat. Softw. 2010, 33, 22. [Google Scholar] [CrossRef]
- Singtoroj, T.; Tarning, J.; Annerberg, A.; Ashton, M.; Bergqvist, Y.; White, N.J.; Lindegardh, N.; Day, N.P. A new approach to evaluate regression models during validation of bioanalytical assays. J. Pharm. Biomed. Anal. 2006, 41, 219–227. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Median (µmol/L) | ||||
|---|---|---|---|---|
| 95% Credibility Interval | ||||
| Class and Metabolite Name | HMDB Id | Estimate | Lower | Upper |
| 1. Alpha Amino Acids and Derivatives | ||||
| 2-Aminoisobutyrate | HMDB0001906 | 1.128 | 0.809 | 1.509 |
| 4-l-Hydroxyproline | HMDB0000725 | 15.895 | 11.300 | 21.184 |
| 5-Hydroxytryptophan | HMDB0000472 | 0.043 | 0.031 | 0.058 |
| ADMA | HMDB0001539 | 0.963 | 0.248 | 2.230 |
| Alanine | HMDB0000161 | 477.946 | 339.667 | 635.383 |
| Aminoadipate | HMDB0000510 | 2.121 | 1.502 | 2.818 |
| Arginine | HMDB0000517 | 84.902 | 60.795 | 113.172 |
| Asparagine | HMDB0000168 | 47.204 | 33.635 | 62.727 |
| Aspartate | HMDB0000191 | 26.932 | 18.786 | 35.424 |
| Betaine | HMDB0000043 | 100.045 | 72.251 | 134.758 |
| Citrulline | HMDB0000904 | 27.815 | 19.812 | 36.972 |
| Creatine | HMDB0000064 | 61.180 | 44.481 | 82.161 |
| Creatinine | HMDB0000562 | 66.085 | 47.095 | 88.066 |
| Cystathionine | HMDB0000099 | 0.131 | 0.093 | 0.174 |
| Dimethylglycine | HMDB0000092 | 3.541 | 2.521 | 4.733 |
| GABA | HMDB0000112 | 0.195 | 0.138 | 0.259 |
| G-Glutamylcysteine | HMDB0001049 | 2.966 | 2.116 | 3.946 |
| Glutamate | HMDB0000148 | 53.446 | 37.691 | 70.613 |
| Glutamine | HMDB0000641 | 791.142 | 560.999 | 1050.605 |
| Glutathione | HMDB0000125 | 0.021 | 0.015 | 0.028 |
| Glycine | HMDB0000123 | 243.717 | 172.929 | 325.417 |
| Guanidoacetate | HMDB0000128 | 2.635 | 1.880 | 3.498 |
| Histidine | HMDB0000177 | 88.616 | 62.718 | 117.848 |
| Homocysteine | HMDB0000742 | 0.480 | 0.124 | 1.153 |
| Homoserine | HMDB0000719 | 0.337 | 0.240 | 0.448 |
| Hydroxykynurenine | HMDB0000732 | 0.096 | 0.068 | 0.128 |
| Isoleucine | HMDB0000172 | 83.316 | 58.352 | 110.232 |
| Kynurenine | HMDB0000684 | 1.146 | 0.813 | 1.520 |
| Leucine | HMDB0000687 | 126.072 | 90.139 | 167.353 |
| Lysine | HMDB0000182 | 176.519 | 127.474 | 236.393 |
| Methionine | HMDB0000696 | 29.201 | 21.081 | 39.249 |
| Ornithine | HMDB0000214 | 90.177 | 65.043 | 120.796 |
| Phenylalanine | HMDB0000159 | 88.326 | 63.916 | 117.977 |
| Proline | HMDB0000162 | 251.689 | 180.529 | 335.079 |
| SDMA | HMDB0003334 | 2.862 | 2.060 | 3.834 |
| Serine | HMDB0000187 | 148.482 | 107.012 | 198.507 |
| Threonine | HMDB0000167 | 152.013 | 108.810 | 203.651 |
| Tryptophan | HMDB0000929 | 33.961 | 24.104 | 45.261 |
| Tyrosine | HMDB0000158 | 65.558 | 45.778 | 86.262 |
| Valine | HMDB0000883 | 394.932 | 282.460 | 526.776 |
| 2. Benzoic Acids and Derivatives | ||||
| 3-Hydroxanthranilate | HMDB0001476 | 0.188 | 0.134 | 0.251 |
| Hippurate | HMDB0000714 | 6.101 | 4.362 | 8.124 |
| 3. Beta Amino Acids and Derivatives | ||||
| Carnosine | HMDB0000033 | 0.014 | 0.010 | 0.019 |
| Pantothenate | HMDB0000210 | 0.310 | 0.220 | 0.411 |
| 4. Bile Acids, Alcohols and Derivatives | ||||
| Chenodeoxycholate | HMDB0000518 | 53.661 | 38.588 | 71.987 |
| Cholate | HMDB0000619 | 0.676 | 0.483 | 0.900 |
| Glycocholate | HMDB0000138 | 0.373 | 0.267 | 0.498 |
| Taurochenodesoxycholate | HMDB0000951 | 0.507 | 0.359 | 0.673 |
| 5. Carbohydrates and Carbohydrate Conjugates | ||||
| d-Ribose-5-P | HMDB0001548 | 1.273 | 0.919 | 1.710 |
| Glyceraldehyde | HMDB0001051 | 239.946 | 172.630 | 322.782 |
| Sucrose | HMDB0000258 | 1.417 | 1.015 | 1.886 |
| 6. Dialkylamines | ||||
| Spermidine | HMDB0001257 | 33.601 | 23.958 | 44.759 |
| 7. Dicarboxylic Acids and Derivatives | ||||
| Succinate | HMDB0000254 | 7.912 | 5.597 | 10.491 |
| 8. Fatty Acyls | ||||
| Acetylcarnitine | HMDB0000201 | 9.709 | 2.323 | 22.264 |
| Decanoylcarnitine | HMDB0000651 | 0.305 | 0.087 | 0.720 |
| Hexanoylcarnitine | HMDB0000705 | 0.055 | 0.015 | 0.129 |
| Isobutyrylcarnitine | HMDB0000736 | 0.248 | 0.061 | 0.578 |
| Isovalerylcarnitine | HMDB0000688 | 0.102 | 0.027 | 0.240 |
| Octanoylcarnitine | HMDB0000791 | 0.297 | 0.076 | 0.691 |
| Propionylcarnitine | HMDB0000824 | 0.423 | 0.119 | 0.998 |
| 9. Folates | ||||
| Folate | HMDB0000121 | 0.011 | 0.003 | 0.027 |
| 10. Glucuronic Acid and Derivatives | ||||
| Glucuronate | HMDB0000127 | 1.960 | 1.404 | 2.627 |
| 11. Imidazoles | ||||
| 1-Methylhistamine | HMDB0000898 | 0.006 | 0.004 | 0.008 |
| Allantoin | HMDB0000462 | 2.447 | 1.741 | 3.254 |
| 12. Indoles and Derivatives | ||||
| 5-Hydroxyindoleacetate | HMDB0000763 | 0.074 | 0.053 | 0.099 |
| 13. Keto Acids and Derivatives | ||||
| Acetoacetate | HMDB0000060 | 6.713 | 4.798 | 8.988 |
| 14. Organic Phosphoric Acids and Derivatives | ||||
| Phosphoethanolamine | HMDB0000224 | 3.316 | 2.356 | 4.420 |
| 15. Organosulfonic Acids | ||||
| Taurine | HMDB0000251 | 221.359 | 156.971 | 294.936 |
| Taurocholate | HMDB0000036 | 0.083 | 0.060 | 0.112 |
| 16. Oxides | ||||
| Trimethylamine N-oxide | HMDB0000925 | 1.477 | 1.055 | 1.973 |
| 17. Phenols | ||||
| Homogentisate | HMDB0000130 | 0.115 | 0.082 | 0.153 |
| Normetanephrine | HMDB0000819 | 0.0010 | 0.0007 | 0.0014 |
| 18. Pteridines and Derivatives | ||||
| Neopterin | HMDB0000845 | 0.005 | 0.004 | 0.007 |
| 19. Purines and Derivatives | ||||
| Adenine | HMDB0000034 | 0.007 | 0.005 | 0.009 |
| Adenosine | HMDB0000050 | 0.008 | 0.006 | 0.011 |
| AMP | HMDB0000045 | 0.106 | 0.075 | 0.140 |
| cAMP | HMDB0000058 | 0.005 | 0.003 | 0.006 |
| cGMP | HMDB0001314 | 0.009 | 0.002 | 0.026 |
| Guanosine | HMDB0000133 | 0.445 | 0.315 | 0.593 |
| Hypoxanthine | HMDB0000157 | 58.838 | 41.908 | 78.087 |
| IMP | HMDB0000175 | 0.212 | 0.151 | 0.282 |
| Inosine | HMDB0000195 | 36.061 | 25.798 | 48.008 |
| Xanthine | HMDB0000292 | 3.926 | 2.790 | 5.211 |
| Xanthosine | HMDB0000299 | 0.352 | 0.249 | 0.466 |
| 20. Pyridines and Derivatives | ||||
| 4-Pyridoxate | HMDB0000017 | 0.057 | 0.042 | 0.077 |
| Cotinine | HMDB0001046 | 0.531 | 0.382 | 0.710 |
| NAD | HMDB0000902 | 0.015 | 0.011 | 0.020 |
| Niacinamide | HMDB0001406 | 0.395 | 0.279 | 0.524 |
| Nicotinate | HMDB0001488 | 0.012 | 0.009 | 0.017 |
| Pyridoxine | HMDB0000239 | 0.0007 | 0.0005 | 0.0009 |
| 21. Pyrimidines and Derivatives | ||||
| Cytidine | HMDB0000089 | 0.003 | 0.002 | 0.004 |
| Cytosine | HMDB0000630 | 0.080 | 0.056 | 0.106 |
| Deoxycytidine | HMDB0000014 | 0.871 | 0.440 | 1.461 |
| Deoxyuridine | HMDB0000012 | 0.543 | 0.391 | 0.726 |
| Orotate | HMDB0000226 | 0.036 | 0.006 | 0.096 |
| UDP Glucose | HMDB0000286 | 0.232 | 0.165 | 0.308 |
| Uracil | HMDB0000300 | 0.058 | 0.042 | 0.078 |
| 22. Quaternary Ammonium Salts | ||||
| Carnitine | HMDB0000062 | 85.669 | 61.210 | 114.060 |
| Choline | HMDB0000097 | 95.170 | 67.565 | 126.279 |
| 23. Quinolines and Derivatives | ||||
| Kynurenate | HMDB0000715 | 0.044 | 0.031 | 0.058 |
| 24. Sugar Alcohols | ||||
| Myo-inositol | HMDB0000211 | 16.989 | 12.075 | 22.606 |
| Sorbitol | HMDB0000247 | 3.692 | 2.617 | 4.904 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nandania, J.; Peddinti, G.; Pessia, A.; Kokkonen, M.; Velagapudi, V. Validation and Automation of a High-Throughput Multitargeted Method for Semiquantification of Endogenous Metabolites from Different Biological Matrices Using Tandem Mass Spectrometry. Metabolites 2018, 8, 44. https://doi.org/10.3390/metabo8030044
Nandania J, Peddinti G, Pessia A, Kokkonen M, Velagapudi V. Validation and Automation of a High-Throughput Multitargeted Method for Semiquantification of Endogenous Metabolites from Different Biological Matrices Using Tandem Mass Spectrometry. Metabolites. 2018; 8(3):44. https://doi.org/10.3390/metabo8030044
Chicago/Turabian StyleNandania, Jatin, Gopal Peddinti, Alberto Pessia, Meri Kokkonen, and Vidya Velagapudi. 2018. "Validation and Automation of a High-Throughput Multitargeted Method for Semiquantification of Endogenous Metabolites from Different Biological Matrices Using Tandem Mass Spectrometry" Metabolites 8, no. 3: 44. https://doi.org/10.3390/metabo8030044
APA StyleNandania, J., Peddinti, G., Pessia, A., Kokkonen, M., & Velagapudi, V. (2018). Validation and Automation of a High-Throughput Multitargeted Method for Semiquantification of Endogenous Metabolites from Different Biological Matrices Using Tandem Mass Spectrometry. Metabolites, 8(3), 44. https://doi.org/10.3390/metabo8030044