LC-HRMS Metabolomics for Untargeted Diagnostic Screening in Clinical Laboratories: A Feasibility Study

Abstract
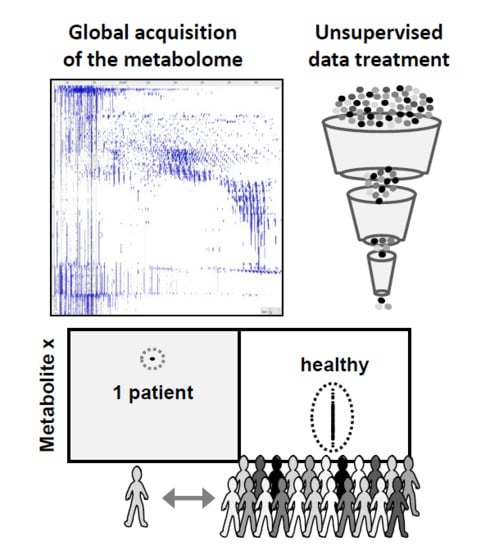
1. Introduction
2. Materials and Methods
2.1. Biomatrix and Sample Preparation
2.2. LC-HRMS System, Parameters, and Analysis
2.3. Data Representation and Data Treatment
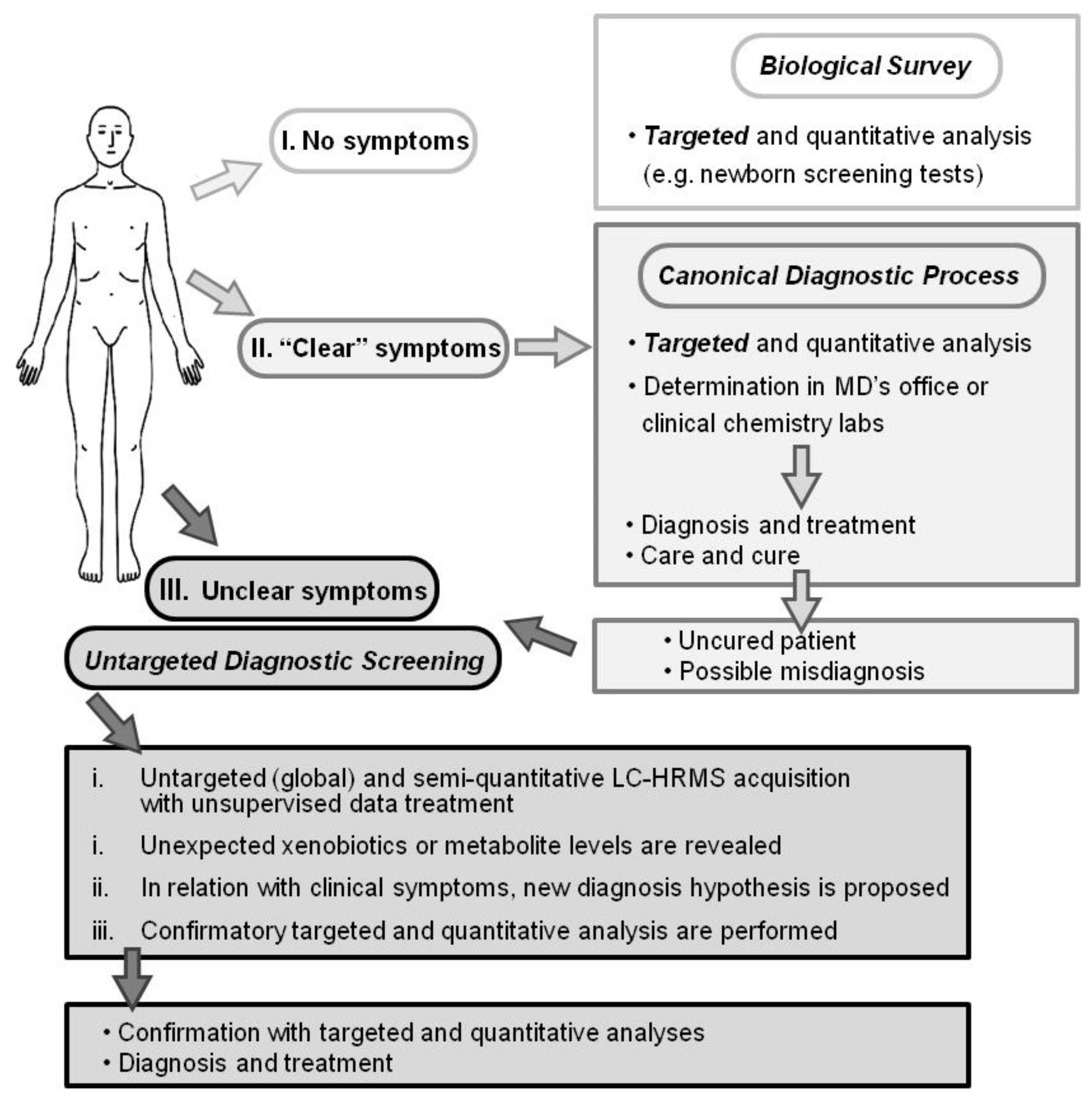
2.4. Evaluation of Untargeted Diagnostics Screening
2.5. Identification of Revealed Features
3. Results and Discussion
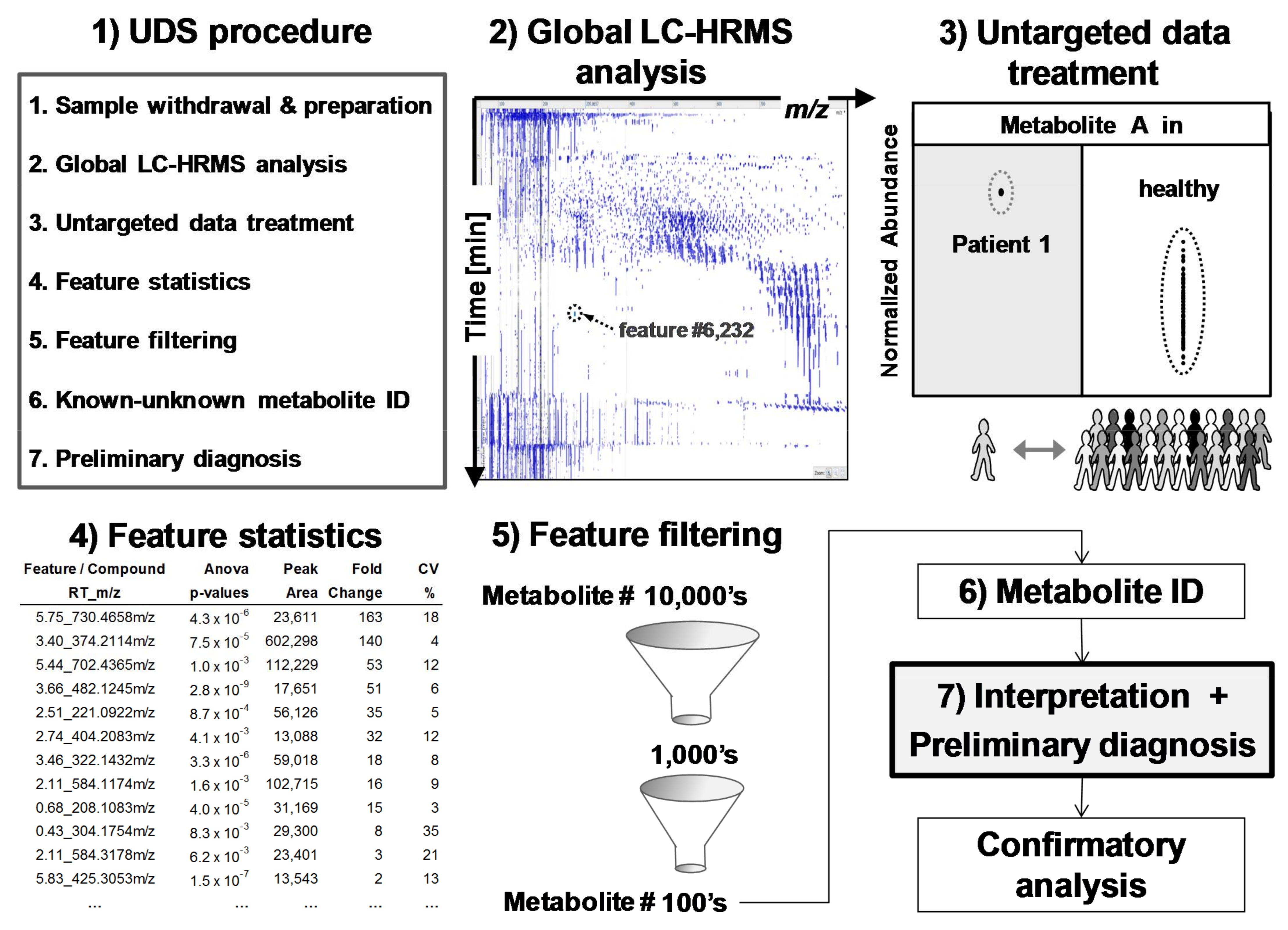
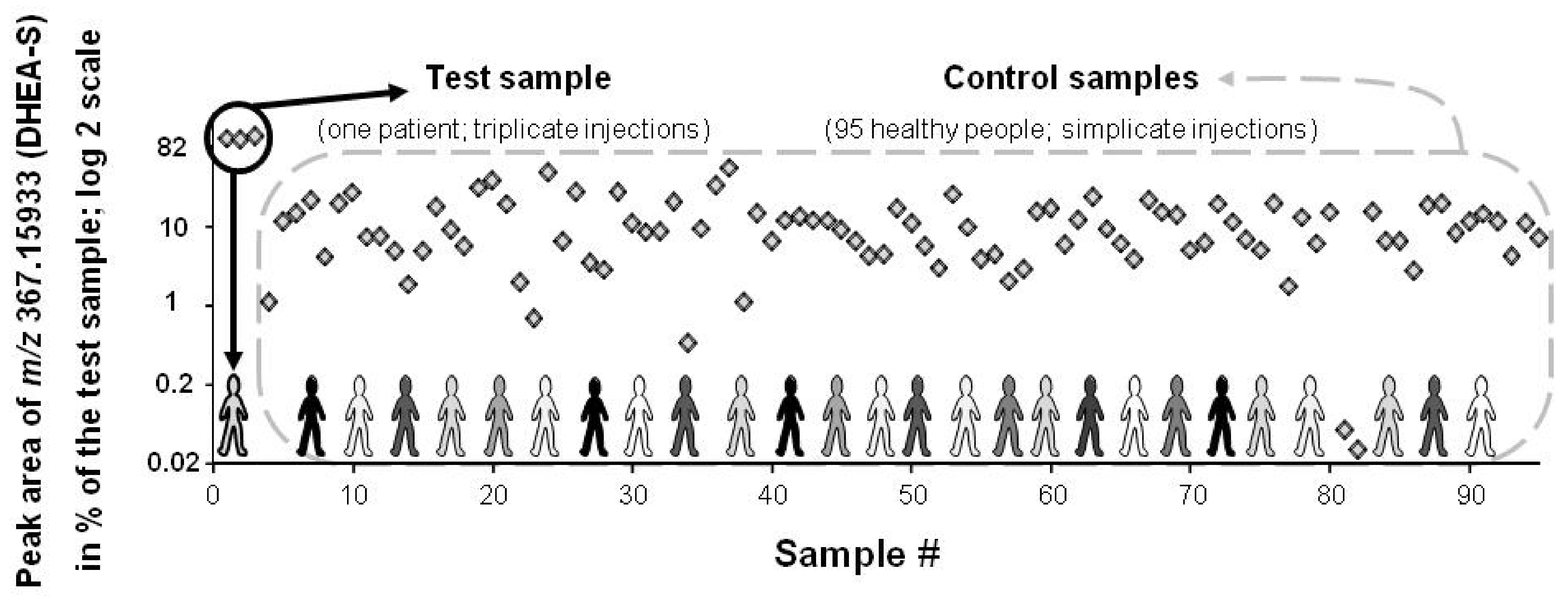
3.1. Data Treatment and Reliability
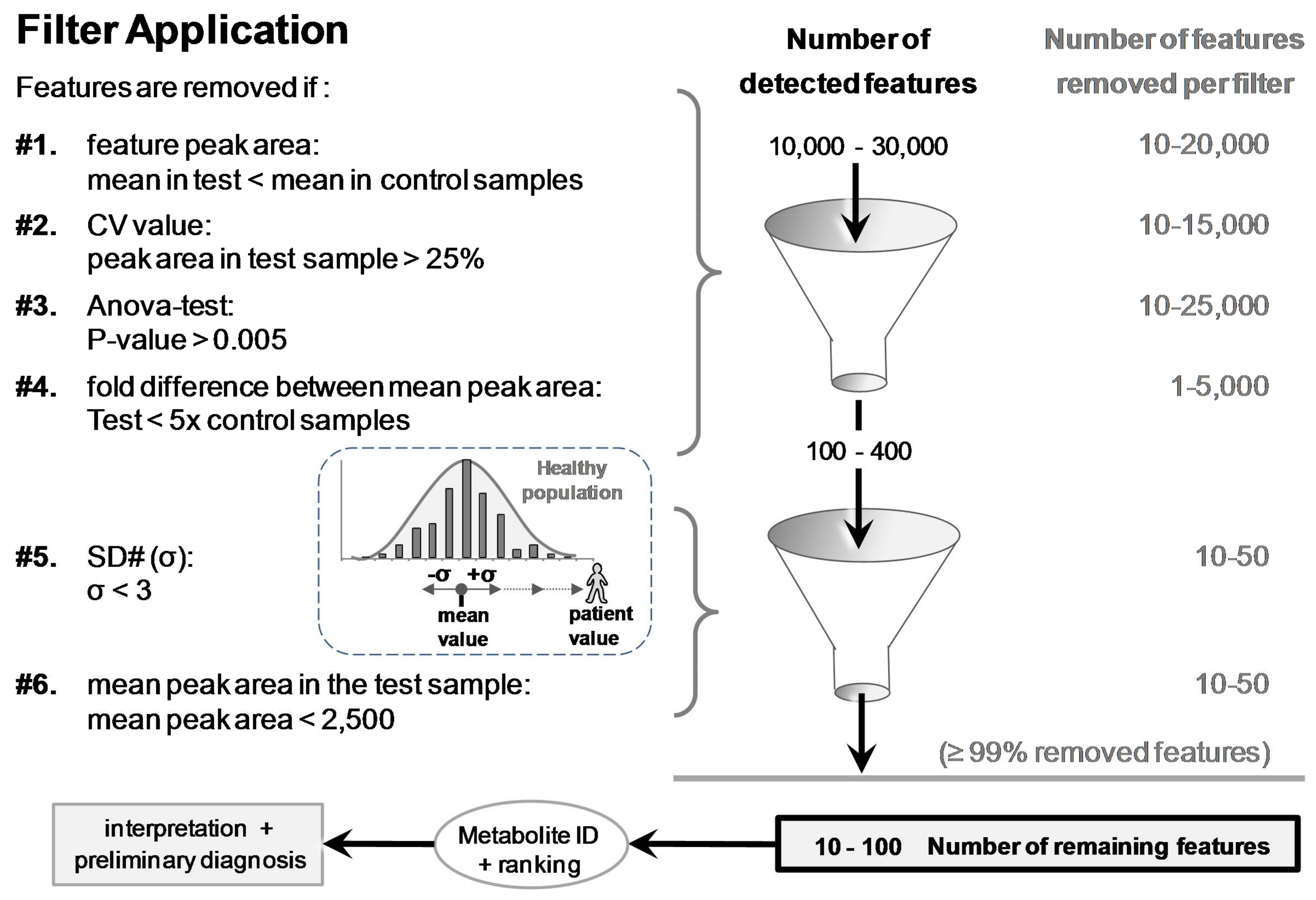
3.2. Applied Filters for Feature Removal and Revealed Metabolites
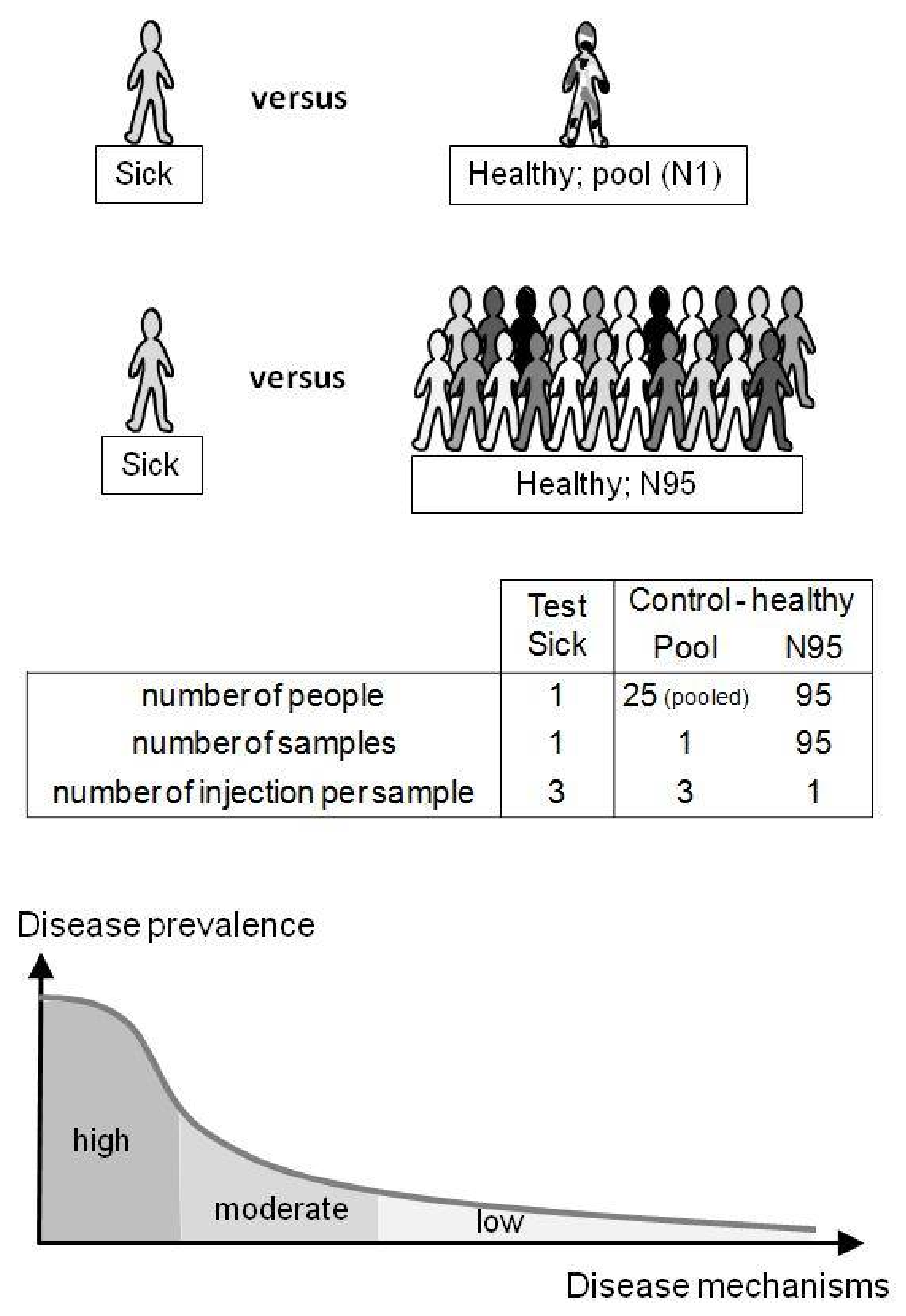
3.3. Spiked Compound Revealed with Pool or N95 as Control Groups
3.4. Feature Ranking Based on SD# or Peak Area
3.5. Metabolite Identification
4. Conclusive Remarks
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| HRMS | high resolution mass spectrometry |
| LC | liquid chromatography |
| UDS | untargeted diagnostic screening |
| DHEA-sulfate | dehydroepiandrosterone-sulfate |
| ID | identification |
References
- Pagana, K.D.; Pagana, T.J.P. Mosby’s Manual of Diagnostic and Laboratory Tests, 5th ed.; Elsevier: New York, NY, USA, 2014; 1264p. [Google Scholar]
- Stern, S.D.C.; Cifu, A.S.; Altkorn, D. Symptom to Diagnosis, an Evidence-Based Guide, 3rd ed.; McGraw-Hill Education Lange: New York, NY, USA, 2015. [Google Scholar]
- The National Academies of Sciences, Engineering, and Medicine, USA. Available online: www.nationalacademies.org/hmd/~/media/Files/Report%20Files/2015/Improving-Diagnosis/DiagnosticError_ReportBrief.pdf (accessed on 5 March 2018).
- James, J.T. A new, evidence-based estimate of patient harms associated with hospital care. J. Patient Saf. 2013, 9, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Gangoiti, J.A.; Barshop, B.A.; Siuzdak, G. Metabolomics identifies perturbations in human disorders of propionate metabolism. Clin. Chem. 2007, 53, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Abily-Donval, L.; Afonso, C.; Marret, S.; Bekri, S. Clinical Metabolomics, The New Metabolic Window for Inborn Errors of Metabolism Investigations in the Post-Genomic Era. Int. J. Mol. Sci. 2016, 17, 1167. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.; Colby, J. High-Resolution Mass Spectrometry for Untargeted Drug Screening. Methods Mol. Biol. 2016, 1383, 153–166. [Google Scholar] [PubMed]
- Thoren, K.L.; Colby, J.M.; Shugarts, S.B.; Wu, A.H.; Lynch, K.L. Comparison of Information-Dependent Acquisition on a Tandem Quadrupole TOF vs. a Triple Quadrupole Linear Ion Trap Mass Spectrometer for Broad-Spectrum Drug Screening. Clin. Chem. 2016, 62, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef] [PubMed]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E.; Engelke, U.F.H.; de Boer, S.; Hoegen, B.; Kwast, H.J.T.; van de Vorst, M.; Huigen, M.C.D.G.; Keularts, I.M.L.W.; et al. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Schork, N.J. Personalized medicine: Time for one-person trials. Nature 2015, 520, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Abrahamyan, L.; Feldman, B.M.; Tomlinson, G.; Faughnan, M.E.; Johnson, S.R.; Diamond, I.R.; Gupta, S. Alternative designs for clinical trials in rare diseases. Am. J. Med. Genet. C Semin. Med. Genet. 2016, 172, 313–331. [Google Scholar] [CrossRef] [PubMed]
- Merlo, J.; Mulinari, S.; Wemrell, M.; Subramanian, S.V.; Hedblad, B. The tyranny of the averages and the indiscriminate use of risk factors in public health: The case of coronary heart disease. SSM Popul. Health 2017, 3, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Tabery, J. Commentary: Hogben vs. the Tyranny of averages. Int. J. Epidemiol. 2011, 40, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Ziegelstein, R.C. Personomics and Precision Medicine. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 160–168. [Google Scholar] [PubMed]
- Ziegelstein, R.C. Personomics: The Missing Link in the Evolution from Precision Medicine to Personalized Medicine. J. Pers. Med. 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Wilson, I.D.; Lindon, J.C. Pharmacometabonomics as an effector for personalized medicine. Pharmacogenomics 2011, 12, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- Adamski, J.; Suhre, K. Metabolomics platforms for genome wide association studies—Linking the genome to the metabolome. Curr. Opin. Biotechnol. 2013, 1, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Daughton, C.G. Illicit drugs: Contaminants in the environment and utility in forensic epidemiology. Rev. Environ. Contam. Toxicol. 2011, 210, 59–110. [Google Scholar] [PubMed]
- Junot, C.; Fenaille, F.; Colsch, B.; Bécher, F. High resolution mass spectrometry based techniques at the crossroads of metabolic pathways. Mass Spectrom. Rev. 2014, 33, 471–500. [Google Scholar] [CrossRef] [PubMed]
- Ríos Peces, S.; Díaz Navarro, C.; Márquez López, C.; Caba, O.; Jiménez-Luna, C.; Melguizo, C.; Prados, J.C.; Genilloud, O.; Vicente Pérez, F.; Pérez Del Palacio, J. Untargeted LC-HRMS-Based Metabolomics for Searching New Biomarkers of Pancreatic Ductal Adenocarcinoma: A Pilot Study. SLAS Discov. 2017, 22, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Rochat, B. From targeted quantification to untargeted metabolomics. Why LC-high-resolution-MS will become a key instrument in clinical labs. Trends Anal. Chem. 2016, 84, 151–164. [Google Scholar] [CrossRef]
- Oberacher, H.; Arnhard, K. Current status of non-targeted liquid chromatography-tandem mass spectrometry in forensic toxicology. Trends Anal. Chem. 2016, 84, 94–105. [Google Scholar] [CrossRef]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug. Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Grund, B.; Marvin, L.; Rochat, B. Quantitative performance of a quadrupole-orbitrap-MS in targeted LC-MS determinations of small molecules. J. Pharm. Biomed. Anal. 2016, 124, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Henry, H.; Sobhi, H.R.; Scheibner, O.; Bromirski, M.; Nimkar, S.B.; Rochat, B. Comparison between a high-resolution single-stage Orbitrap and a triple quadrupole mass spectrometer for quantitative analyses of drugs. Rapid Commun. Mass Spectrom. 2012, 26, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Rochat, B.; Kottelat, E.; McMullen, J. The future key role of LC-high-resolution-MS analyses in clinical laboratories: A focus on quantification. Bioanalysis 2012, 4, 2939–2958. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Reliability of veterinary drug residue confirmation: High resolution mass spectrometry versus tandem mass spectrometry. Anal. Chim. Acta 2015, 856, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, G.; Randak, T.; Lindberg, R.H.; Grabic, R. Comparison of the quantitative performance of a Q-Exactive high-resolution mass spectrometer with that of a triple quadrupole tandem mass spectrometer for the analysis of illicit drugs in wastewater. Rapid Commun. Mass Spectrom. 2013, 27, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Weinshilboum, R. Pharmacometabolomics Research Network. Metabolomic Signatures for Drug Response Phenotypes: Pharmacometabolomics Enables Precision Medicine. Clin. Pharmacol. Ther. 2015, 98, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Beck, O. Can Pharmacometabolomics and LC-HRMS develop a new Concept for Therapeutic Drug Monitoring? J. Appl. Bioanal. 2015, 1, 42–45. [Google Scholar] [CrossRef]
- Rochat, B. Is there a future for metabotyping in clinical laboratories? Bioanalysis 2015, 7, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Hicks, L.C.; Ralphs, S.J.; Williams, H.R. Metabonomics and diagnostics. Methods Mol. Biol. 2015, 1277, 233–244. [Google Scholar] [PubMed]
- Dénes, J.; Szabó, E.; Robinette, S.L.; Szatmári, I.; Szőnyi, L.; Kreuder, J.G.; Rauterberg, E.W.; Takáts, Z. Metabonomics of newborn screening dried blood spot samples: A novel approach in the screening and diagnostics of inborn errors of metabolism. Anal. Chem. 2012, 84, 10113–10120. [Google Scholar] [CrossRef] [PubMed]
- Gertsman, I.; Gangoiti, J.A.; Barshop, B.A. Validation of a dual LC-HRMS platform for clinical metabolic diagnosis in serum, bridging quantitative analysis and untargeted metabolomics. Metabolomics 2014, 10, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations. Int. J. Mol. Sci. 2016, 17, 1555. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.; Kidd, R. Editorial: ChemSpider—A tool for Natural Products research. Nat. Prod. Rep. 2015, 32, 1163–1164. [Google Scholar]
- Little, J.L.; Cleven, C.D.; Brown, S.D. Identification of “known unknowns” utilizing accurate mass data and chemical abstracts service databases. J. Am. Soc. Mass Spectrom. 2011, 22, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; Shoemaker, B.A.; Wang, J.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Rochat, B. Proposed confidence scale and ID score in the identification of known-unknown compounds using high resolution MS data. J. Am. Soc. Mass Spectrom. 2017, 28, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Advances in metabolite identification. Bioanalysis 2011, 3, 1769–1782. [Google Scholar] [CrossRef] [PubMed]
- Commission Decision 2002/657/EC Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32002D0657&from=EN (accessed on 5 March 2018).
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.; Varesio, E.; Ivosev, G.; Burton, L.; Bonner, R.; Hopfgartner, G. Comprehensive analytical strategy for biomarker identification based on liquid chromatography coupled to mass spectrometry and new candidate confirmation tools. Anal. Chem. 2009, 81, 7677–7694. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gardinali, P.R. Comparison of multiple API techniques for the simultaneous detection of microconstituents in water by on-line SPE-LC-MS/MS. J. Mass Spectrom. 2012, 47, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.; Rudnitskaya, A.; Blackburn, G.J.; Mohd Fauzi, N.; Pitt, A.R.; Spickett, C.M. A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. J. Lipid Res. 2013, 54, 1812–1824. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Xenobiotic | Formula | Adducts | m/ztheor [Da] | [μg/mL] | LC Area ◊ | RT [min] | MA [ppm] | CV [%] |
| methamphetamine | C10H15N | +H+ | 150.12773 | 0.005 | 1 | 0.4 | 0.7 | 97 |
| methadone | C21H27NO | +H+ | 310.21654 | 0.005 | 64 | 3.2 | −0.2 | 22 |
| dextromethorphan | C18H25NO | +H+ | 272.20089 | 0.005 | 76 | 2.6 | −0.2 | 5 |
| endoxifen | C25H27NO2 | +H+ | 374.21146 | 5/0.5 | 6778/602 | 3.4 | −0.1/0.5 | 6/4 |
| imatinib | C29H31N7O | +2H+ | 247.63678 | 5.0 | 159,359 | 1.9 | 0.3 | 10 |
| Endogenous | Formula | Adducts | m/ztheor [Da] | [μM] | LC Area ◊ | RT [min] | MA [ppm] | CV [%] |
| DHEA-S | C19H28O5S | −[H+]− | 367.15847 | 20 ○ | 61,361 | 3.2 | 2.4 | 5 |
| testosterone | C19H28O2 | +H+ | 289.21621 | 0.07 ○ | 91 | 3.4 | −0.4 | 6 |
| Control | Spiked: Endoxifen (Xenobiotic) | Fold Difference ** | Ranking # Based on | |||||
| Group | [μg/mL] | LC Area * | CV [%] | Anova (p) | LC Area * | SD# | SD# | LC Area * |
| N95 | 0.5 | 602 | 3.7 | 0.0001 | 140 | 101 | 3 | 11 |
| Pool | 0.5 | 509 | 4.9 | 0.0048 | 186 | 118 | 127 | 12 |
| N95 | 5 | 6778 | 5.5 | <0.0001 | 1205 | 900 | 13 | 1 |
| Pool | 5 | 6365 | 4.6 | 0.0012 | 1927 | 1259 | 55 | 1 |
| Control | Spiked: DHEAS (Endogenous) | Fold Difference ** | Ranking # Based on | |||||
| Group | [μM] | LC Area * | CV [%] | Anova (p) | LC Area * | SD# | SD# | LC Area * |
| N95 | 20 | 61,361 | 5.0 | 0.0002 | 9.2 | 10.7 | 25 | 1 |
| Pool | 20 | 56,819 | 5.6 | 0.0082 | 6.2 | 6.7 | 64 | 1 |
| Xenobiotic | Spiked Levels | Fold Difference Related to | Ranking # Based on | ||||
|---|---|---|---|---|---|---|---|
| [μg/mL] | CV [%] * | p-Value | LC Area ** | SD# | SD# | LC Area (10−3) | |
| methamphetamine | 0.005 | 97 ◊ | 0.0001 | 105 | 45 | 33 | 191 |
| methadone | 0.005 | 22 | <0.0001 | 14,162 | 3615 | 6 | 29 |
| dextrometorphan | 0.005 | 5 | <0.0001 | 50 | 12,480 | 1 | 6 |
| endoxifen | 5.0 | 6 | <0.0001 | 1205 | 900 | 13 | 1 |
| imatinib | 5.0 | 10 | <0.0001 | 40,417 | 34,189 | 4 | 1 |
| Endogenous | Endo. + μM | ||||||
| DHEA-S | 20 | 5 | 0.0002 | 9 | 11 | 25 | 1 |
| testosterone | 0.07 | 6 | 0.0020 | 5 | 4 | 44 | 12 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rochat, B.; Mohamed, R.; Sottas, P.-E. LC-HRMS Metabolomics for Untargeted Diagnostic Screening in Clinical Laboratories: A Feasibility Study. Metabolites 2018, 8, 39. https://doi.org/10.3390/metabo8020039
Rochat B, Mohamed R, Sottas P-E. LC-HRMS Metabolomics for Untargeted Diagnostic Screening in Clinical Laboratories: A Feasibility Study. Metabolites. 2018; 8(2):39. https://doi.org/10.3390/metabo8020039
Chicago/Turabian StyleRochat, Bertrand, Rayane Mohamed, and Pierre-Edouard Sottas. 2018. "LC-HRMS Metabolomics for Untargeted Diagnostic Screening in Clinical Laboratories: A Feasibility Study" Metabolites 8, no. 2: 39. https://doi.org/10.3390/metabo8020039
APA StyleRochat, B., Mohamed, R., & Sottas, P.-E. (2018). LC-HRMS Metabolomics for Untargeted Diagnostic Screening in Clinical Laboratories: A Feasibility Study. Metabolites, 8(2), 39. https://doi.org/10.3390/metabo8020039