Quantification of Stable Isotope Traces Close to Natural Enrichment in Human Plasma Metabolites Using Gas Chromatography-Mass Spectrometry

 , ,
, ,
Abstract
:1. Introduction
2. Results
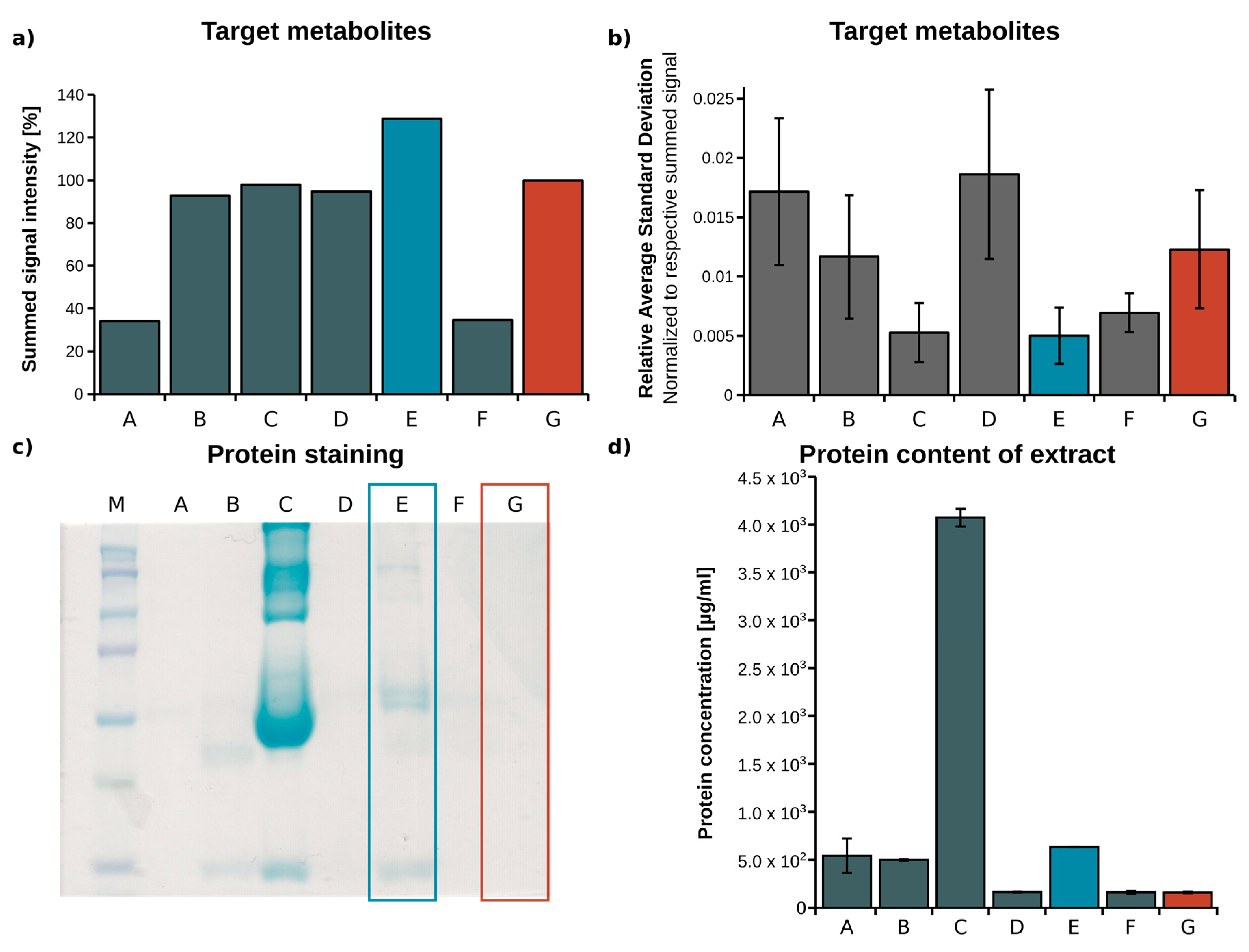
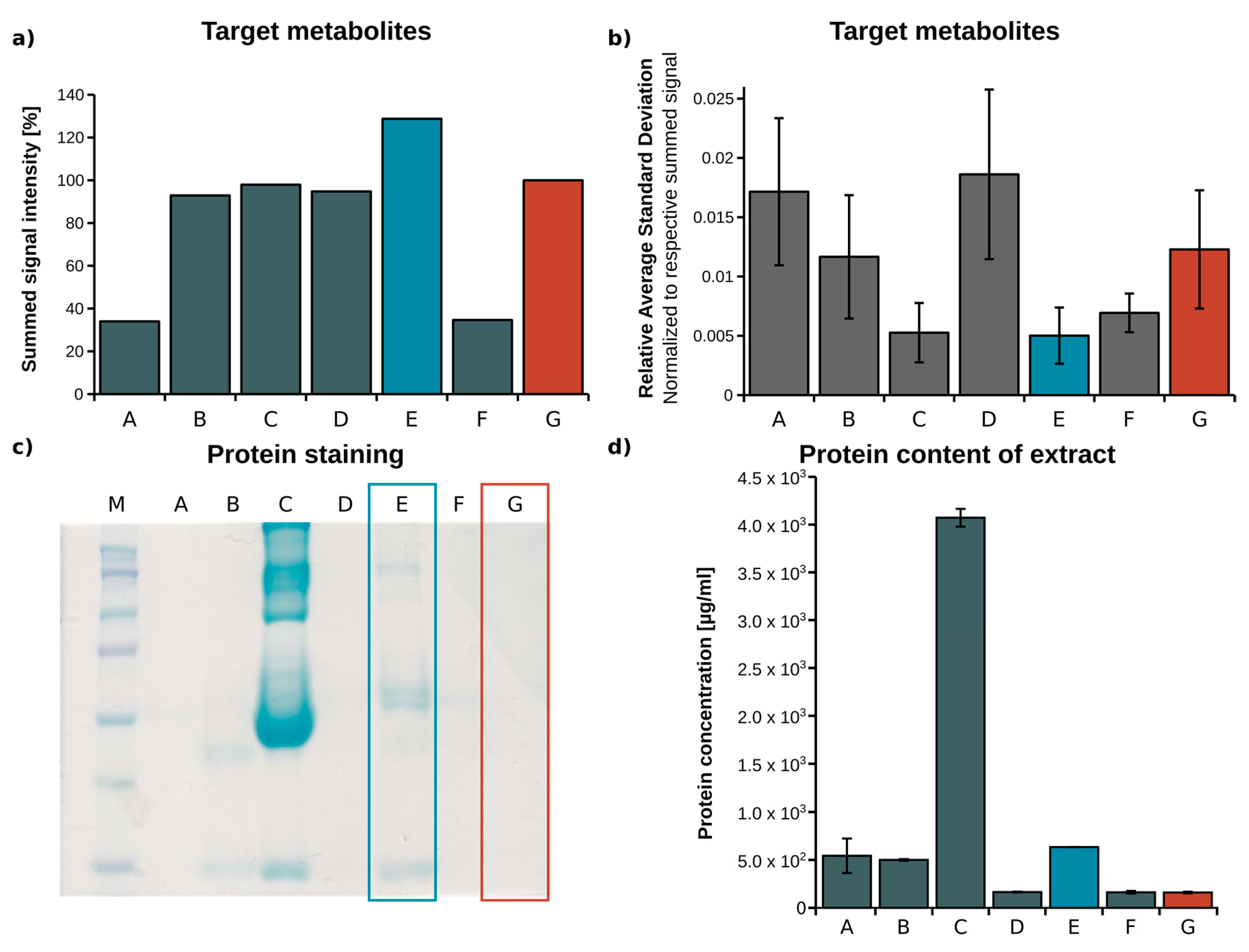
2.1. Optimization of Metabolite Extraction from Plasma
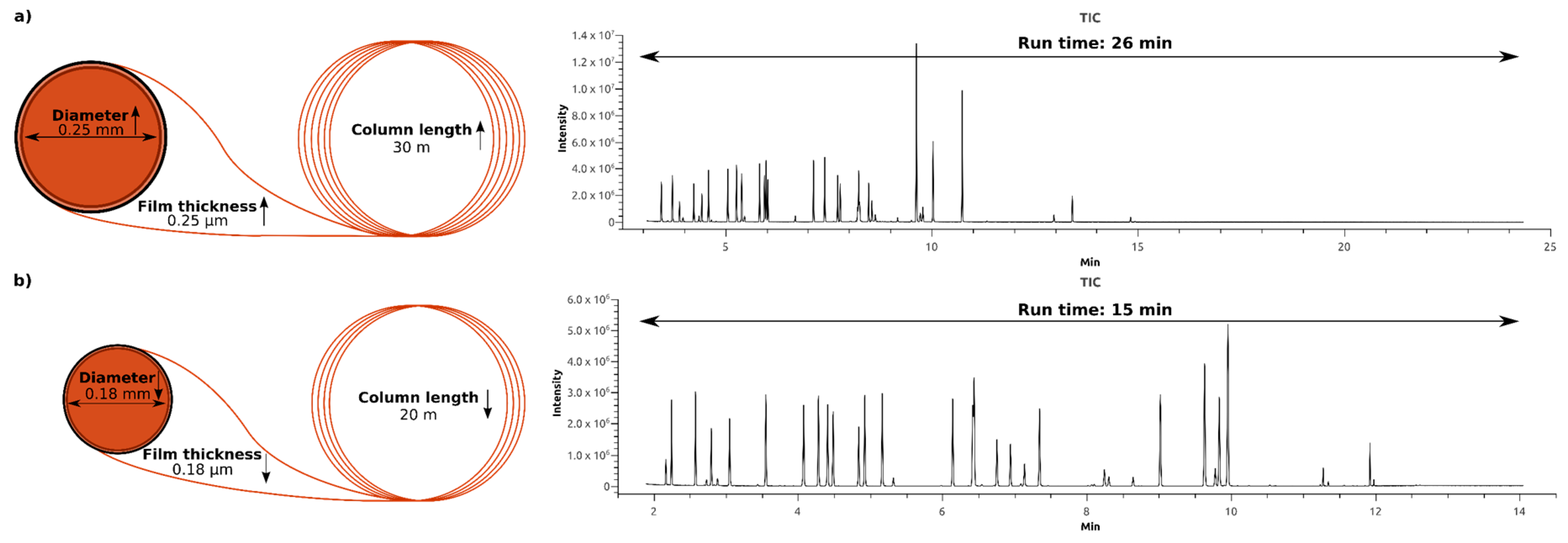
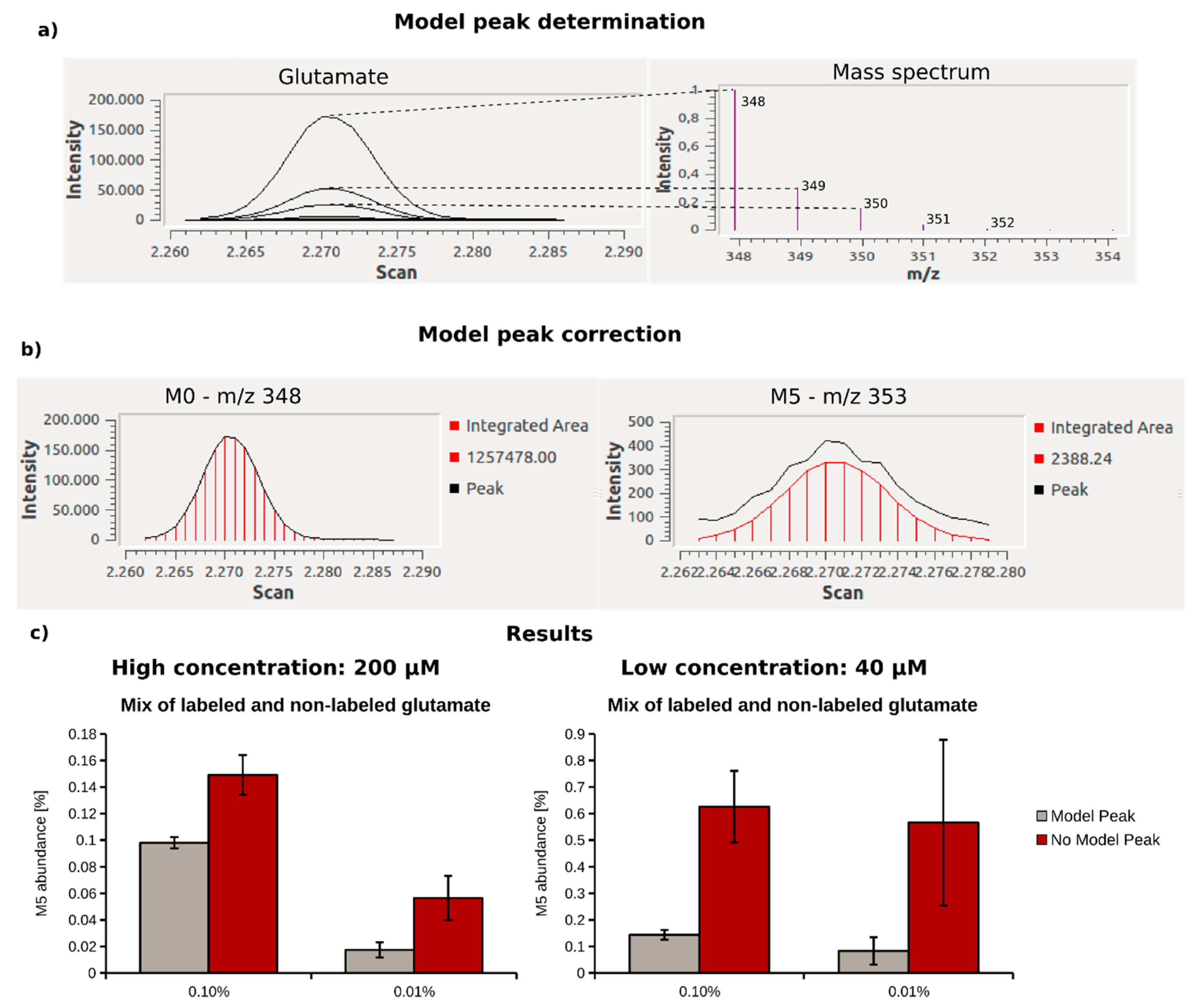
2.2. Optimization of the Gas Chromatography Measurements
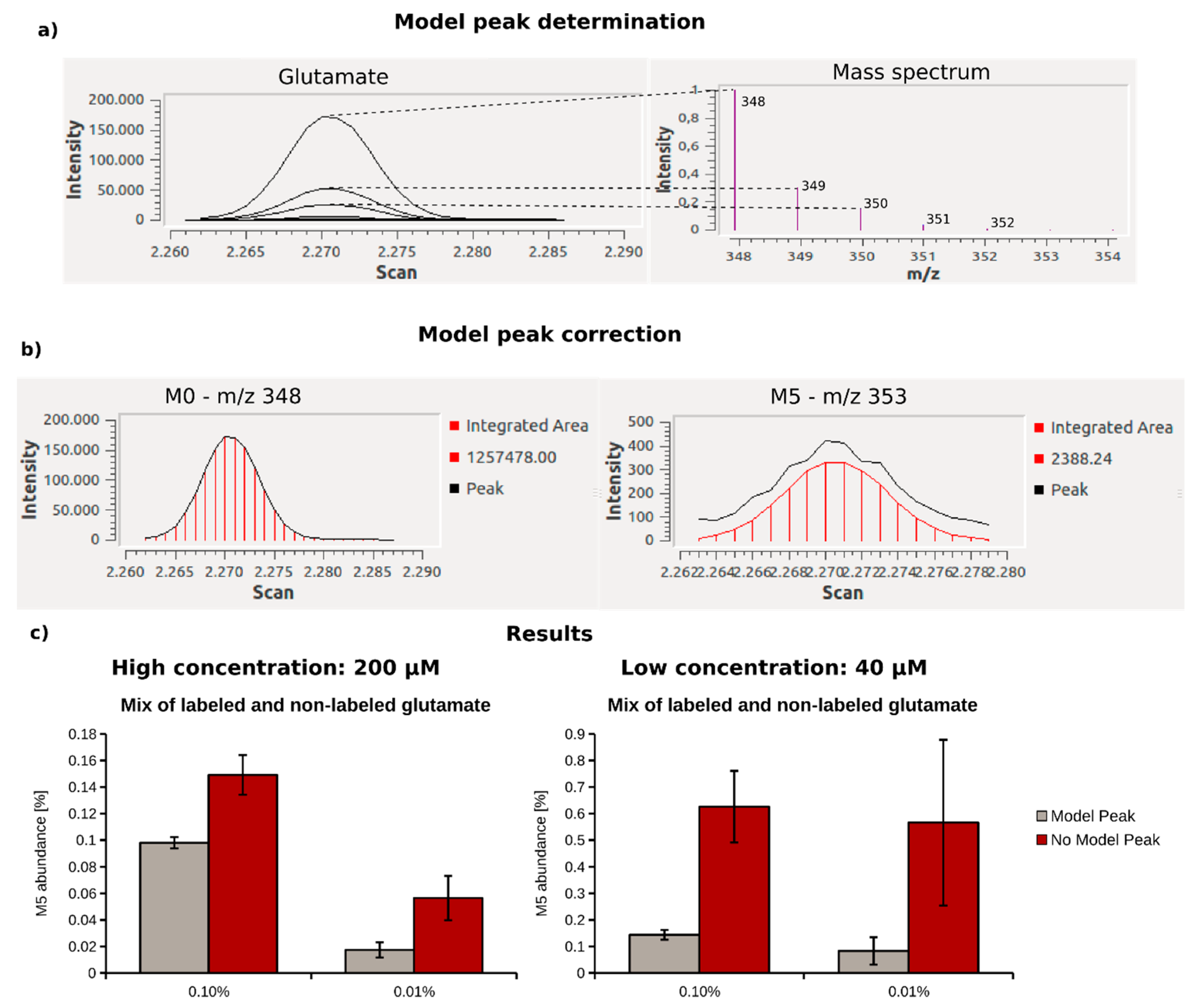
2.3. Optimization of Data Analysis with Metabolite Detector
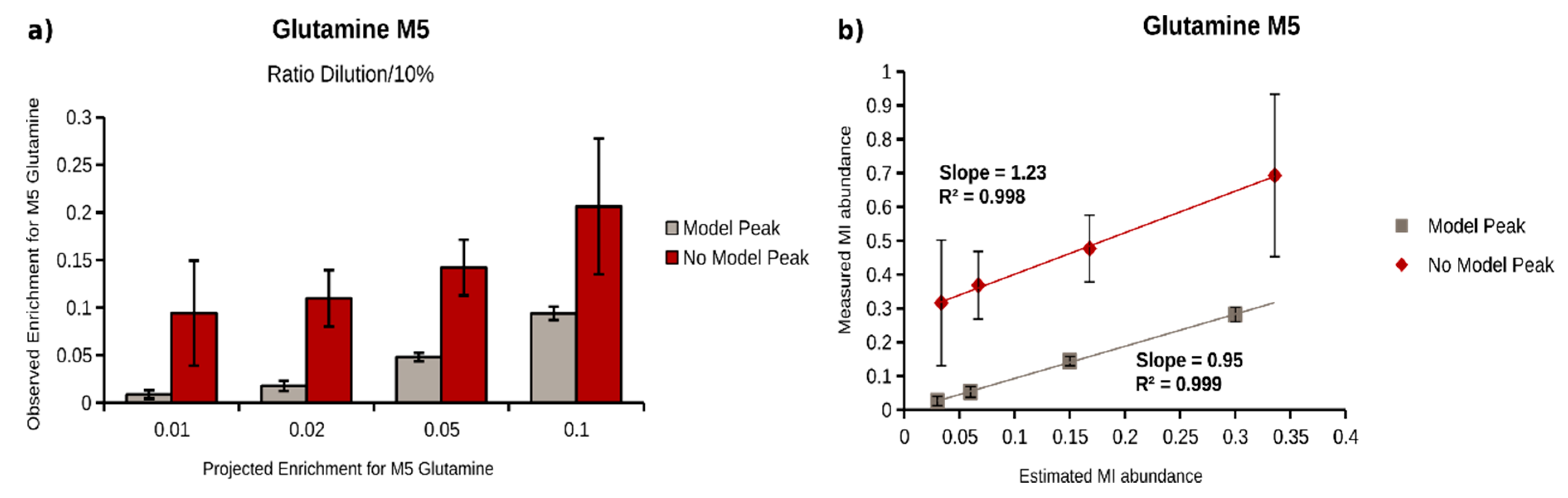
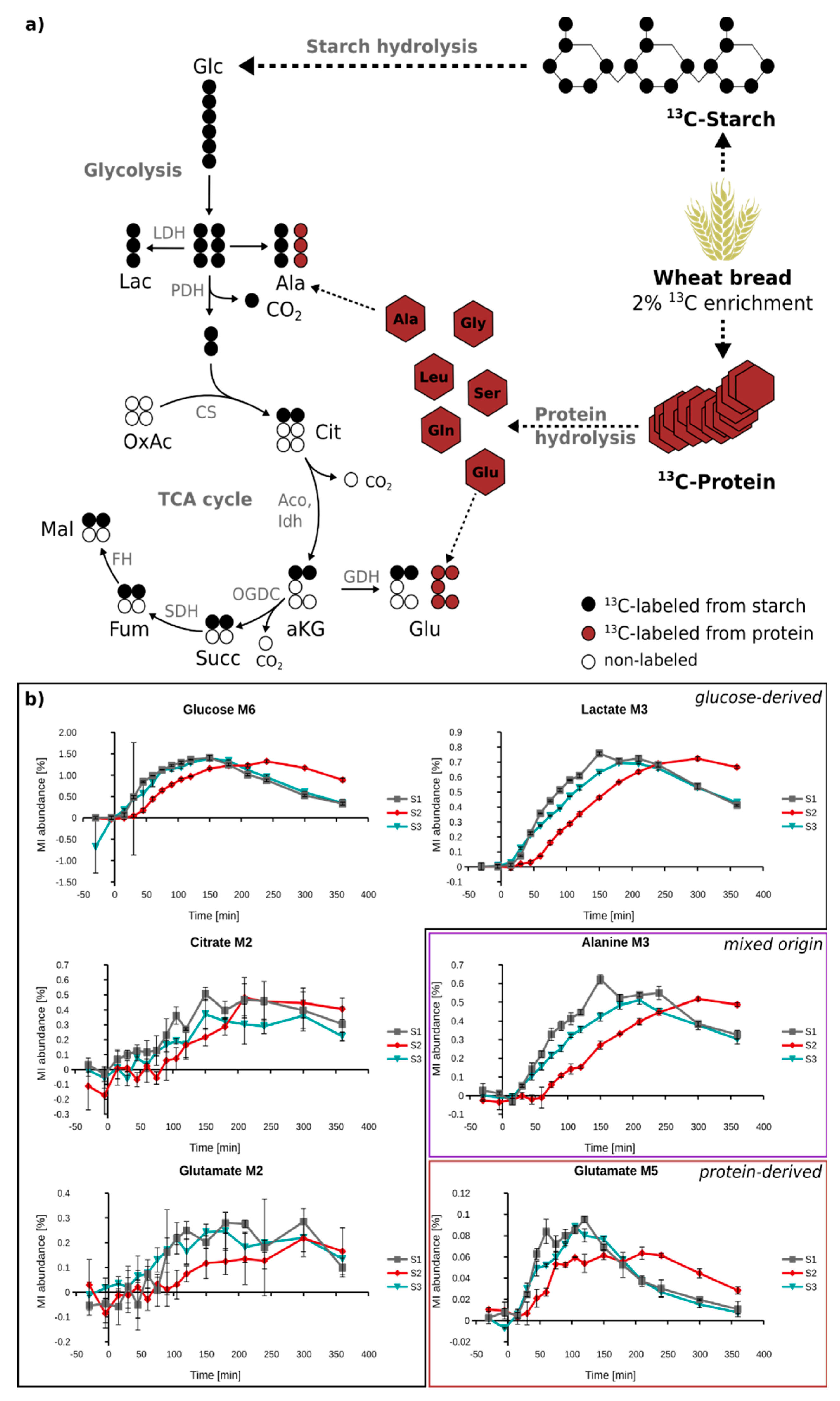
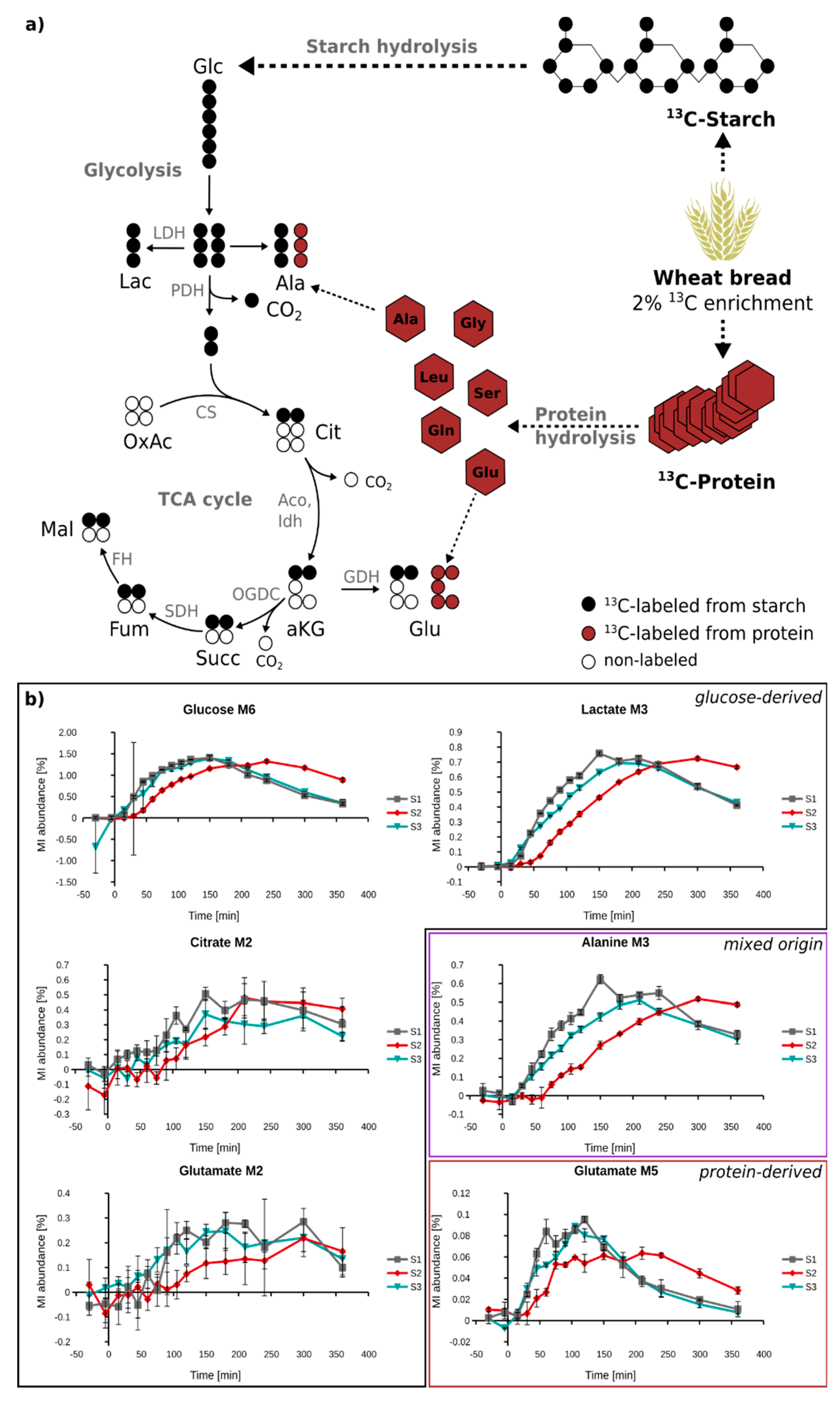
2.4. Quantification of 13C Enrichment in Plasma
2.5. In Vivo Metabolism of Fully 13C-Labeled Wheat Flour
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Plasma Samples
4.3. Metabolite Extraction from Plasma
4.4. Coomassie Staining
4.5. Protein Quantification
4.6. 13C-Enrichment Assay of Glutamate
4.7. 13C-Enrichment Assay of 13C-Labeled Cell Extract
4.8. Metabolite Extraction of Plasma Samples from a Nutritional Intervention Study
4.9. Metabolite Derivatization and GC-MS Measurement
4.10. Data Processing
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brennan, L. Metabolomics in nutrition research: Current status and perspectives. Biochem. Soc. Trans. 2013, 41, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Bondia-Pons, I.; Nordlund, E.; Mattila, I.; Katina, K.; Aura, A.-M.; Kolehmainen, M.; Orešič, M.; Mykkänen, H.; Poutanen, K. Postprandial differences in the plasma metabolome of healthy Finnish subjects after intake of a sourdough fermented endosperm rye bread versus white wheat bread. Nutr. J. 2011, 10, 116. [Google Scholar] [CrossRef] [PubMed]
- Van Velzen, E.J.J.; Westerhuis, J.A.; Van Duynhoven, J.P.M.; Van Dorsten, F.A.; Grün, C.H.; Jacobs, D.M.; Duchateau, G.S.M.J.E.; Vis, D.J.; Smilde, A.K. Phenotyping Tea Consumers by Nutrikinetic Analysis of Polyphenolic End-Metablites. J. Proteome Res. 2008, 8, 3317–3330. [Google Scholar] [CrossRef] [PubMed]
- Van Ommen, B.; Van Den Broek, T.; De Hoogh, I.; Van Erk, M.; Van Someren, E.; Rouhani-Rankouhi, T.; Anthony, J.C.; Hogenelst, K.; Pasman, W.; Boorsma, A.; et al. Systems Biology of personalized nutrition. Nutr. Rev. 2017, 75, 579–599. [Google Scholar] [CrossRef] [PubMed]
- Urpi-Sarda, M.; Boto-Ordóñez, M.; Queipo-Ortuño, M.I.; Tulipani, S.; Corella, D.; Estruch, R.; Tinahones, F.J.; Andres-Lacueva, C. Phenolic and microbial-targeted metabolomics to discovering and evaluating wine intake biomarkers in human urine and plasma. Electrophoresis 2015, 36, 2259–2268. [Google Scholar] [CrossRef] [PubMed]
- Ostertag, L.M.; Philo, M.; Colquhoun, I.J.; Tapp, H.S.; Saha, S.; Duthie, G.G.; Kemsley, E.K.; De Roos, B.; Kroon, P.A.; Lle Le Gall, G. Acute consumption of Flavan-3-ol-Enriched Dark Chocolate Affects Human Endogenous Metabolism. J. Proteome Res. 2017, 16, 2516–2526. [Google Scholar] [CrossRef] [PubMed]
- Chokkathukalam, A.; Kim, D.-H.; Barrett, M.P.; Breitling, R.; Creek, D.J. Stable isotope-labeling studies in metabolomics: New insights into structure and dynamics of metabolic networks. Bioanalysis 2014, 6, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Hiller, K.; Metallo, C.; Stephanopoulos, G. Elucidation of cellular metabolism via metabolomics and stable-isotope assisted metabolomics. Curr. Pharm. Biotechnol. 2011, 12, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Bier, D.M.; Arnold, K.J.; Sherman, W.R.; Holland, W.H.; Holmes, W.F.; Kipnis, D.M. In-vivo measurement of glucose and alanine metabolism with stable isotopic tracers. Diabetes 1977, 26, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hoene, M.; Zhao, X.; Chen, S.; Wei, H.; Häring, H.U.; Lin, X.; Zeng, Z.; Weigert, C.; Lehmann, R.; Xu, G. Stable isotope-assisted lipidomics combined with nontargeted isotopomer filtering, a tool to unravel the complex dynamics of lipid metabolism. Anal. Chem. 2013, 85, 4651–4657. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.K. New stable isotope-mass spectrometric techniques for measuring fluxes through intact metabolic pathways in mammalian systems: Introduction of moving pictures into functional genomics and phenotyping. Metab. Eng. 2004, 6, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Eelderink, C.; Moerdijk-Poortvliet, T.C.W.; Wang, H.; Schepers, M.; Preston, T.; Boer, T.; Vonk, R.J.; Schierbeek, H.; Priebe, M.G. The glycemic response does not reflect the in vivo starch digestibility of fiber-rich wheat products in healthy men. J. Nutr. 2012, 142, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Eelderink, C.; Schepers, M.; Preston, T.; Vonk, R.J.; Oudhuis, L.; Priebe, M.G. Slowly and rapidly digestible starchy foods can elicit a similar glycemic response because of differential tissue glucose uptake in healthy men. Am. J. Clin. Nutr. 2012, 96, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Korach-André, M.; Roth, H.; Barnoud, D.; Péan, M.; Péronnet, F.; Leverve, X. Glucose appearance in the peripheral circulation and liver glucose output in men after a large 13C starch meal. Am. J. Clin. Nutr. 2004, 80, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.; Rizza, R.A. Application of Isotopic Techniques Using Constant Specific Activity or Enrichment to the Study of Carbohydrate Metabolism. Diabetes 2009, 58, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Di Camillo, B.; Toffolo, G.; Basu, A.; Shah, P.; Vella, A.; Rizza, R.; Cobelli, C. Use of a novel triple-tracer approach to assess postprandial glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E55–E69. [Google Scholar] [CrossRef] [PubMed]
- Bos, C.; Mahé, S.; Gaudichon, C.; Benamouzig, R.; Gausserès, N.; Luengo, C.; Ferrière, F.; Rautureau, J.; Tomé, D. Assessment of net postprandial protein utilization of 15 N-labelled milk nitrogen in human subjects. Br. J. Nutr. 1999, 81, 221–226. [Google Scholar] [PubMed]
- Kasumov, T.; Ilchenko, S.; Li, L.; Rachdaoui, N.; Sadigov, R.; Willard, B.; Mccullough, A.J.; Previs, S. Measuring of protein synthesis using metabolic 2 H-labeling, high-resolution mass spectrometry and an algorithm. Anal. Biochem. 2011, 412, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Chavez Jauregui, R.; Mattes, R.D.; Parks, E.J. Dynamics of fat absorption and impact of sham feeding on postprandial lipema. Gastroenterology 2010, 139, 1528–1548. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Klein, S.; Wolfe, R.R.; Dupre, J.; Kell, J.M.; Sönksen, P.H.; Jones, R.H.; Eagles, F.; Dennis, J.; Parker, I.; et al. Insulin resistance: The shape of things to come. Lancet 1994, 344, 521–524. [Google Scholar] [CrossRef]
- Butler, R.N.; Kosek, M.; Krebs, N.F.; Loechl, C.U.; Loy, A.; Owino, V.O.; Zimmermann, M.B.; Morrison, D.J.; Morrison, D. Stable isotope techniques for the assessment of host and microbiota response during gastrointestinal dysfunction. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Van Der Borght, A.; Goesaert, H.; Veraverbeke, W.S.; Delcour, J.A. Fractionation of wheat and wheat flour into starch and gluten: Overview of the main processes and the factors involved. J. Cereal Sci. 2005, 41, 221–237. [Google Scholar] [CrossRef]
- Katz, J.; Lee, W.N.P.; Wals, P.A.; Bergner, E.A. Studies of glycogen synthesis and the Krebs cycle by mass isotopomer analysis with [U-13C]glucose in rates. J. Biol. Chem. 1989, 264, 12994–13001. [Google Scholar] [PubMed]
- Hootman, K.C.; Trezzi, J.-P.; Kraemer, L.; Burwell, L.S.; Dong, X.; Guertin, K.A.; Jaeger, C.; Stover, P.J.; Hiller, K.; Cassano, P.A.; et al. Erythritol is a pentose phosphate pathway metabolite and associated with adiposity gain in young adults. PNAS 2017, 23, 4233–4240. [Google Scholar] [CrossRef] [PubMed]
- Jiye, A.; Trygg, J.; Gullberg, J.; Johansson, A.I.; Jonsson, P.; Antti, H.; Marklund, S.L.; Moritz, T. Extraction and GC/MS analysis of the human blood plasma metabolome. Anal. Chem. 2005, 77, 8086–8094. [Google Scholar]
- Hiller, K.; Hangebrauk, J.; Jäger, C.; Spura, J.; Schreiber, K.; Schomburg, D. Metabolite Detector: Comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Anal. Chem. 2009, 81, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Boers, H.M.; Macaulay, K.; Murray, P.; Dobriyal, R.; Mela, D.J.; Spreeuwenberg, M.A.M. Efficacy of fibre additions to flatbread flour mixes for reducing post-meal glucose and insulin responses in healthy Indian subjects. Br. J. Nutr. 2017, 117, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Kind, T. Metabolite Profiling in Blood Plasma. Metabol. Methods Protoc. 2007, 358, 3–17. [Google Scholar]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Boernsen, K.O.; Gatzek, S.; Imbert, G. Controlled Protein Precipitation in Combination with Chip-Based Nanospray Infusion Mass Spectrometry. An Approach for Metabolomics Profiling of Plasma. Anal. Chem. 2005, 77, 7255–7264. [Google Scholar] [CrossRef] [PubMed]
- Heinzle, E.; Yuan, Y.; Kumar, S.; Wittmann, C.; Gehre, M.; Richnow, H.H.; Wehrung, P.; Adam, P.; Albrecht, P. Analysis of 13C labeling enrichment in microbial culture applying metabolic tracer experiments using gas chromatography-combustion-isotope ratio mass spectrometry. Anal. Biochem. 2008, 380, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; Deberardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13 C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Zabielski, P.; Ford, G.C.; Mai Persson, X.; Jaleel, A.; Dewey, J.D.; Sreekumaran Nair, K. Comparison of different mass spectrometry techniques in the measurement of L-[ring-13C6]phenylalanine incorporation into mixed muscle proteins. J. Mass Spectrom. 2013, 48, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Croyal, M.; Bourgeois, R.; Ouguerram, K.; Billon-Crossouard, S.; Aguesse, A.; Nguyen, P.; Krempf, M.; Ferchaud-Roucher, V.; Nobécourt, E. Comparison of gas chromatography-mass spectrometry and gas chromatography-combution-isotope ratio mass spectrometry analysis for in vivo estimates of metabolic fluxes. Anal. Biochem. 2016, 500, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Maštovská, K.; Lehotay, S.J. Pracitcal approaches to fast gas chromatography-mass spectrometry. J. Chromatogr. A 2003, 1000, 153–180. [Google Scholar] [CrossRef]
- Dromey, R.G.; Stefik, M.J.; Rindfleisch, T.C.; Duffield, A.M. Extraction of Mass Spectra Free of Background and Neighboring Component Contributions from Gas Chromatography/Mass Spectrometry Data. Anal. Chem. 1976, 48, 1368–1375. [Google Scholar] [CrossRef]
- Stein, S.E. An integrated method for spectrum extraction and compound identification from gas chromatography/mass spectrometry data. J. Am. Soc. Mass Spectrom. 1999, 10, 770–781. [Google Scholar] [CrossRef]
- Tissot, S.; Normand, S.; Guilluy, R.; Pachiaudi, C.; Beylot, M.; Laville, M.; Cohen, R.; Mornex, R.; Riou, J.P. Use of a new gas chromatograph isotope ratio mass spectrometer to trace exogenous 13C labeled glucose at a very low level of enrichment in man. Diabetologia 1990, 33, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.W.; Zhang, X.-J.; Chen, Y.; Klein, S.; Wolfe, R.R. Measurement of very low stable isotope enrichments by gas chromatography/mass spectrometry: Application to measurement of muscle protein synthesis. Metabolism 1997, 46, 943–948. [Google Scholar] [CrossRef]
- Huck, J.H.J.; Struys, E.A.; Verhoeven, N.M.; Jakobs, C.; Van Der Knaap, M.S. Profiling of Pentose Phosphate Pathway Intermediates in Blood Spots by Tandem Mass Spectrometry: Application to Transaldolase Deficiency. Clin. Chem. 2003, 49, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Ingraham, L.; Li, M.; Renfro, J.L.; Parker, S.; Vapurcuyan, A.; Hanna, I.; Pelis, R.M. A plasma concentration of α-ketoglutarate influences the kinetic interaction of ligands with organic anion transporter 1. Mol. Pharmacol. Mol. Pharmacol. 2014, 86, 86–95. [Google Scholar] [PubMed]
- Cynober, L.A. Plasma amino acid levels with a note on membrane transport: Characteristics, regulation, and metabolic significance. Nutrition 2002, 18, 761–766. [Google Scholar] [CrossRef]
- Moros, G.; Chatziioannou, A.C.; Gika, H.G.; Raikos, N.; Theodoridis, G. Investigation of the derivatization conditions for GC-MS metabolomics of biological samples. Bioanalysis 2017, 9, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.; Tayek, J.A. Recycling of glucose and determination of the Cori Cycle and gluconeogenesis. Am. J. Physiol. 1999, 277, E401–E407. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Nielsen, S.; Burguera, B.; Jensen, M.D. Free fatty acid turnover measured using ultralow doses of [U-13C]palmitate. J. Lipid Res. 1997, 38, 1888–1895. [Google Scholar] [PubMed]
- Korem, T.; Zeevi, D.; Zmora, N.; Weissbrod, O.; Bar, N.; Lotan-Pompan, M.; Avnit-Sagi, T.; Kosower, N.; Malka, G.; Rein, M.; et al. Bread Affects Clinical Paramters and Induces Gut Microbiome-Associated Personal Glycemic Responses. Cell Metab. 2017, 25, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Sapcariu, S.C.; Kanashova, T.; Weindl, D.; Ghelfi, J.; Dittmar, G.; Hiller, K. Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX. 2014, 1, 74–80. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Concentration Range (µM) | MI | Model Peak | No Model Peak | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Min MI Abundance (%) | Accuracy (n = 3) | Reproducibility (n = 3)/Stdev | Slope | R2 | Min MI Abundance (%) | Accuracy (n = 3) | Reproducibility (n = 3)/Stdev | Slope | R2 | |||
| Serine | 87.9–96.2 | M3 | 0.0207 | 0.0263 | 0.0054 | 0.76 | 0.98 | 0.5800 | 0.0728 | 0.0400 | 0.27 | 0.81 |
| Alanine | 302.0–330.0 | M3 | 0.0165 | 0.0059 | 0.0089 | 0.82 | 0.99 | 0.8950 | 0.0565 | 0.1293 | 0.18 | 0.65 |
| Lactate | 1540–1620 | M3 | 0.0054 | 0.0022 | 0.0013 | 1.02 | 1 | 0.0410 | 0.0062 | 0.0031 | 0.97 | 1 |
| Erythronate | 15.0–16.0 | M4 | 0.2317 | no data | 0.1545 | 1.01 | 0.99 | 4.7448 | 1.0821 | 0.3720 | 0.48 | 0.82 |
| Fumarate | 2.1–2.3 | M4 | 0.4698 | 2.4931 | 0.1227 | 0.9 | 0.92 | 5.2324 | 0.3983 | 1.8185 | 0.59 | 0.96 |
| Glutamine | 409.8–469.3 | M5 | 0.0260 | 0.0022 | 0.0135 | 0.95 | 1 | 0.3162 | 0.1518 | 0.1856 | 1.22 | 1 |
| Glucose | 6330–6631 | M6 | 0.2337 | no data | 0.0167 | 0.6 | 0.98 | 0.8812 | no data | 0.0471 | 0.34 | 0.9 |
| Glutamate | 540.1–657.0 | M5 | 0.0293 | 0.0015 | 0.0038 | 1 | 1 | 0.1024 | 0.0094 | 0.0095 | 0.98 | 1 |
| Succinate | 9.6–10.4 | M4 | 0.1001 | 0.0831 | 0.0639 | 0.73 | 0.95 | 1.8842 | 0.1708 | 0.3157 | 0.3 | 0.95 |
| Identifier | Extraction Fluid | Volume EF (µL) | Centrifugation (min) | Volume Vial (µL) |
|---|---|---|---|---|
| A [27] | Acetone/Isopropanol, 1:2 | 267 | 5 | 200 |
| B [28] | Methanol (MeOH) | 60 | 15 | 56 |
| C | Acetonitrile (ACN)/H2O, 3:1 | 180 | 5 | 140 |
| D | H2O/MeOH/ACN, 1:2:2 | 180 | 5 | 140 |
| E | MeOH/H2O, 5:1 | 180 | 5 | 140 |
| F | ACN | 77.5 | 20 | 68.25 |
| G [24] | MeOH/H2O mixture, 8:1 | 180 | 5 | 140 |
| Metabolite | Derivatization | Dwell Time (ms) | Fragment | Sum Formula | m/z |
|---|---|---|---|---|---|
| Pyruvate | 1MeOX 1TMS | 10 | M-15 | C6H12O3NSi | 174.1 |
| Lactate | 2TMS | 15 | M-15 | C8H19O3Si2 | 219.1 |
| Alanine | 2TMS | 15 | M-15 | C8H20O2NSi2 | 218.1 |
| 2-Hydroxybutyrate | 2TMS | 15 | M-15 | C9H21O3Si2 | 233.1 |
| Oxalate | 2TMS | 15 | M-15 | C7H15O4Si2 | 219.1 |
| 3-Hydroxybutyrate | 2TMS | 20 | M-15 | C9H21O3Si2 | 233.1 |
| Valine | 2TMS | 15 | M-15 | C10H24O2NSi2 | 246.1 |
| Leucine | 2TMS | 15 | M-15 | C11H26O2NSi2 | 260.2 |
| Isoleucine | 2TMS | 15 | M-15 | C11H26O2NSi2 | 260.1 |
| Glycine | 3TMS | 20 | M-15 | C10H26O2NSi3 | 276.1 |
| Succinate | 2TMS | 15 | M-15 | C9H19O4Si2 | 247.1 |
| Glycerate | 3TMS | 15 | M-30 | C10H24O4Si3 | 292.1 |
| Fumarate | 2TMS | 15 | M-15 | C9H17O4Si2 | 245.1 |
| Serine | 3TMS | 20 | M-15 | C11H28O3NSi3 | 306.1 |
| Threonine | 3TMS | 15 | M-15 | C12H30O3NSi3 | 320.2 |
| Malate | 3TMS | 15 | M-15 | C12H27O5Si3 | 335.1 |
| Erythritol | 4TMS | 20 | M-90 | C10H24O4Si4 | 320.2 |
| Aspartate | 3TMS | 15 | M-15 | C12H28O4NSi3 | 334.1 |
| Erythronate | 4TMS | 20 | M-15 | C15H37O5Si4 | 409.2 |
| α-Ketoglutarate | 1MeOX 2TMS | 10 | M-15 | C11H22O5NSi2 | 304.1 |
| Glutamate | 3TMS | 15 | M-15 | C13H30O4NSi3 | 348.1 |
| Glutamine | 3TMS | 15 | M-15 | C13H31O3N2Si3 | 347.2 |
| Citrate | 4TMS | 15 | M-15 | C17H37O7Si4 | 465.2 |
| Glucose | 1MeOX 5TMS | 15 | M-15 | C21H52O6NSi5 | 554.3 |
| Tyrosine | 3TMS | 10 | M-15 | C17H32O3NSi3 | 382.2 |
| Internal Standard (IS) | Derivatization | Ions |
|---|---|---|
| Norleucine | 2TMS | 158.1, 232.1, 260.1 |
| Pentanedioic-acid-d6 | 2TMS | 206.1, 239.1, 267.1 |
| U13C-Ribitol | 2TMS | 220.1, 310.1, 323.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krämer, L.; Jäger, C.; Trezzi, J.-P.; Jacobs, D.M.; Hiller, K. Quantification of Stable Isotope Traces Close to Natural Enrichment in Human Plasma Metabolites Using Gas Chromatography-Mass Spectrometry. Metabolites 2018, 8, 15. https://doi.org/10.3390/metabo8010015
Krämer L, Jäger C, Trezzi J-P, Jacobs DM, Hiller K. Quantification of Stable Isotope Traces Close to Natural Enrichment in Human Plasma Metabolites Using Gas Chromatography-Mass Spectrometry. Metabolites. 2018; 8(1):15. https://doi.org/10.3390/metabo8010015
Chicago/Turabian StyleKrämer, Lisa, Christian Jäger, Jean-Pierre Trezzi, Doris M. Jacobs, and Karsten Hiller. 2018. "Quantification of Stable Isotope Traces Close to Natural Enrichment in Human Plasma Metabolites Using Gas Chromatography-Mass Spectrometry" Metabolites 8, no. 1: 15. https://doi.org/10.3390/metabo8010015
APA StyleKrämer, L., Jäger, C., Trezzi, J.-P., Jacobs, D. M., & Hiller, K. (2018). Quantification of Stable Isotope Traces Close to Natural Enrichment in Human Plasma Metabolites Using Gas Chromatography-Mass Spectrometry. Metabolites, 8(1), 15. https://doi.org/10.3390/metabo8010015