
Metabolic Portraits of Breast Cancer by HR MAS MR Spectroscopy of Intact Tissue Samples


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
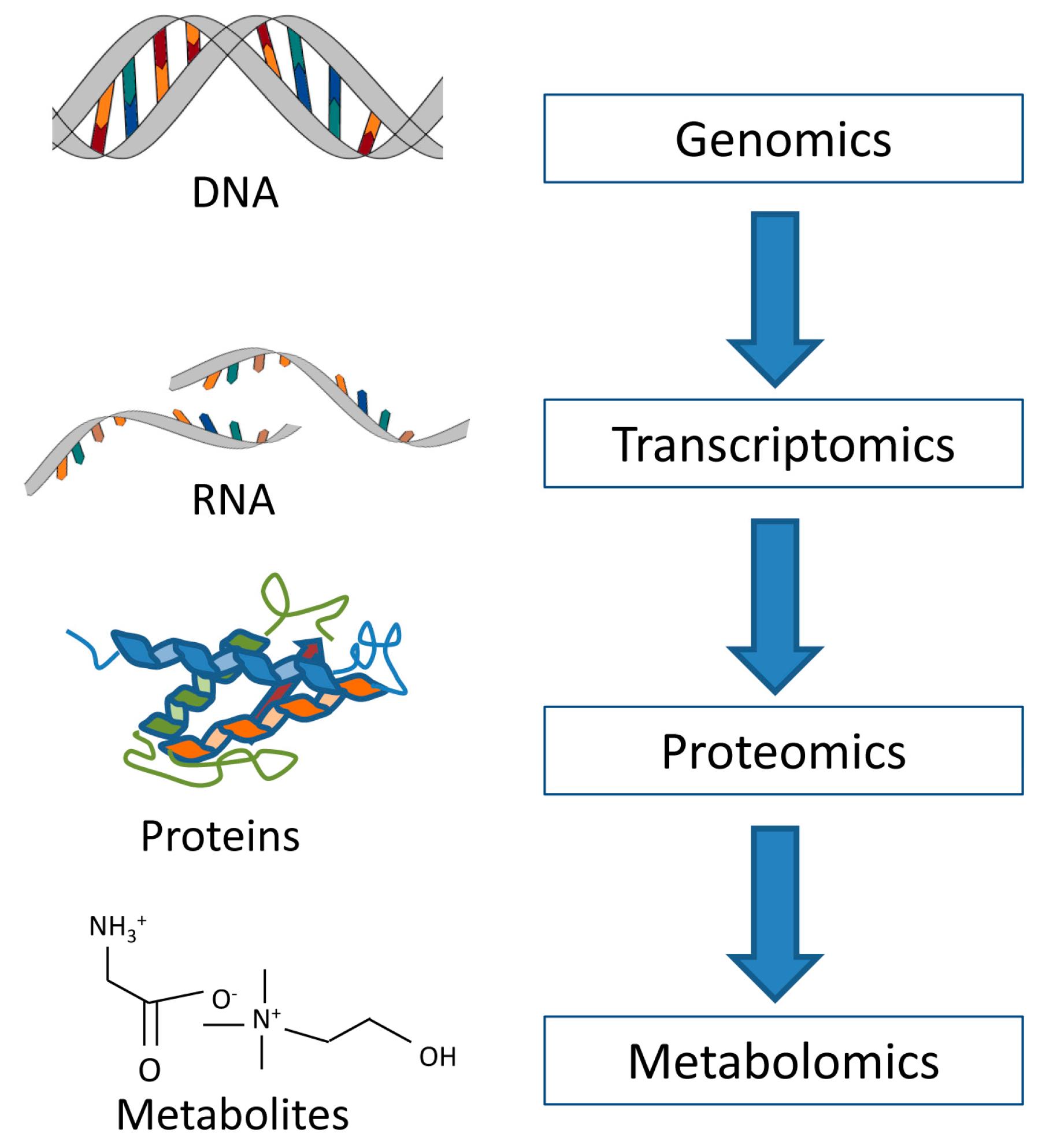
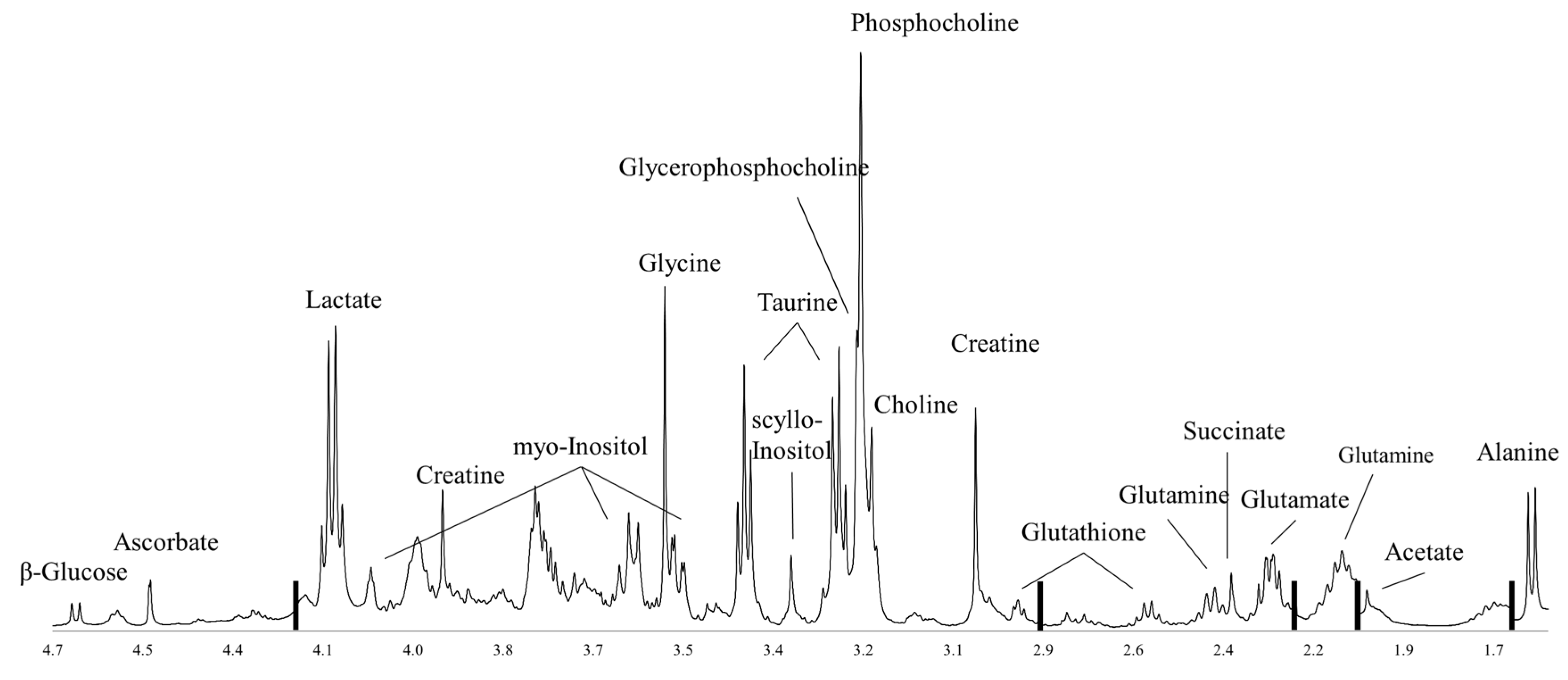
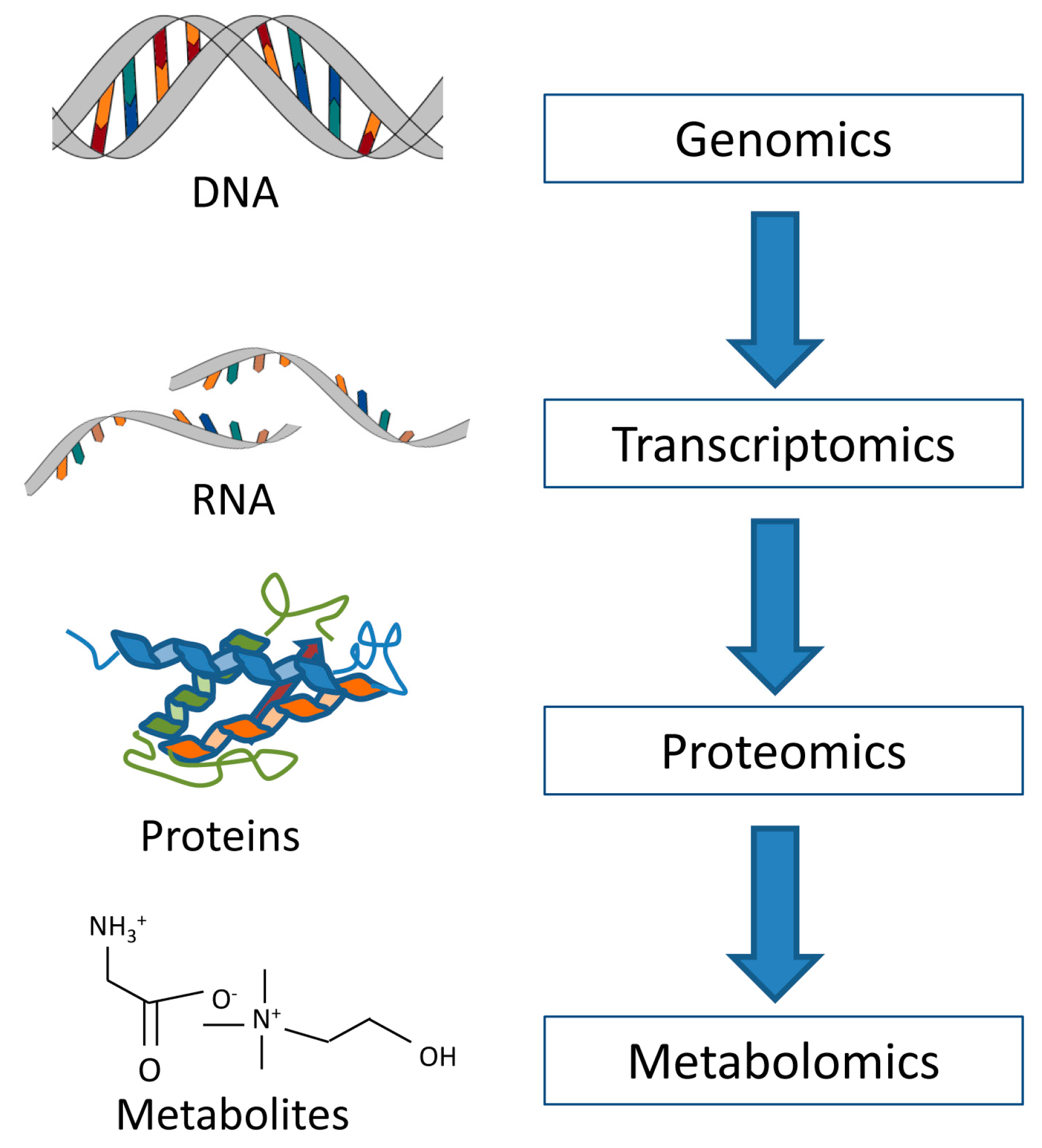
:1. Introduction
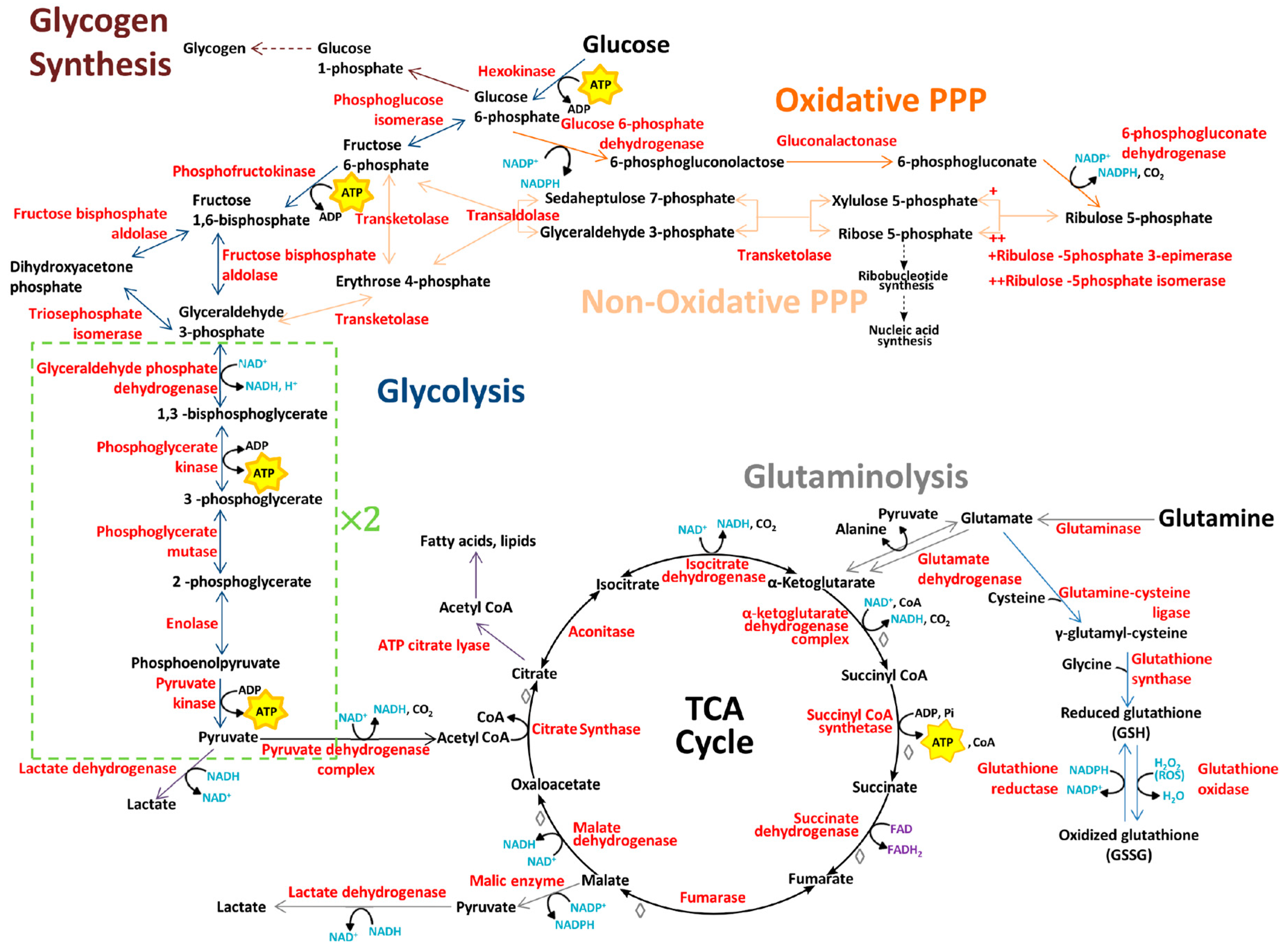
2. Glucose Metabolism
3. Amino Acid Metabolism
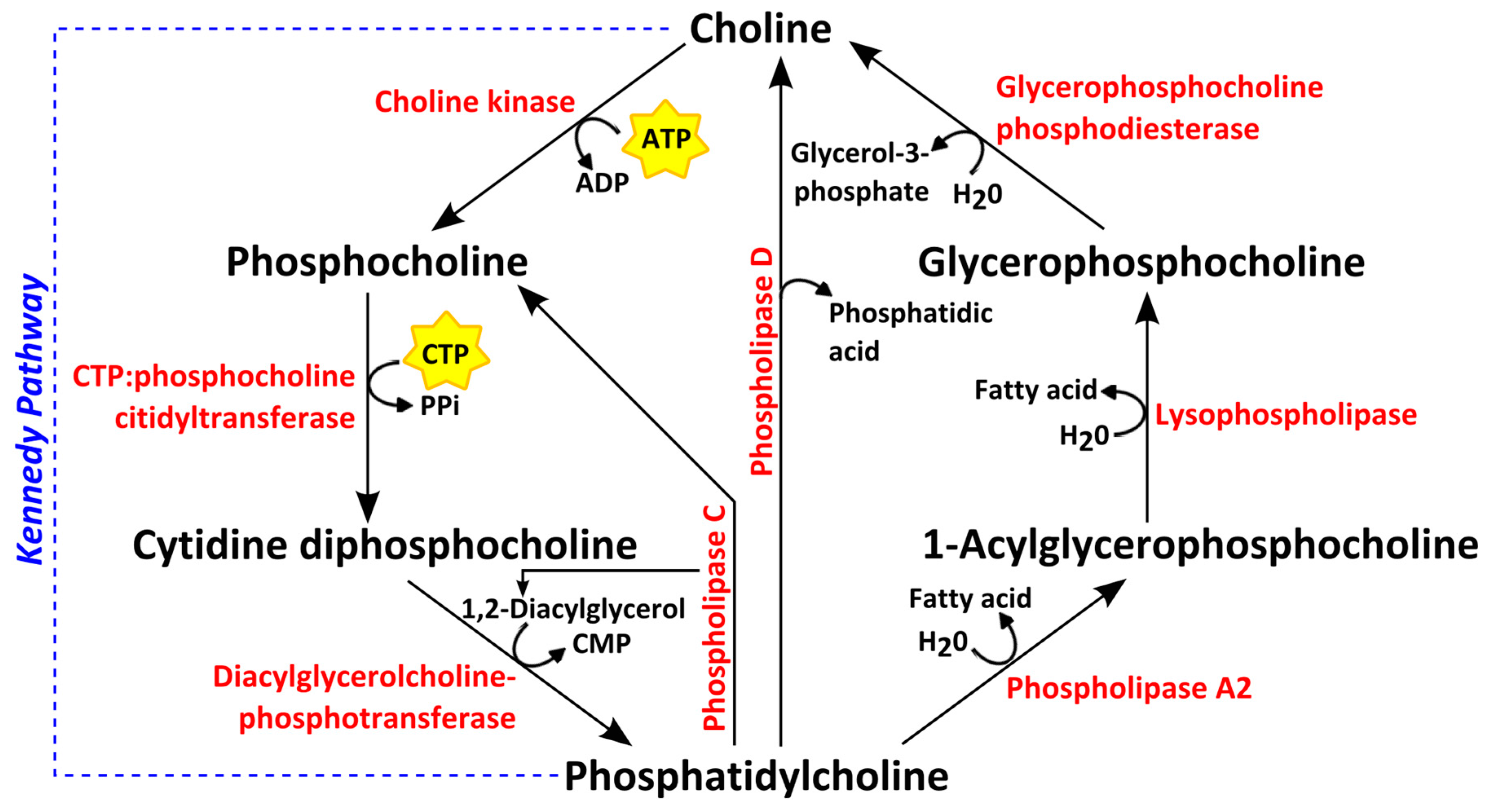
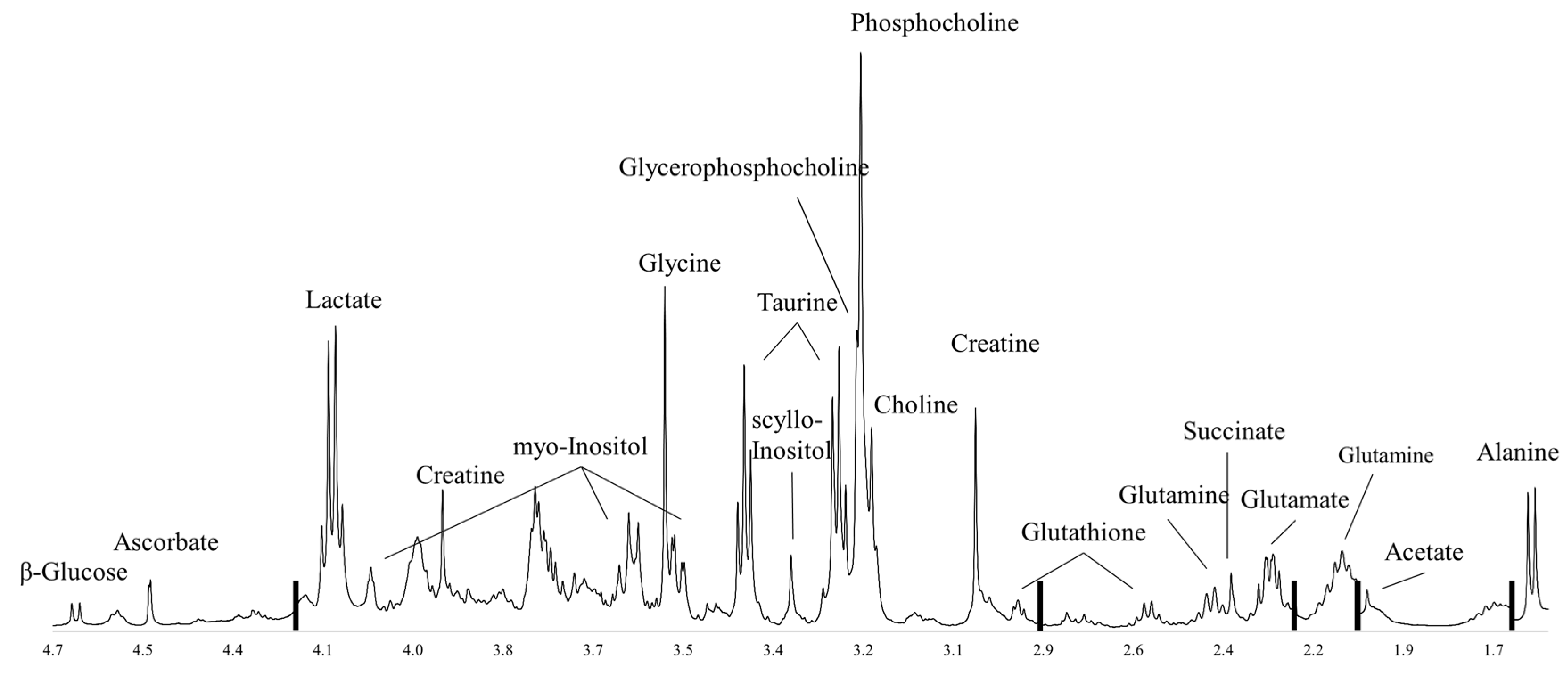
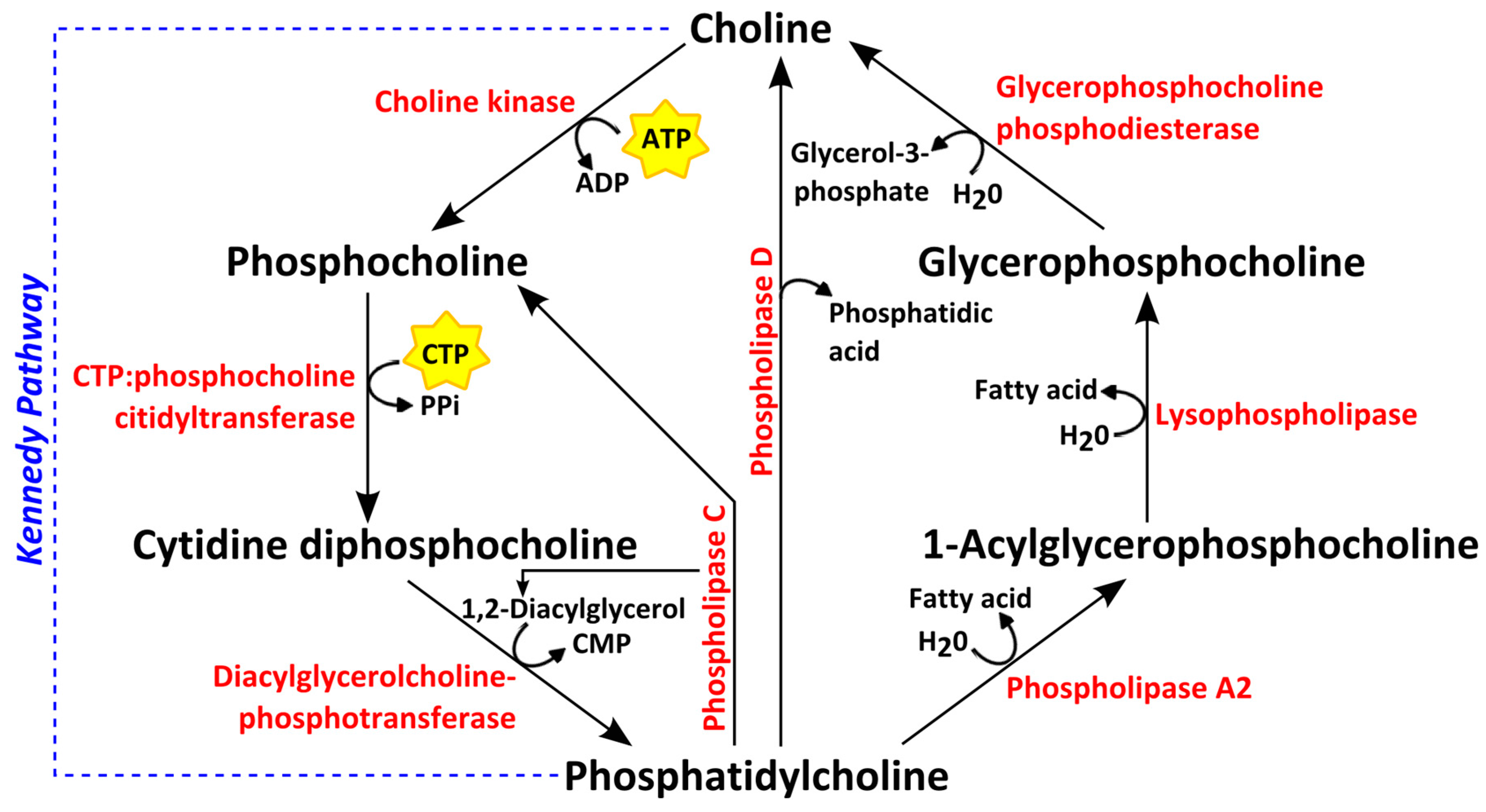
4. Choline Phospholipid Metabolism
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Cancer Registry of Norway. Cancer In Norway 2015—Cancer Incidence, Mortality, Survival and Prevalence In Norway; Cancer Registry of Norway: Oslo, Norway, 2016. [Google Scholar]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, L.; Press, M. Does estrogen receptor expression in normal breast tissue predict breast cancer risk? J. Natl. Cancer Inst. 1998, 90, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Weigel, M.T.; Dowsett, M. Current and emerging biomarkers in breast cancer: Prognosis and prediction. Endocr. Relat. Cancer 2010, 17, R245–R262. [Google Scholar] [CrossRef] [PubMed]
- Dunnwald, L.; Rossing, M.; Li, C. Hormone receptor status, tumor characteristics, and prognosis: A prospective cohort of breast cancer patients. Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef] [PubMed]
- Engstrøm, M.; Opdahl, S.; Hagen, A.; Romundstad, P.; Akslen, L.; Haugen, O.; Vatten, L.; Bofin, A. Molecular subtypes, histopathological grade and survival in a historic cohort of breast cancer patients. Breast Cancer Res. Treat. 2013, 140, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lei, Y.; Mei, J.; Wang, C. Recent progress in HER2 associated breast cancer. Asian Pac. J. Cancer Prev. 2014, 16, 2591–2600. [Google Scholar] [CrossRef]
- Figueroa-Magalhães, M.C.; Jelovac, D.; Connolly, R.M.; Wolff, A.C. Treatment of HER2-positive breast cancer. Breast 2014, 23, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Yerushalmi, R.; Woods, R.; Ravdin, P.M.; Hayes, M.M.; Gelmon, K.A. Ki67 in breast cancer: Prognostic and predictive potential. Lancet Oncol. 2010, 11, 174–183. [Google Scholar] [CrossRef]
- Metzger-Filho, O.; Tutt, A.; de Azambuja, E.; Saini, K.S.; Viale, G.; Loi, S.; Bradbury, I.; Bliss, J.M.; Azim, H.A.; Ellis, P.; et al. Dissecting the heterogeneity of triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 1879–1887. [Google Scholar] [CrossRef] [PubMed]
- Ismail-Khan, R.; Bui, M.M. A review of triple-negative breast cancer. Cancer Control 2010, 17, 173–176. [Google Scholar] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Hennessy, B.; Lu, Y.; Gonzalez-Angulo, A.; Carey, M.; Myhre, S.; Ju, Z.; Davies, M.; Liu, W.; Coombes, K.; Meric-Bernstam, F. A technical assessment of the utility of reverse phase protein arrays for the study of the functional proteome in non-microdissected human breast cancers. Clin. Proteom. 2010, 6, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Birnbaum, D. Reasons for breast cancer heterogeneity. J. Biol. 2008, 7. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim. Biophys. Acta 2012, 1826, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.; Thompson, C. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Bathen, T.F.; Sitter, B.; Sjøbakk, T.E.; Tessem, M.-B.; Gribbestad, I.S. Magnetic resonance metabolomics of intact tissue: A biotechnological tool in cancer diagnostics and treatment evaluation. Cancer Res. 2010, 70, 6692–6696. [Google Scholar] [CrossRef] [PubMed]
- Andrew, E.R. The narrowing of NMR spectra of solids by high-speed specimen rotation and the resolution of chemical shift and spin multiplet structures for solids. Prog. Nucl. Magn. Reson. Spectrosc. 1971, 8, 1–39. [Google Scholar] [CrossRef]
- Haukaas, T.H.; Moestue, S.A.; Vettukattil, R.; Sitter, B.; Lamichhane, S.; Segura, R.; Giskeødegård, G.F.; Bathen, T.F. Impact of freezing delay time on tissue samples for metabolomic studies. Front. Oncol. 2016, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Moestue, S.A.; Sitter, B.; Bathen, T.F.; Tessem, M.-B.; Gribbestad, I.S. HR MAS MR spectroscopy in metabolic characterization of cancer. Curr. Top. Med. Chem. 2011, 11, 2–26. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.J.; Fellows, G.A.; Griffiths, J.R.; Wilson, M.; Bell, B.A.; Howe, F.A. Ex-vivo HRMAS of adult brain tumours: Metabolite quantification and assignment of tumour biomarkers. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Emir, U.E.; Deelchand, D.; Henry, P.G.; Terpstra, M. Noninvasive quantification of T2 and concentrations of ascorbate and glutathione in the human brain from the same double-edited spectra. NMR Biomed. 2011, 24, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Yoon, D.; Yun, M.; Choi, J.S.; Park, V.Y.; Kim, E.-K.; Jeong, J.; Koo, J.S.; Yoon, J.H.; Moon, H.J.; et al. Metabolomics of breast cancer using high-resolution magic angle spinning magnetic resonance spectroscopy: Correlations with 18F-FDG positron emission tomography-computed tomography, dynamic contrast-enhanced and diffusion-weighted imaging MRI. PLoS ONE 2016, 11, e0159949. [Google Scholar] [CrossRef] [PubMed]
- Sitter, B.; Sonnewald, U.; Spraul, M.; Fjøsne, H.E.; Gribbestad, I.S. High-resolution magic angle spinning MRS of breast cancer tissue. NMR Biomed. 2002, 15, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E. P53 guards the metabolic pathway less travelled. Nat. Cell Biol. 2011, 13, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Bellot, G.; Pouyssegur, J. Hypoxia and energetic tumour metabolism. Curr. Opin. Genet. Dev. 2011, 21, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Zois, C.E.; Favaro, E.; Harris, A.L. Glycogen metabolism in cancer. Biochem. Pharmacol. 2014, 92, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E. Colorectal cancer: Lactate dehydrogenase (LDH) activity as a prognostic marker. In Methods of Cancer Diagnosis, Therapy, and Prognosis; Hayat, M.A., Ed.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Warburg, O. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology, Translated by Frank Dickens; Constable & Co. Ltd.: London, UK, 1930. [Google Scholar]
- Hirschhaeuser, F.; Sattler, U.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.; Gillies, R. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Stokkel, M.P.; Draisma, A.; Pauwels, E.K. Positron emission tomography with 2-[18F]-fluoro-2-deoxy-D-glucose in oncology. Part IIIb: Therapy response monitoring in colorectal and lung tumours, head and neck cancer, hepatocellular carcinoma and sarcoma. J. Cancer Res. Clin. Oncol. 2001, 127, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Beckonert, O.; Monnerjahn, J.; Bonk, U.; Leibfritz, D. Visualizing metabolic changes in breast-cancer tissue using 1H-NMR spectroscopy and self-organizing maps. NMR Biomed. 2003, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sitter, B.; Bathen, T.F.; Singstad, T.; Fjøsne, H.E.; Lundgren, S.; Halgunset, J.; Gribbestad, I.S. Quantification of metabolites in breast cancer patients with different clinical prognosis using HR MAS MR spectroscopy. NMR Biomed. 2010, 23, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Grinde, M.T.; Moestue, S.A.; Borgan, E.; Risa, Ø.; Engebraaten, O.; Gribbestad, I.S. 13C high-resolution-magic angle spinning MRS reveals differences in glucose metabolism between two breast cancer xenograft models with different gene expression patterns. NMR Biomed. 2011, 24, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Borgan, E.; Sitter, B.; Lingjærde, O.; Johnsen, H.; Lundgren, S.; Bathen, T.F.; Sørlie, T.; Børresen-Dale, A.-L.; Gribbestad, I.S. Merging transcriptomics and metabolomics-advances in breast cancer profiling. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Haukaas, T.H.; Euceda, L.R.; Giskeødegård, G.F.; Lamichhane, S.; Krohn, M.; Jernström, S.; Aure, M.R.; Lingjærde, O.C.; Schlichting, E.; Garred, Ø.; et al. Metabolic clusters of breast cancer in relation to gene- and protein expression subtypes. Cancer Metab. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Giskeødegård, G.F.; Bathen, T.F.; Sitter, B.; Bofin, A.; Lønning, P.E.; Lundgren, S.; Gribbestad, I.S. Prognostic value of metabolic response in breast cancer patients receiving neoadjuvant chemotherapy. BMC Cancer 2012, 12, 12–39. [Google Scholar] [CrossRef] [PubMed]
- Euceda, L.R.; Haukaas, T.H.; Giskeødegård, G.F.; Vettukattil, R.; Engel, J.; Silwal-Pandit, L.; Lundgren, S.; Borgen, E.; Garred, Ø.; Postma, G.; et al. Evaluation of metabolomic changes during neoadjuvant chemotherapy combined with bevacizumab in breast cancer using MR spectroscopy. Metabolomics 2017, 13. [Google Scholar] [CrossRef]
- Cao, M.D.; Sitter, B.; Bathen, T.F.; Bofin, A.; Lønning, P.E.; Lundgren, S.; Gribbestad, I.S. Predicting long-term survival and treatment response in breast cancer patients receiving neoadjuvant chemotherapy by MR metabolic profiling. NMR Biomed. 2012, 25, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Walenta, S.; Schroeder, T.; Mueller-Klieser, W. Lactate in solid malignant tumors: Potential basis of a metabolic classification in clinical oncology. Curr. Med. Chem. 2004, 11, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Giskeødegård, G.F.; Grinde, M.T.; Sitter, B.; Axelson, D.; Lundgren, S.; Fjøsne, H.E.; Dahl, S.; Gribbestad, I.S.; Bathen, T.F. Multivariate modeling and prediction of breast cancer prognostic factors using MR metabolomics. J. Proteom. Res. 2010, 9, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Lin, C.-C.; Spasojevic, I.; Iversen, E.S.; Chi, J.-T.; Marks, J.R. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Giskeødegård, G.F.; Lundgren, S.; Sitter, B.; Fjøsne, H.E.; Postma, G.; Buydens, L.M.C.; Gribbestad, I.S.; Bathen, T.F. Lactate and glycine—potential MR biomarkers of prognosis in estrogen receptor-positive breast cancers. NMR Biomed. 2012, 25, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Mehta, A.; Seenu, V.; Jagannathan, N. Biochemical characterization of metastatic lymph nodes of breast cancer patients by in vitro 1H magnetic resonance spectroscopy: A pilot study. Magn. Reson. Imaging 2004, 22, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Seenu, V.; Kumar, M.; Sharma, U.; Gupta, S.; Mehta, S.; Jagannathan, N. Potential of magnetic resonance spectroscopy to detect metastasis in axillary lymph nodes in breast cancer. Magn. Reson. Imaging 2005, 23, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Elf, S.E.; Chen, J. Targeting glucose metabolism in patients with cancer. Cancer 2014, 120, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Islamian, J.; Aghaee, F.; Farajollahi, A.; Baradaran, B.; Fazel, M. Combined treatment with 2-deoxy-D-glucose and doxorubicin enhances the in vitro efficiency of breast cancer radiotherapy. Asian Pac. J. Cancer Prev. 2014, 16, 8431–8438. [Google Scholar] [CrossRef]
- Ma, S.; Jia, R.; Li, D.; Shen, B. Targeting cellular metabolism chemosensitizes the doxorubicin-resistant human breast adenocarcinoma cells. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.-Y.; Zhang, Q.-W.; Zhao, S.-R.; Wu, C.-Z.; Cheng, X.; Jiang, C.-C.; Jiang, Z.-W.; Liu, H. 3-Bromopyruvate induces apoptosis in breast cancer cells by downregulating Mcl-1 through the PI3K/Akt signaling pathway. Anti-Cancer Drugs 2014, 25, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Y.; Zhang, P.; Chao, Z.; Xia, F.; Jiang, C.; Zhang, X.; Jiang, Z.; Liu, H. Hexokinase II inhibitor, 3-BrPa induced autophagy by stimulating ROS formation in human breast cancer cells. Genes Cancer 2014, 5. [Google Scholar] [CrossRef]
- Champe, P.C.; Harvey, R.A.; Ferrier, D.R. Biochemistry; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 2016, 6, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.; Thompson, C. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Cassago, A.; Ferreira, A.P.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.; Greene, K.S.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.; Ambrosio, A.L. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Lamichhane, S.; Lundgren, S.; Bofin, A.; Fjøsne, H.E.; Giskeødegård, G.F.; Bathen, T.F. Metabolic characterization of triple negative breast cancer. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cioce, M.; Valerio, M.; Casadei, L.; Pulito, C.; Sacconi, A.; Mori, F.; Biagioni, F.; Manetti, C.; Muti, P.; Strano, S. Metformin-induced metabolic reprogramming of chemoresistant ALDHbright breast cancer cells. Oncotarget 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Sitter, B.; Lundgren, S.; Bathen, T.F.; Halgunset, J.; Fjøsne, H.E.; Gribbestad, I.S. Comparison of HR MAS MR spectroscopic profiles of breast cancer tissue with clinical parameters. NMR Biomed. 2006, 19, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Song, Y.; Cho, N.; Chang, J.; Koo, H.; Yi, A.; Kim, H.; Park, S.; Moon, W. An HR-MAS MR metabolomics study on breast tissues obtained with core needle biopsy. PLoS ONE 2011, 6, e25563. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Baek, H.-M.; Kim, S.; Kim, M.; Youk, J.; Moon, H.; Kim, E.-K.; Han, K.; Kim, D.-H.; Kim, S. HR-MAS MR spectroscopy of breast cancer tissue obtained with core needle biopsy: Correlation with prognostic factors. PLoS ONE 2012, 7, e51712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, H.; Wang, Y.; Liu, C.; Zhu, W.; Zheng, S.; Wan, F. Taurine induces the apoptosis of breast cancer cells by regulating apoptosis-related proteins of mitochondria. Int. J. Mol. Med. 2015, 35, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Moestue, S.A.; Borgan, E.; Huuse, E.M.; Lindholm, E.M.; Sitter, B.; Børresen-Dale, A.-L.; Engebraaten, O.; Mælandsmo, G.M.; Gribbestad, I.S. Distinct choline metabolic profiles are associated with differences in gene expression for basal-like and luminal-like breast cancer xenograft models. BMC Cancer 2010, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, F.; Smith, T. The kennedy pathway—De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Katz-Brull, R.; Seger, D.; Rivenson-Segal, D.; Rushkin, E.; Degani, H. Metabolic markers of breast cancer enhanced choline metabolism and reduced choline-ether-phospholipid synthesis. Cancer Res. 2002, 62, 1966–1970. [Google Scholar] [PubMed]
- Fagone, P.; Jackowski, S. Phosphatidylcholine and the CDP–choline cycle. Biochim. Biophys. Acta 2013, 1831, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, N.D. The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Katz-Brull, R.; Degani, H. Kinetics of choline transport and phosphorylation in human breast cancer cells; NMR application of the zero trans method. Anticancer Res. 1996, 16, 1375–1380. [Google Scholar] [PubMed]
- Iorio, E.; Mezzanzanica, D.; Alberti, P.; Spadaro, F.; Ramoni, C.; D'Ascenzo, S.; Millimaggi, D.; Pavan, A.; Dolo, V.; Canevari, S.; et al. Alterations of choline phospholipid metabolism in ovarian tumor progression. Cancer Res. 2005, 65, 9369–9376. [Google Scholar] [CrossRef] [PubMed]
- De Molina, A.R.; Rodrı́guez-González, A.; Gutiérrez, R.; Martınez-Pineiro, L.; Sánchez, J.J.; Bonilla, F.; Rosell, R.; Lacal, J.C. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem. Biophys. Res. Commun. 2002, 296, 580–583. [Google Scholar] [CrossRef]
- Granata, A.; Nicoletti, R.; Tinaglia, V.; De Cecco, L.; Pisanu, M.; Ricci, A.; Podo, F.; Canevari, S.; Iorio, E.; Bagnoli, M. Choline kinase-alpha by regulating cell aggressiveness and drug sensitivity is a potential druggable target for ovarian cancer. Br. J. Cancer 2014, 110, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Jie, C.; Bhujwalla, Z.M. Molecular causes of the aberrant choline phospholipid metabolism in breast cancer. Cancer Res. 2004, 64, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Noh, D.Y.; Ahn, S.J.; Lee, R.A.; Park, I.A.; Kim, J.H.; Suh, P.G.; Ryu, S.H.; Lee, K.H.; Han, J.S. Overexpression of phospholipase D1 in human breast cancer tissues. Cancer Lett. 2000, 161, 207–214. [Google Scholar] [CrossRef]
- Cao, M.D.; Döpkens, M.; Krishnamachary, B.; Vesuna, F.; Gadiya, M.; Lønning, P.E.; Bhujwalla, Z.M.; Gribbestad, I.S.; Glunde, K. Glycerophosphodiester phosphodiesterase domain containing 5 (GDPD5) expression correlates with malignant choline phospholipid metabolite profiles in human breast cancer. NMR Biomed. 2012, 25, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.D.; Marchan, R.; Lesjak, M.S.; Lambert, J.; Hergenroeder, R.; Ellis, J.K.; Lau, C.H.; Keun, H.C.; Schmitz, G.; Schiller, J.; et al. Choline-releasing glycerophosphodiesterase EDI3 drives tumor cell migration and metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 8155–8160. [Google Scholar] [CrossRef] [PubMed]
- Grinde, M.T.; Skrbo, N.; Moestue, S.A.; Rødland, E.; Borgan, E.; Kristian, A.; Sitter, B.; Bathen, T.F.; Børresen-Dale, A.-L.; Mælandsmo, G.; et al. Interplay of choline metabolites and genes in patient-derived breast cancer xenografts. Breast Cancer Res. 2014, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lacal, J.C.; Campos, J.M. Preclinical characterization of RSM-932A, a novel anticancer drug targeting the human choline kinase alpha, an enzyme involved in increased lipid metabolism of cancer cells. Mol. Cancer Ther. 2015, 14, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Traslational Cancer Drugs Pharma SL. Study of Intravenous TCD-717 in Patients with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01215864 (accessed on 16 March 2017).
- Aboagye, E.O.; Bhujwalla, Z.M. Malignant transformation alters membrane choline phospholipid metabolism of human mammary epithelial cells. Cancer Res. 1999, 59, 80–84. [Google Scholar] [PubMed]
- Maria, R.M.; Altei, W.F.; Andricopulo, A.D.; Becceneri, A.B.; Cominetti, M.R.; Venâncio, T.; Colnago, L.A. Characterization of metabolic profile of intact non-tumor and tumor breast cells by high-resolution magic angle spinning nuclear magnetic resonance spectroscopy. Anal. Biochem. 2015, 488, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Galons, J.P.; Job, C.; Gillies, R.J. Increase of GPC levels in cultured mammalian cells during acidosis. A 31P MR spectroscopy study using a continuous bioreactor system. Magn. Reson. Med. 1995, 33, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, N.; Kumar, M.; Seenu, V.; Coshic, O.; Dwivedi, S.; Julka, P.; Srivastava, A.; Rath, G. Evaluation of total choline from in-vivo volume localized proton MR spectroscopy and its response to neoadjuvant chemotherapy in locally advanced breast cancer. Br. J. Cancer 2001, 84, 1016. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.M.; Chen, J.H.; Nalcioglu, O.; Su, M.Y. Proton MR spectroscopy for monitoring early treatment response of breast cancer to neo-adjuvant chemotherapy. Ann. Oncol. 2008, 19, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- Moestue, S.A.; Dam, C.G.; Gorad, S.S.; Kristian, A.; Bofin, A.; Maelandsmo, G.M.; Engebraten, O.; Gribbestad, I.S.; Bjørkøy, G. Metabolic biomarkers for response to PI3K inhibition in basal-like breast cancer. Breast Cancer Res. 2013, 15, R16. [Google Scholar] [CrossRef] [PubMed]
- Euceda, L.R.; Hill, D.K.; Stokke, E.; Hatem, R.; Botty, R.E.; Bieche, I.; Marangoni, E.; Bathen, T.F.; Moestue, S.A. Metabolic response to everolimus in patient-derived triple negative breast cancer xenografts. J. Proteom. Res. 2017, 16, 1868–1879. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haukaas, T.H.; Euceda, L.R.; Giskeødegård, G.F.; Bathen, T.F. Metabolic Portraits of Breast Cancer by HR MAS MR Spectroscopy of Intact Tissue Samples. Metabolites 2017, 7, 18. https://doi.org/10.3390/metabo7020018
Haukaas TH, Euceda LR, Giskeødegård GF, Bathen TF. Metabolic Portraits of Breast Cancer by HR MAS MR Spectroscopy of Intact Tissue Samples. Metabolites. 2017; 7(2):18. https://doi.org/10.3390/metabo7020018
Chicago/Turabian StyleHaukaas, Tonje H., Leslie R. Euceda, Guro F. Giskeødegård, and Tone F. Bathen. 2017. "Metabolic Portraits of Breast Cancer by HR MAS MR Spectroscopy of Intact Tissue Samples" Metabolites 7, no. 2: 18. https://doi.org/10.3390/metabo7020018
APA StyleHaukaas, T. H., Euceda, L. R., Giskeødegård, G. F., & Bathen, T. F. (2017). Metabolic Portraits of Breast Cancer by HR MAS MR Spectroscopy of Intact Tissue Samples. Metabolites, 7(2), 18. https://doi.org/10.3390/metabo7020018