
A Simple Method for Measuring Carbon-13 Fatty Acid Enrichment in the Major Lipid Classes of Microalgae Using GC-MS

Abstract
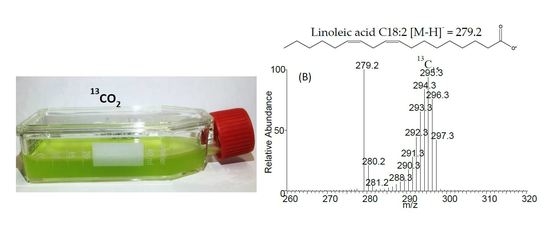
:
1. Introduction
2. Results
2.1. Selection of Optimum Solvent Mixture for Lipid Extraction
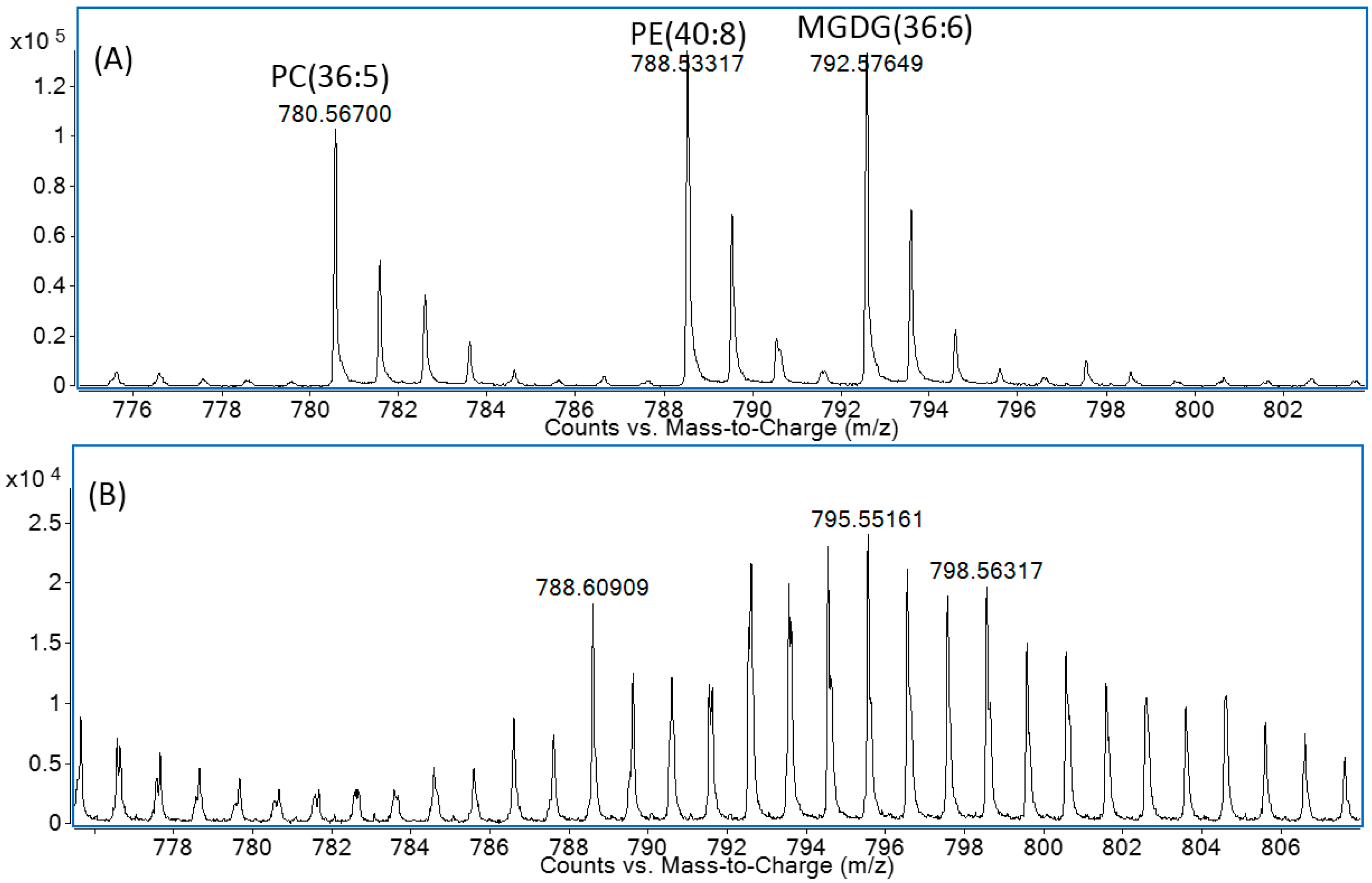
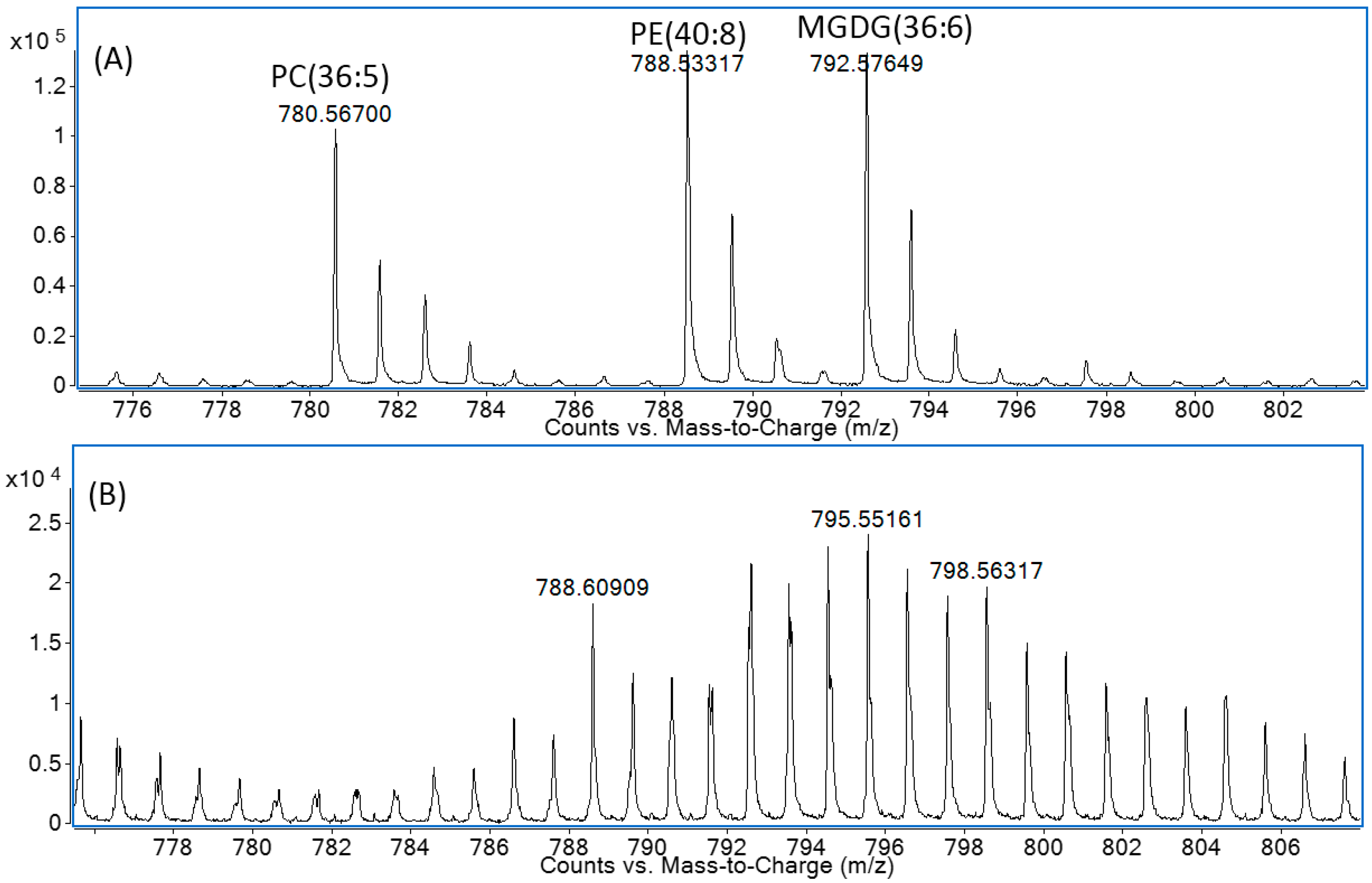
2.2. Solid-Phase Extraction Method
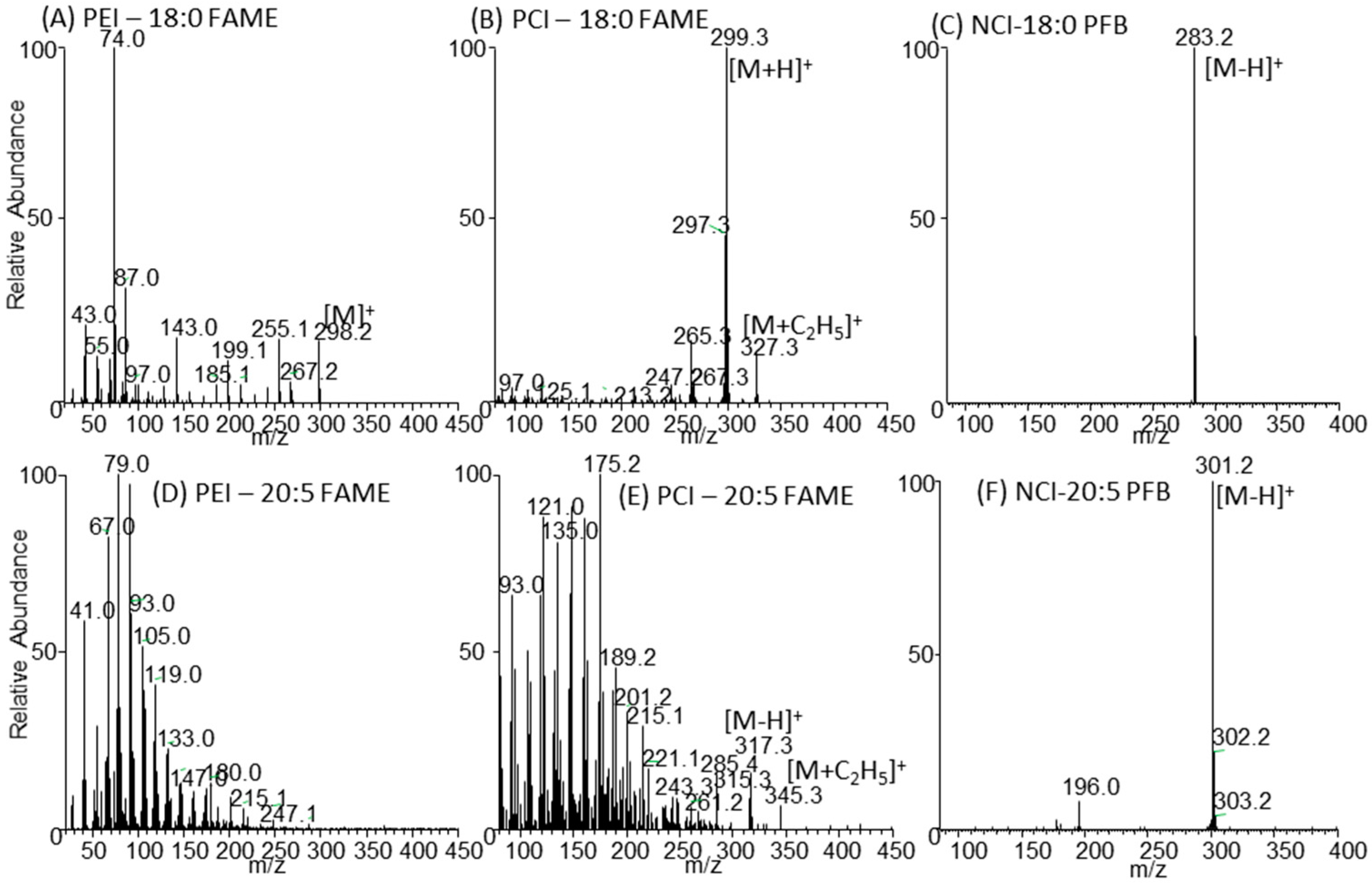
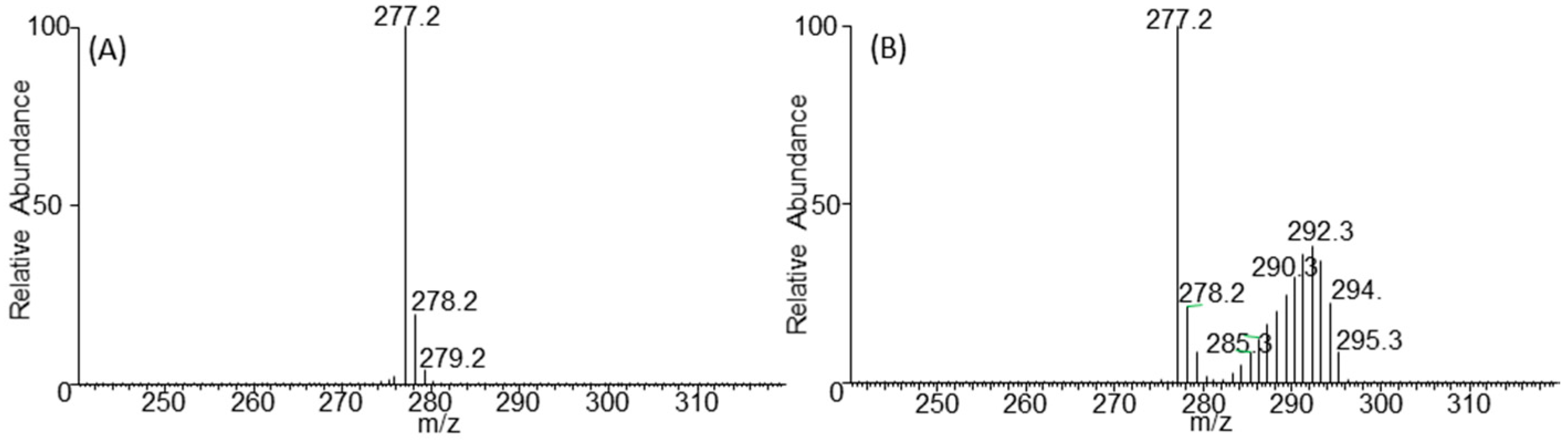
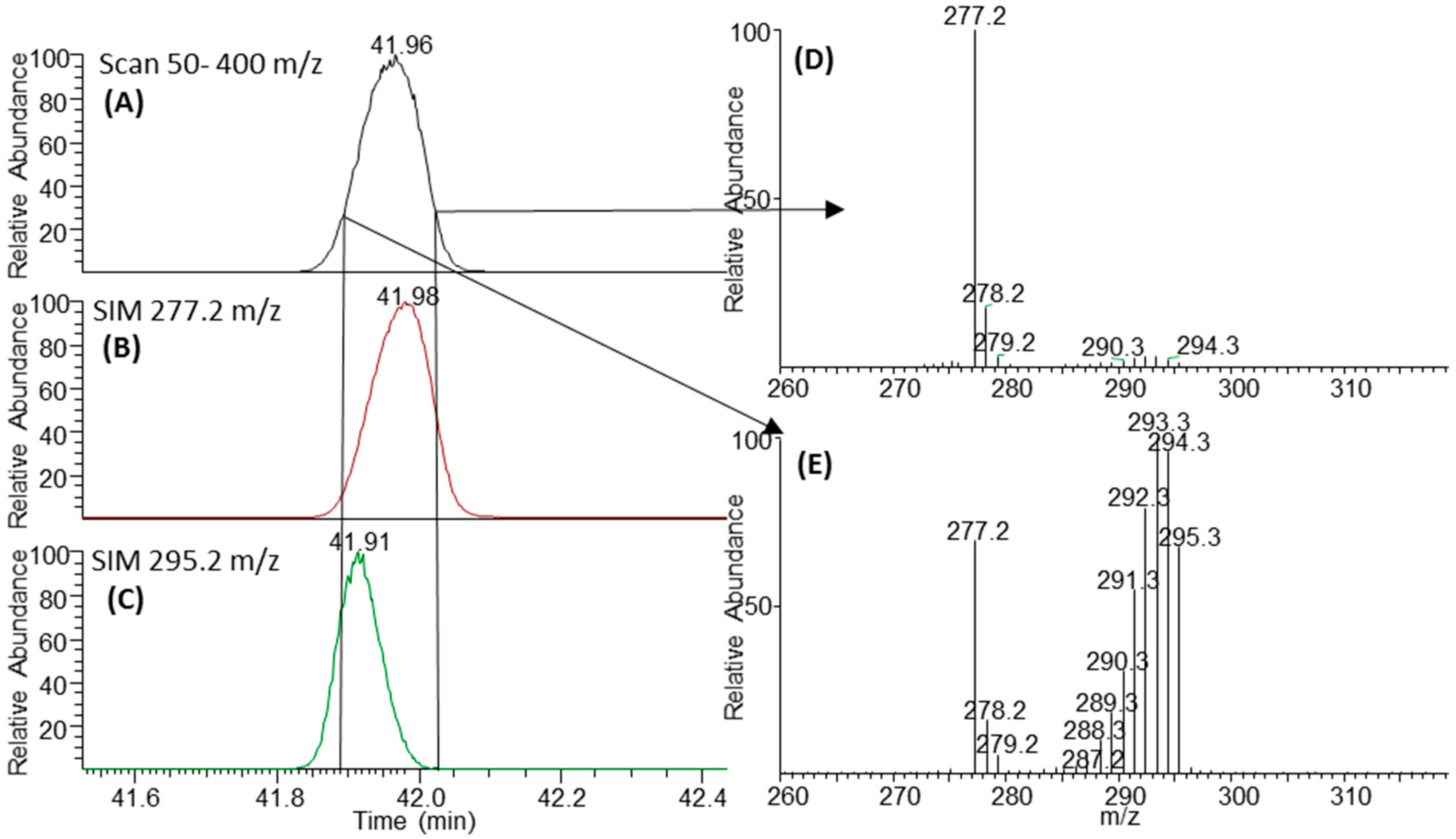
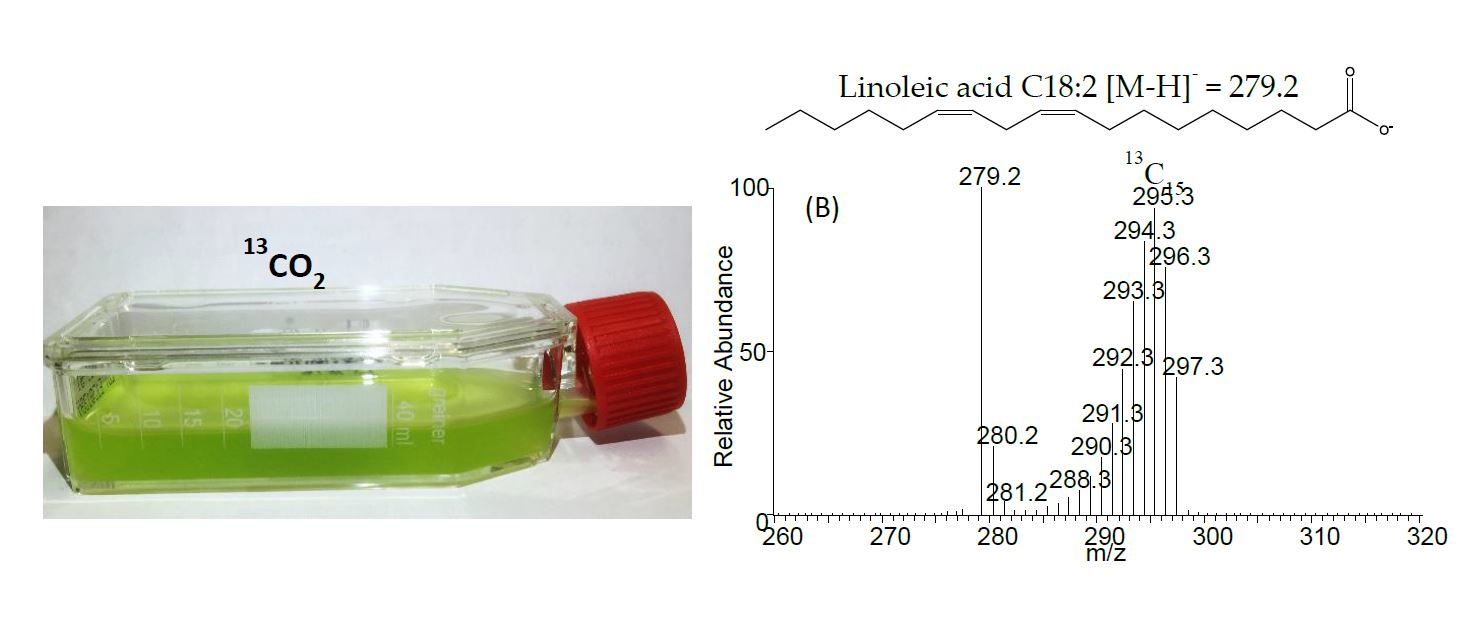
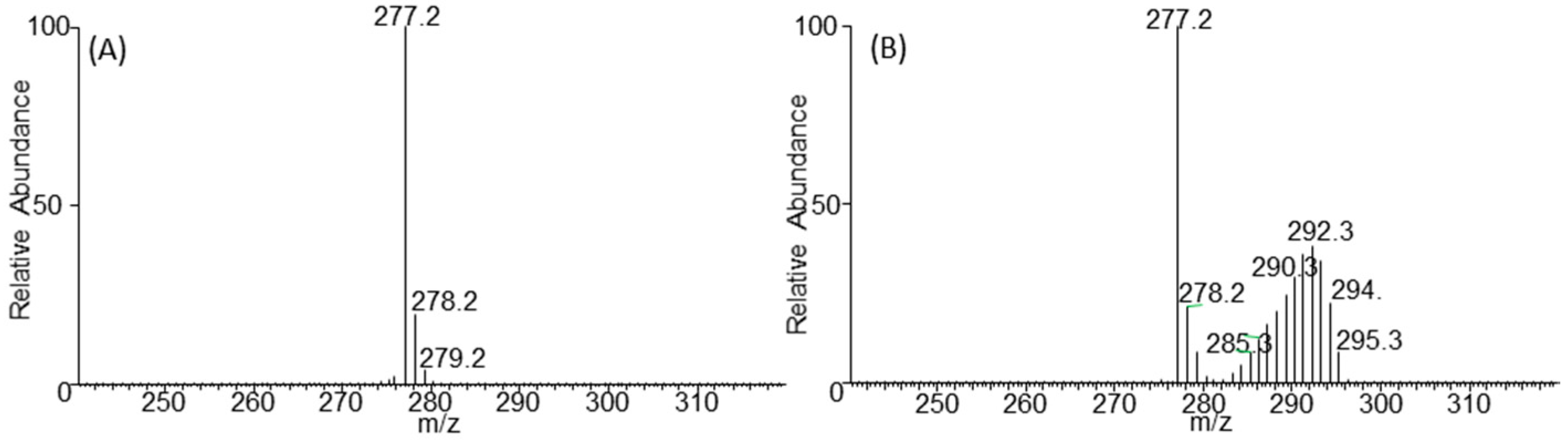
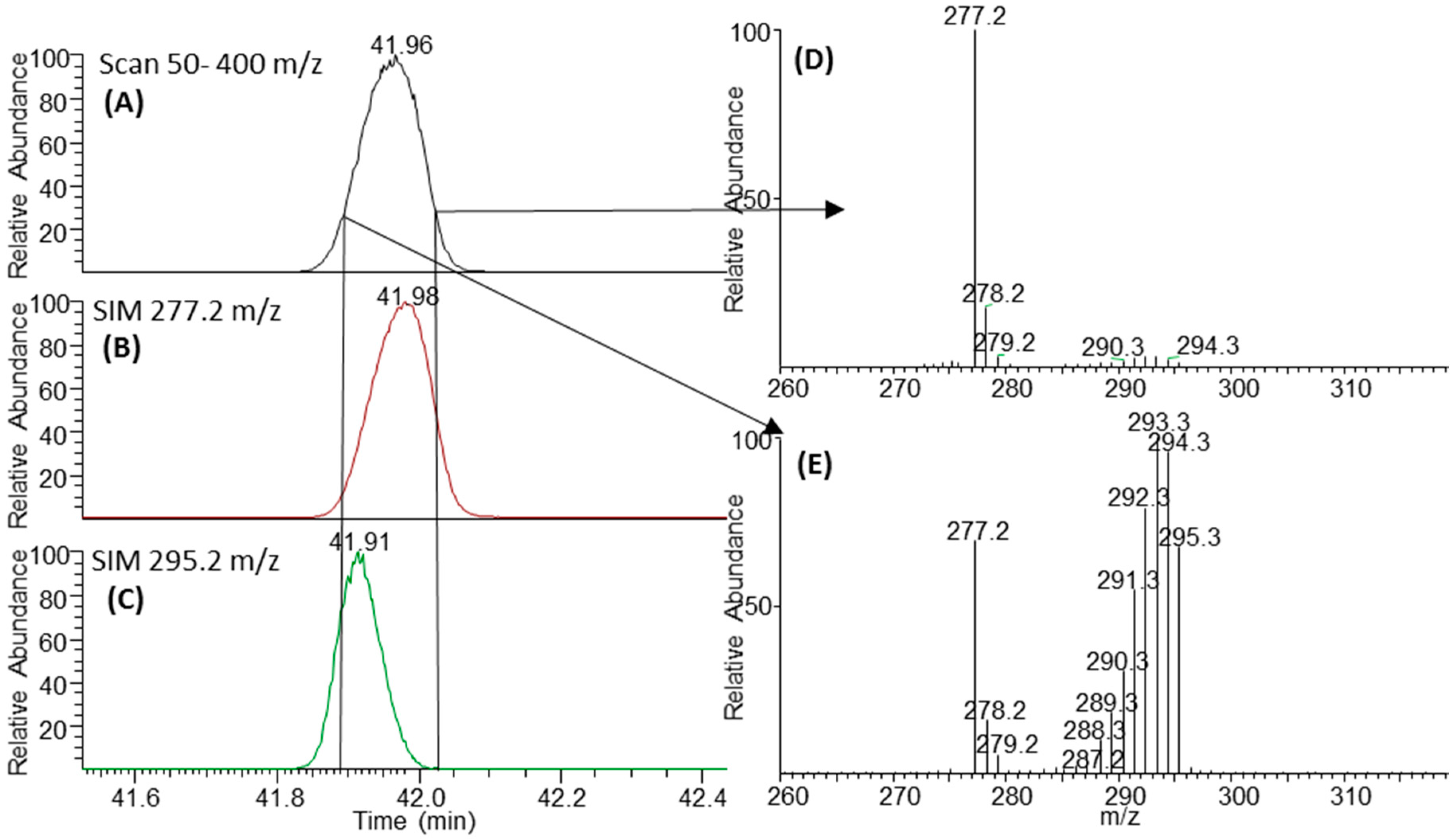
2.3. Negative Ion Chemical Ionisation-Gas Chromotagraphy-Mass Spectrometry (NCI-GC-MS) of 13C-Labelled Fatty Acids
3. Discussion
3.1. Key Considerations
3.2. Retention Time Shift of Heavy Isotopes
3.3. Calculation of Percentage of Label Incorporation
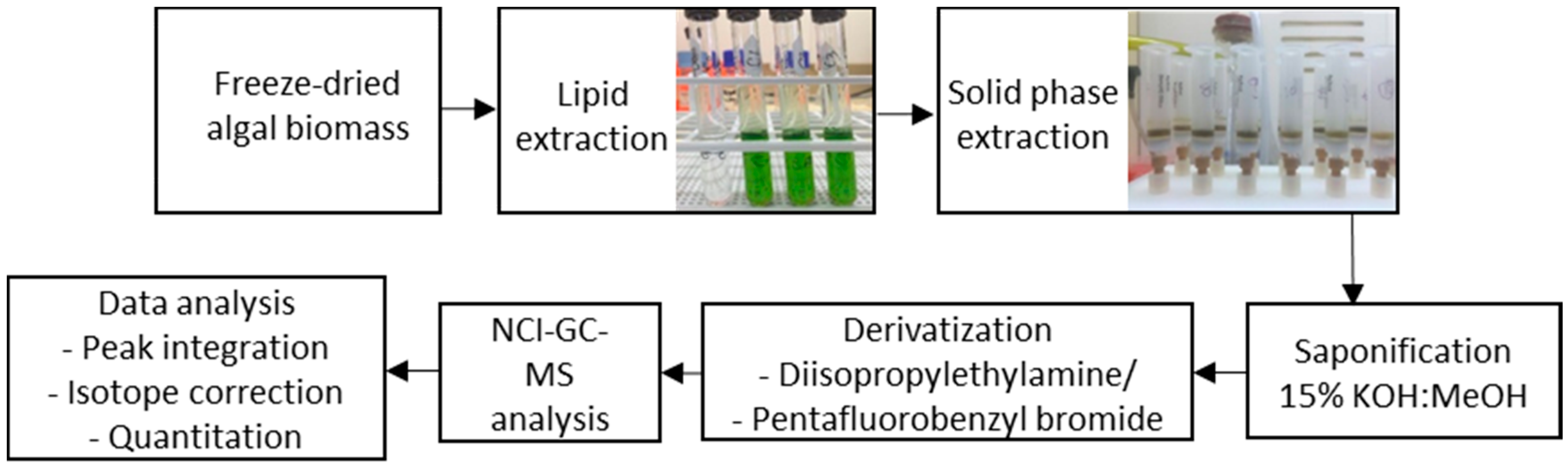
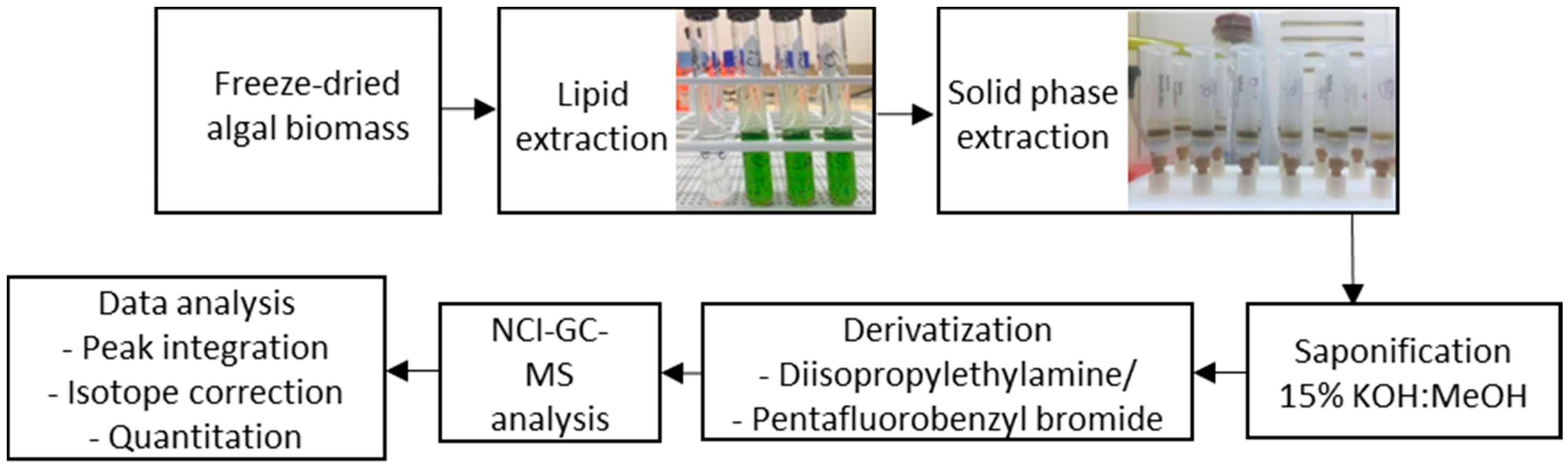
4. Materials and Methods
4.1. Materials
4.2. Microalgae Culture
4.3. Lipid Extraction Optimisation
4.3.1. Lipid Extraction Protocol One: Chloroform:Methanol:Water (Folch)
4.3.2. Lipid Extraction Protocol Two: Methyl-tert-butyl ether:Methanol:Water (MTBE)
4.3.3. Lipid Extraction Protocol Three: Butanol:Methanol (BUME Single-Phase)
4.3.4. Lipid Extraction Protocol Four: Butanol:Methanol:Heptane:Ethylacetate (BUME: Two-Phase)
4.4. Solid-Phase Extraction
4.5. Saponification of Lipids
4.6. Fatty Acid Derivatisation
4.7. GC-MS Method
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wenk, M.R. The emerging field of lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Sommerfeld, M.; Jarvis, E.; Ghirardi, M.; Posewitz, M.; Seibert, M.; Darzins, A. Microalgal triacylglycerols as feedstocks for biofuel production: Perspectives and advances. Plant J. 2008, 54, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Groessl, M.; Graf, S.; Knochenmuss, R. High resolution ion mobility-mass spectrometry for separation and identification of isomeric lipids. Analyst 2015, 140, 6904–6911. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, W.D. A timeline of stable isotopes and mass spectrometry in the life sciences. Mass Spectrom. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wuchter, C.; Schouten, S.; Boschker, H.T.; Damste, J.S.S. Bicarbonate uptake by marine crenarchaeota. FEMS Microbiol. Lett. 2003, 219, 203–207. [Google Scholar] [CrossRef]
- Boschker, H.T.S.; Nold, S.C.; Wellsbury, P.; Bos, D.; de Graaf, W.; Pel, R.; Parkes, R.J.; Cappenberg, T.E. Direct linking of microbial populations to specific biogeochemical processes by 13C-labelling of biomarkers. Nature 1998, 392, 801–805. [Google Scholar] [CrossRef]
- Ecker, J.; Liebisch, G. Application of stable isotopes to investigate the metabolism of fatty acids, glycerophospholipid and sphingolipid species. Prog. Lipid Res. 2014, 54, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Fan, T.W.M.; Xie, Z.; Moseley, H.N.B.; Higashi, R.M. Isotopomer analysis of lipid biosynthesis by high resolution mass spectrometry and nmr. Anal. Chim. Acta 2009, 651, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Sprecher, H.W.; Salem, N., Jr. High sensitivity negative ion GC-MS method for detection of desaturated and chain-elongated products of deuterated linoleic and linolenic acids. J. Lipid Res. 1992, 33, 1711–1717. [Google Scholar] [PubMed]
- Quehenberger, O.; Armando, A.M.; Dennis, E.A. High sensitivity quantitative lipidomics analysis of fatty acids in biological samples by gas chromatography-mass spectrometry. BBBA Mol. Cell Biol. Lipids 2011, 1811, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.G. Chemical Ionization Mass Spectrometry; CRC Press: Boca Raton, FL, USA, 1992. [Google Scholar]
- Lin, Y.H.; Pawlosky, R.J.; Salem, N., Jr. Simultaneous quantitative determination of deuterium- and carbon-13-labeled essential fatty acids in rat plasma. J. Lipid Res. 2005, 46, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; van Dijk, T.H.; Tietge, U.J.F.; Boer, T.; Havinga, R.; Stellaard, F.; Groen, A.K.; Kuipers, F.; Reijngoud, D.-J. High fat feeding induces hepatic fatty acid elongation in mice. PLoS ONE 2009, 4, e6066. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biol. Phys. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Li, Y.; Ghasemi Naghdi, F.; Garg, S.; Adarme-Vega, T.C.; Thurecht, K.J.; Ghafor, W.A.; Tannock, S.; Schenk, P.M. A comparative study: The impact of different lipid extraction methods on current microalgal lipid research. Microb. Cell Fact. 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.; Wang, M.; Frisch-Daiello, J.; Han, X. Improved butanol-methanol (BUME) method by replacing acetic acid for lipid extraction of biological samples. Lipids 2016, 51, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Löfgren, L.; Ståhlman, M.; Forsberg, G.-B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The bume method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Olmstead, I.L.D.; Hill, D.R.A.; Dias, D.A.; Jayasinghe, N.S.; Callahan, D.L.; Kentish, S.E.; Scales, P.J.; Martin, G.J.O. A quantitative analysis of microalgal lipids for optimization of biodiesel and omega-3 production. Biotechnol. Bioeng. 2013, 110, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.A.; Nichols, P.D.; White, D.C. A method for the separation and characterization of archaebacterial signature ether lipids. J. Lipid Res. 1986, 27, 49–56. [Google Scholar] [PubMed]
- Heinzelmann, S.M.; Bale, N.J.; Hopmans, E.C.; Sinninghe Damsté, J.S.; Schouten, S.; van der Meer, M.T.J. Critical assessment of glyco- and phospholipid separation by using silica chromatography. Appl. Environ. Microbiol. 2014, 80, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.J.; Hill, D.R.; Olmstead, I.L.; Bergamin, A.; Shears, M.J.; Dias, D.A.; Kentish, S.E.; Scales, P.J.; Botté, C.Y.; Callahan, D.L. Lipid profile remodeling in response to nitrogen deprivation in the microalgae chlorella sp. (Trebouxiophyceae) and nannochloropsis sp. (Eustigmatophyceae). PLoS ONE 2014, 9, e103389. [Google Scholar] [CrossRef] [PubMed]
- Fauland, A.; Trotzmuller, M.; Eberl, A.; Afiuni-Zadeh, S.; Kofeler, H.; Guo, X.; Lankmayr, E. An improved spe method for fractionation and identification of phospholipids. J. Sep. Sci. 2013, 36, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, A.; Degenhardt, O.S.; McDonald, G.R.; Narang, D.; Paulsen, I.M.; Kozuska, J.L.; Holt, A. On the disruption of biochemical and biological assays by chemicals leaching from disposable laboratory plasticware. Can. J. Physiol. Pharm. 2012, 90, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.-H.; Liu, G.-Y.; Yang, K.; Gross, R.W.; Patti, G.J. Inaccurate quantitation of palmitate in metabolomics and isotope tracer studies due to plastics. Metabolomics 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Meier-Augenstein, W. Stable isotope analysis of fatty acids by gas chromatography–isotope ratio mass spectrometry. Anal. Chim. Acta 2002, 465, 63–79. [Google Scholar] [CrossRef]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opt. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.K.; Bates, P.D.; Tjellström, H. Tracking the metabolic pulse of plant lipid production with isotopic labeling and flux analyses: Past, present and future. Prog. Lipid Res. 2015, 58, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Nanchen, A.; Fuhrer, T.; Sauer, U. Determination of metabolic flux ratios from 13C-experiments and gas chromatography-mass spectrometry data. Metab. Methods Protoc. 2007, 358, 177–197. [Google Scholar]
- Shastri, A.A.; Morgan, J.A. A transient isotopic labeling methodology for 13C metabolic flux analysis of photoautotrophic microorganisms. Phytochemistry 2007, 68, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Alshehry, Z.H.; Barlow, C.K.; Weir, J.M.; Zhou, Y.; McConville, M.J.; Meikle, P.J. An efficient single phase method for the extraction of plasma lipids. Metabolites 2015, 5, 389–403. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | C16:0 | C16:1c | C18:0 | C18:1c | C18:2c | C18:3n3 |
|---|---|---|---|---|---|---|
| CHCl3 | 33 | 8.3 | 5.9 | 44 | 37 | 24 |
| BuOH 2-phases | 24 | 6.2 | 4.0 | 34 | 30 | 19 |
| MTBE | 12 | 3.1 | 2.0 | 15 | 15 | 10 |
| BUME % rel to CHCl3 | 73 | 74 | 67 | 77 | 82 | 79 |
| MTBE % rel to CHCl3 | 37 | 37 | 33 | 35 | 41 | 43 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elahee Doomun, S.N.; Loke, S.; O’Callaghan, S.; Callahan, D.L. A Simple Method for Measuring Carbon-13 Fatty Acid Enrichment in the Major Lipid Classes of Microalgae Using GC-MS. Metabolites 2016, 6, 42. https://doi.org/10.3390/metabo6040042
Elahee Doomun SN, Loke S, O’Callaghan S, Callahan DL. A Simple Method for Measuring Carbon-13 Fatty Acid Enrichment in the Major Lipid Classes of Microalgae Using GC-MS. Metabolites. 2016; 6(4):42. https://doi.org/10.3390/metabo6040042
Chicago/Turabian StyleElahee Doomun, Sheik Nadeem, Stella Loke, Sean O’Callaghan, and Damien L. Callahan. 2016. "A Simple Method for Measuring Carbon-13 Fatty Acid Enrichment in the Major Lipid Classes of Microalgae Using GC-MS" Metabolites 6, no. 4: 42. https://doi.org/10.3390/metabo6040042
APA StyleElahee Doomun, S. N., Loke, S., O’Callaghan, S., & Callahan, D. L. (2016). A Simple Method for Measuring Carbon-13 Fatty Acid Enrichment in the Major Lipid Classes of Microalgae Using GC-MS. Metabolites, 6(4), 42. https://doi.org/10.3390/metabo6040042