
A Conversation on Data Mining Strategies in LC-MS Untargeted Metabolomics: Pre-Processing and Pre-Treatment Steps




Abstract
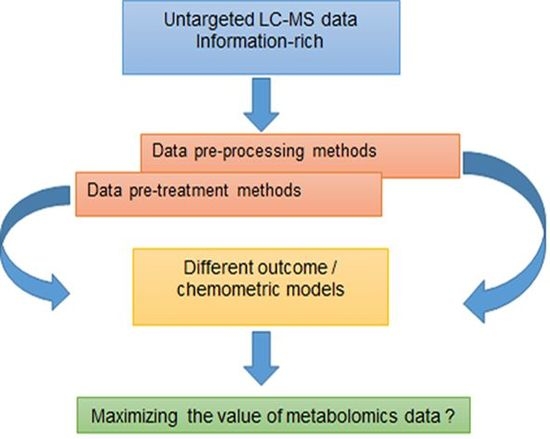
:
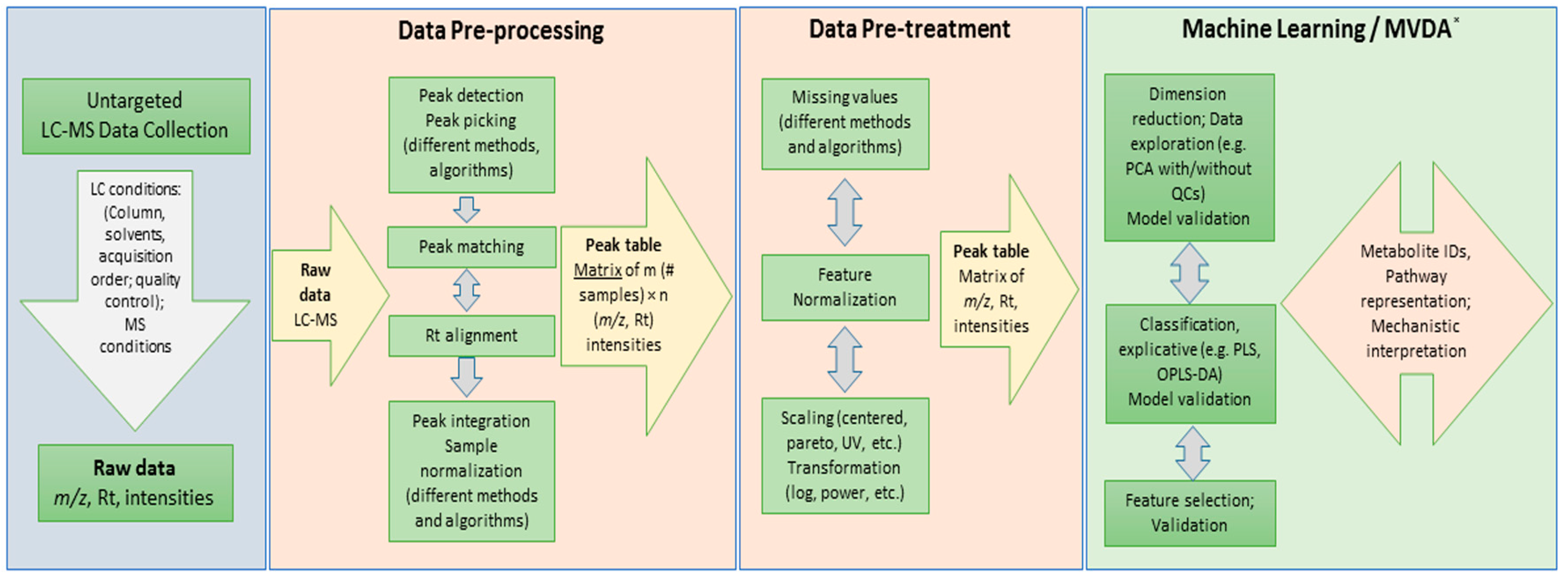
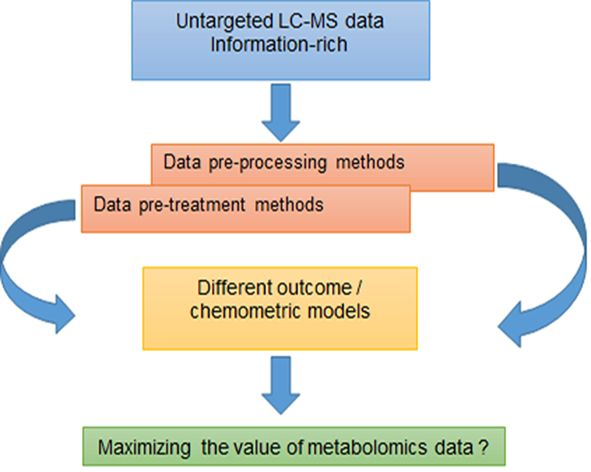
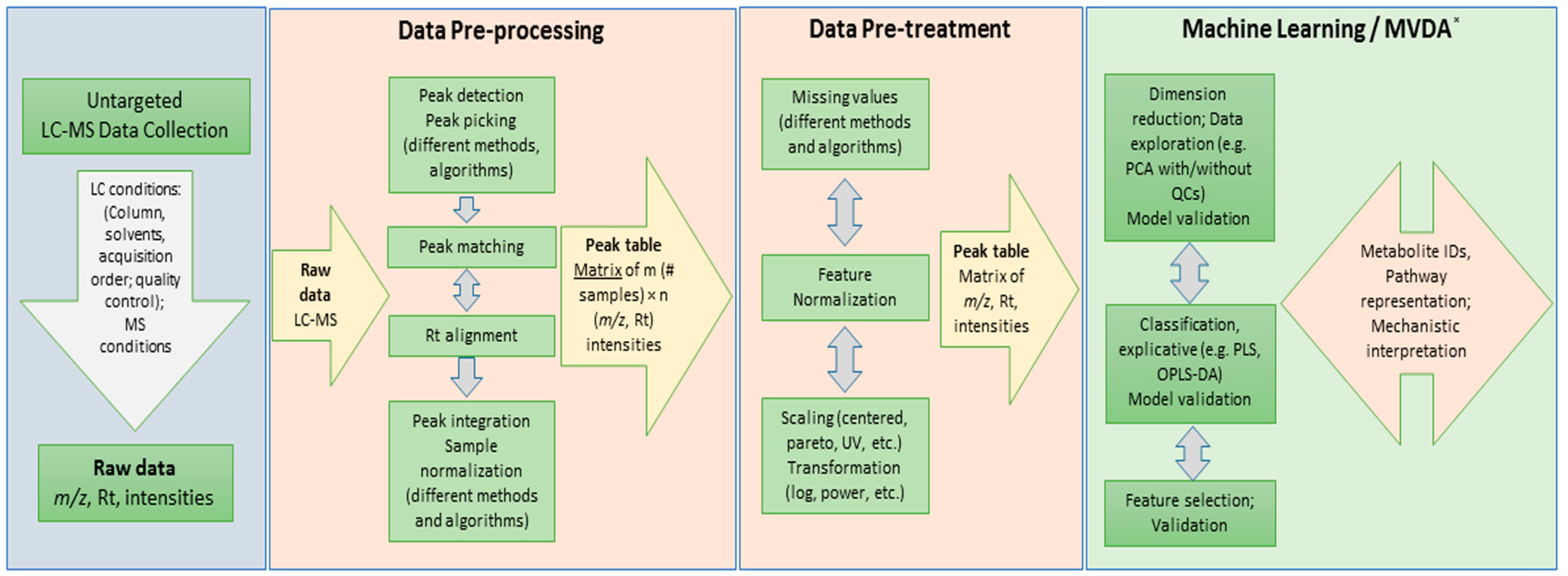
1. Introduction
2. Results and Discussion
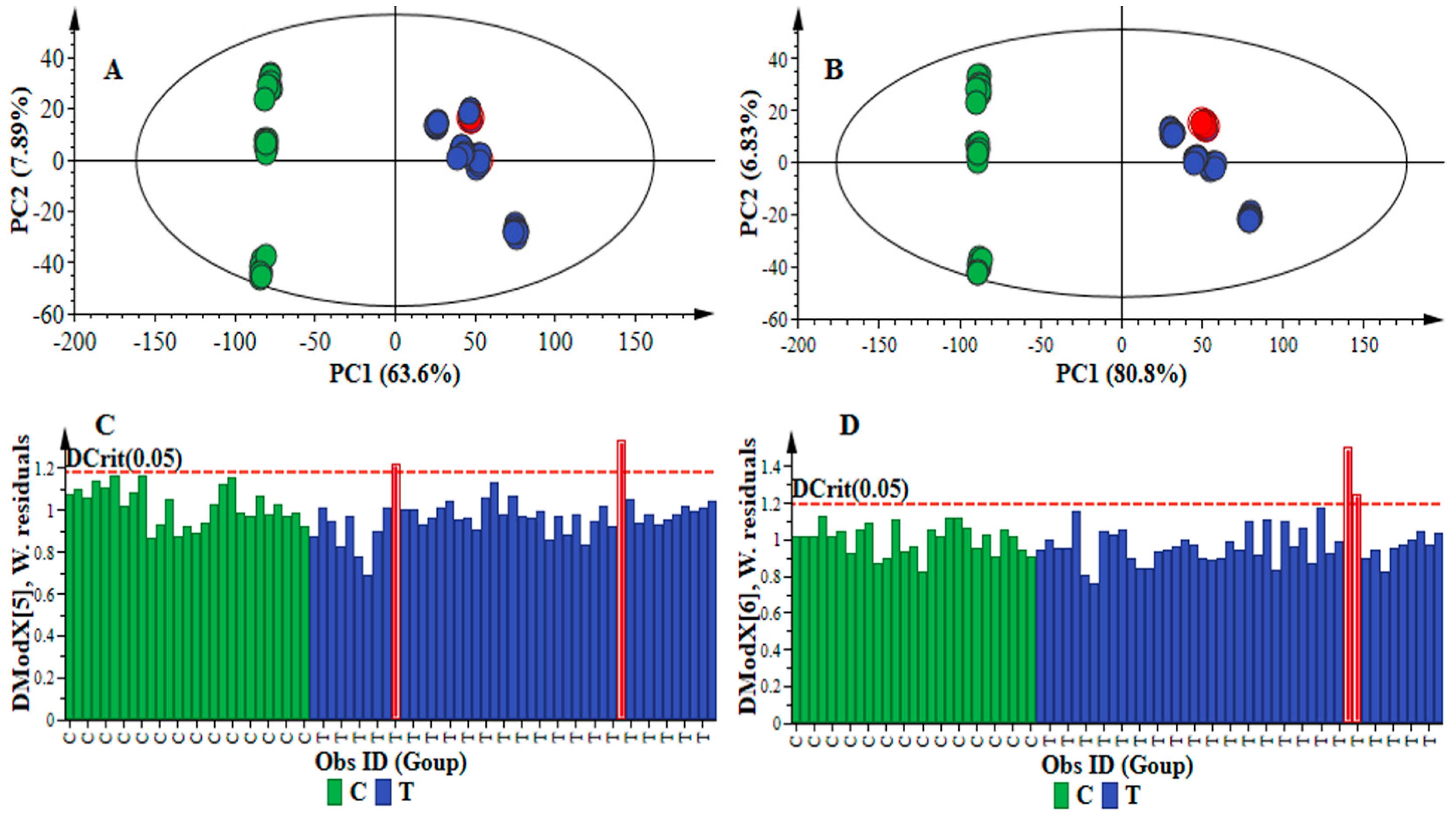
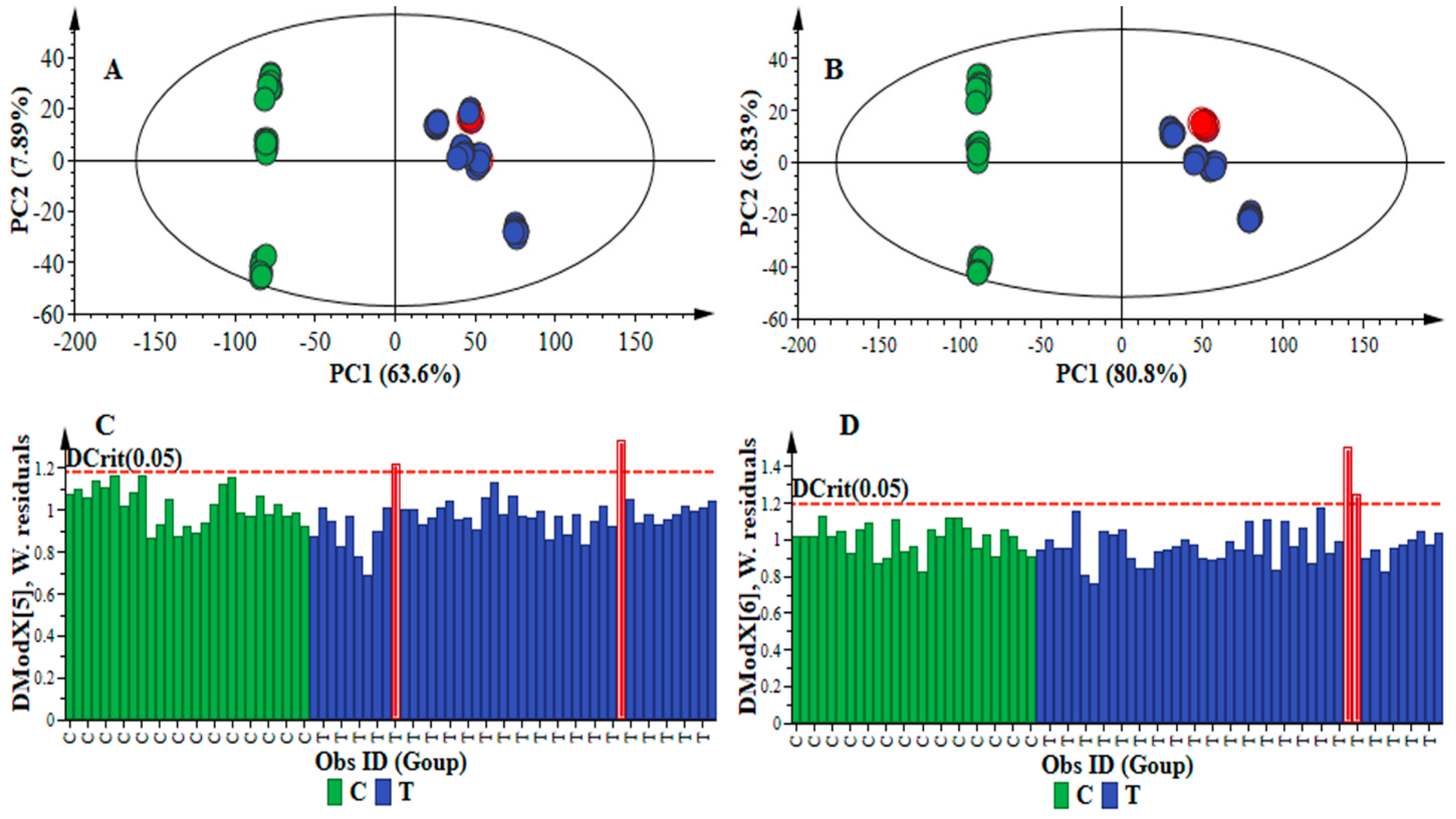
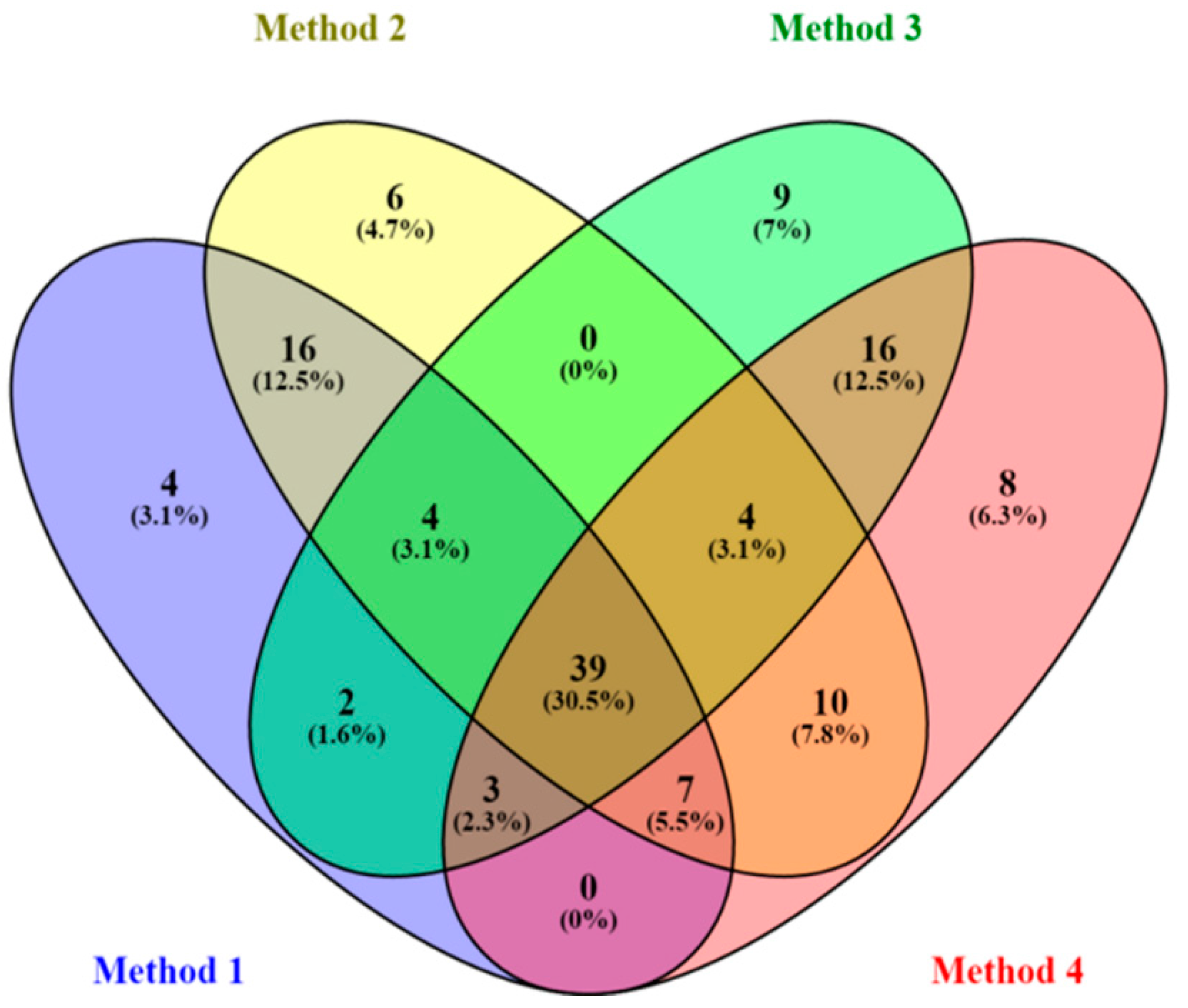
2.1. Data Processing Parameters: Mass Tolerance and Intensity Threshold
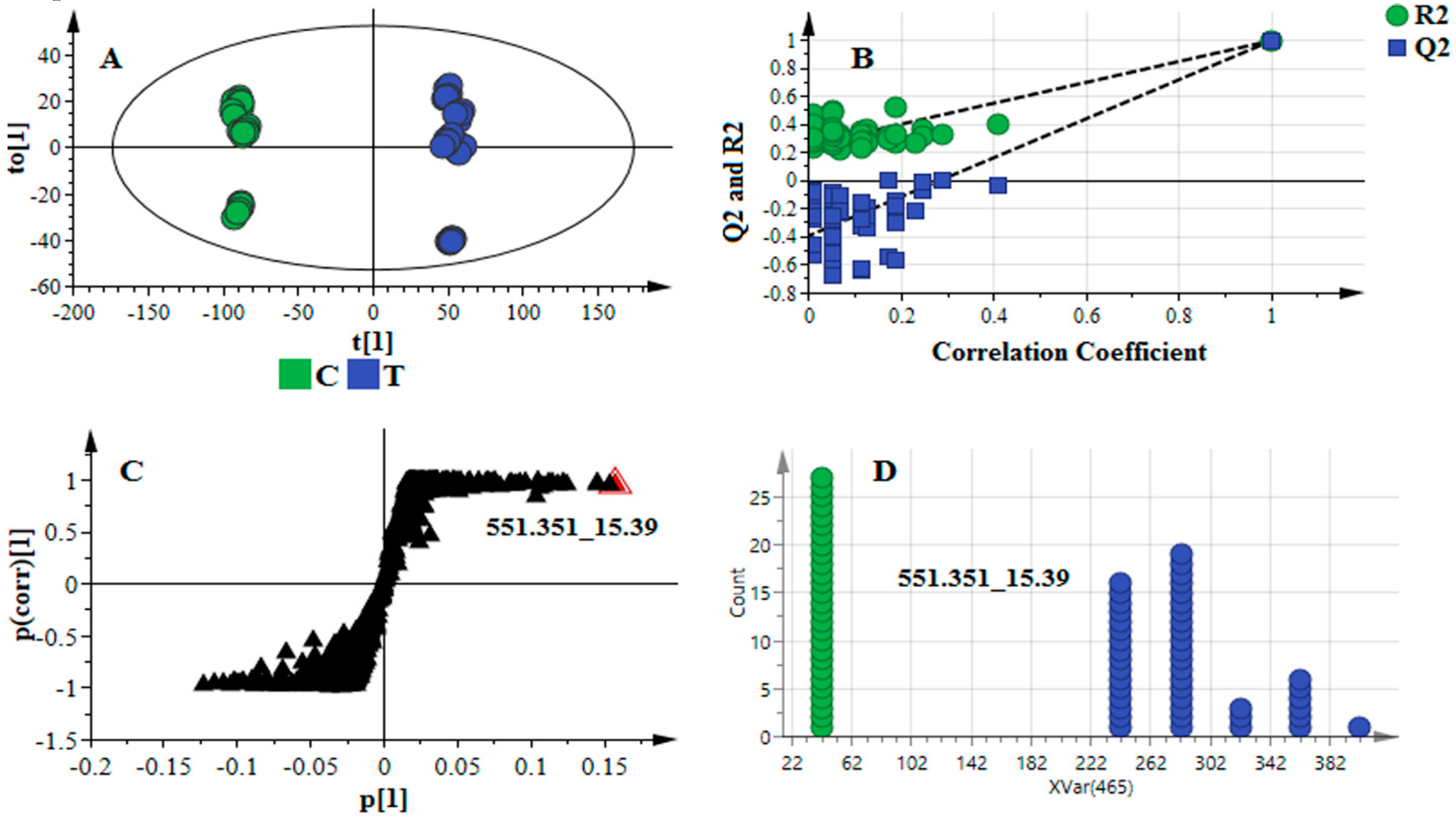
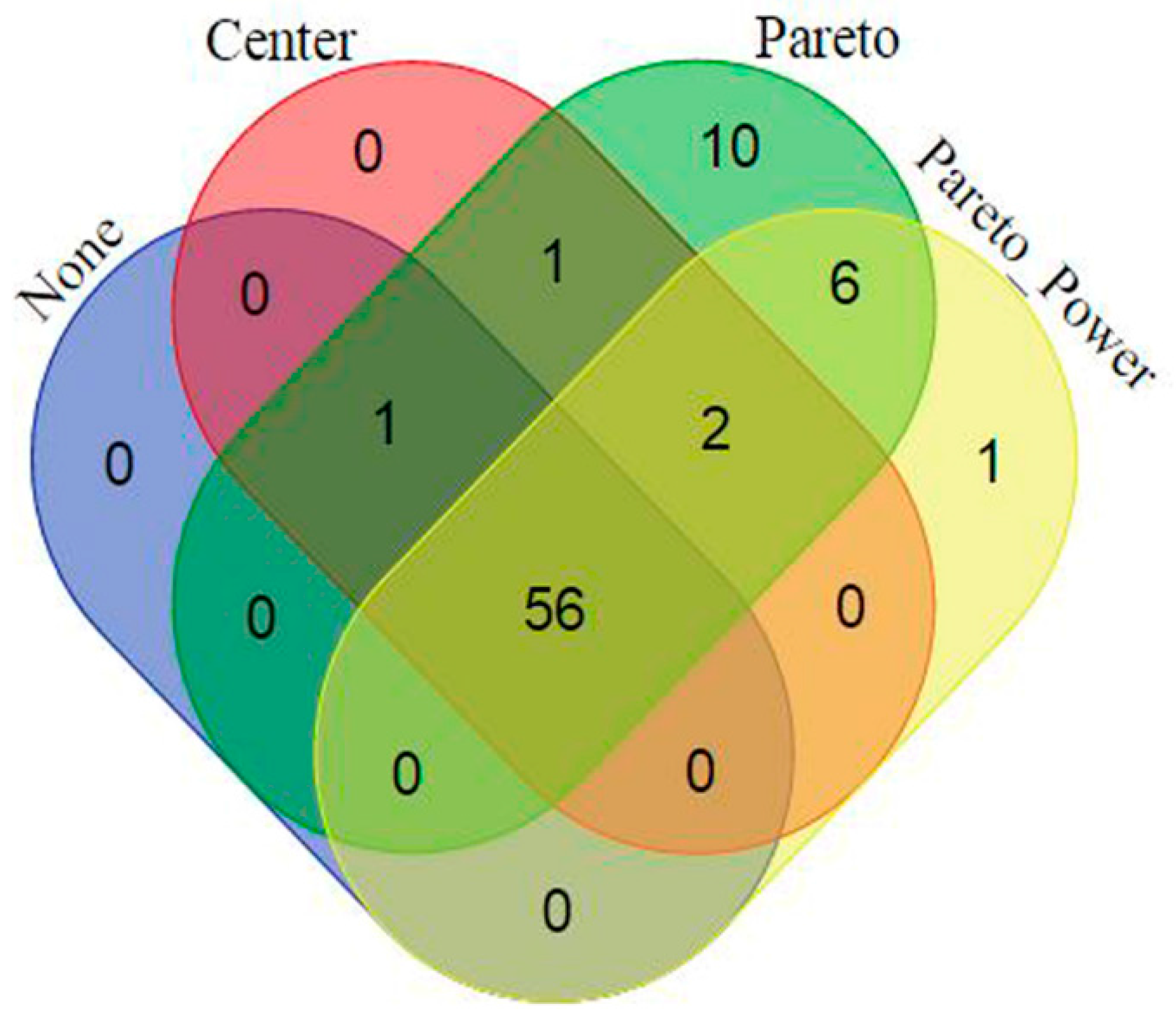
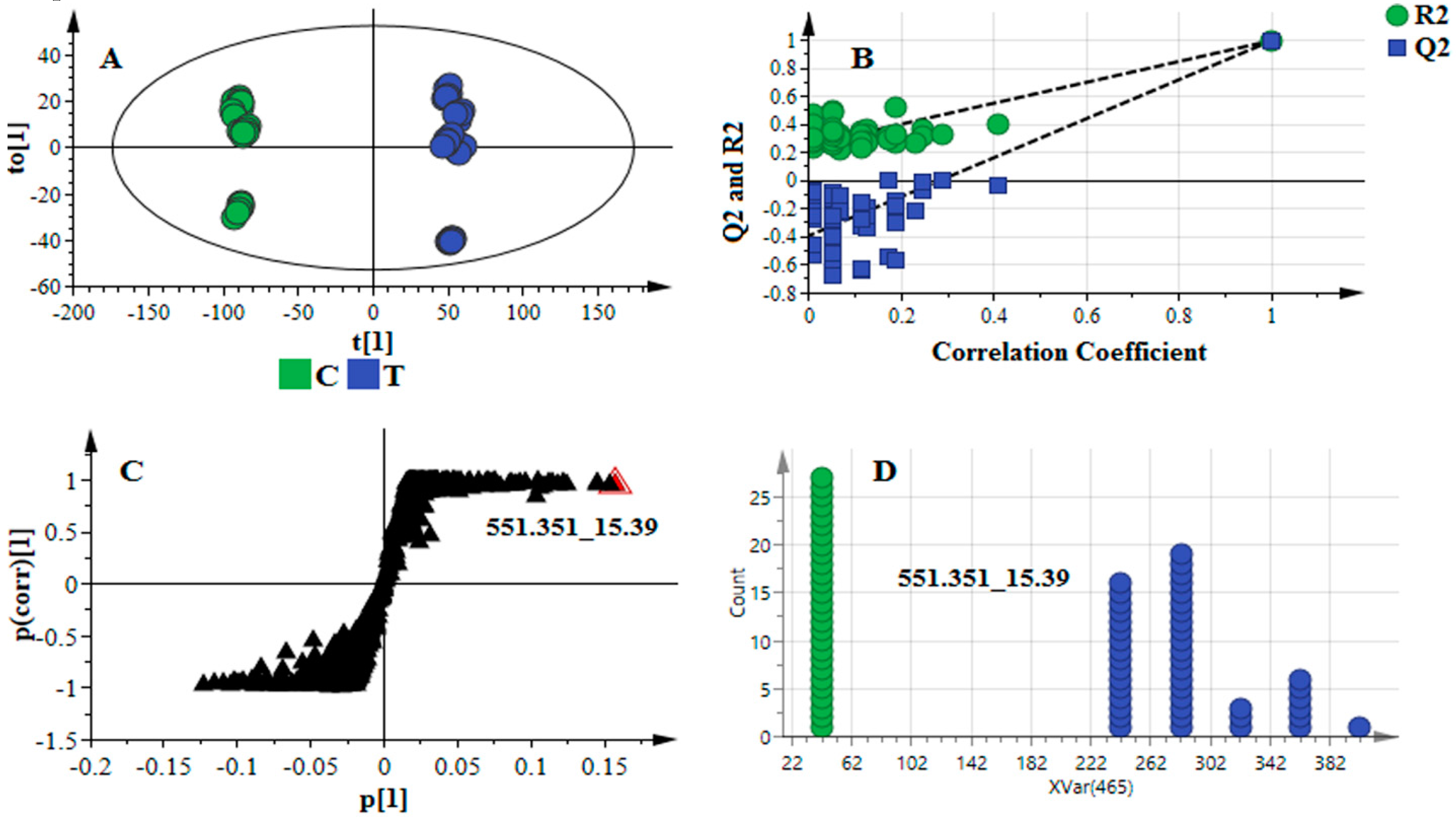
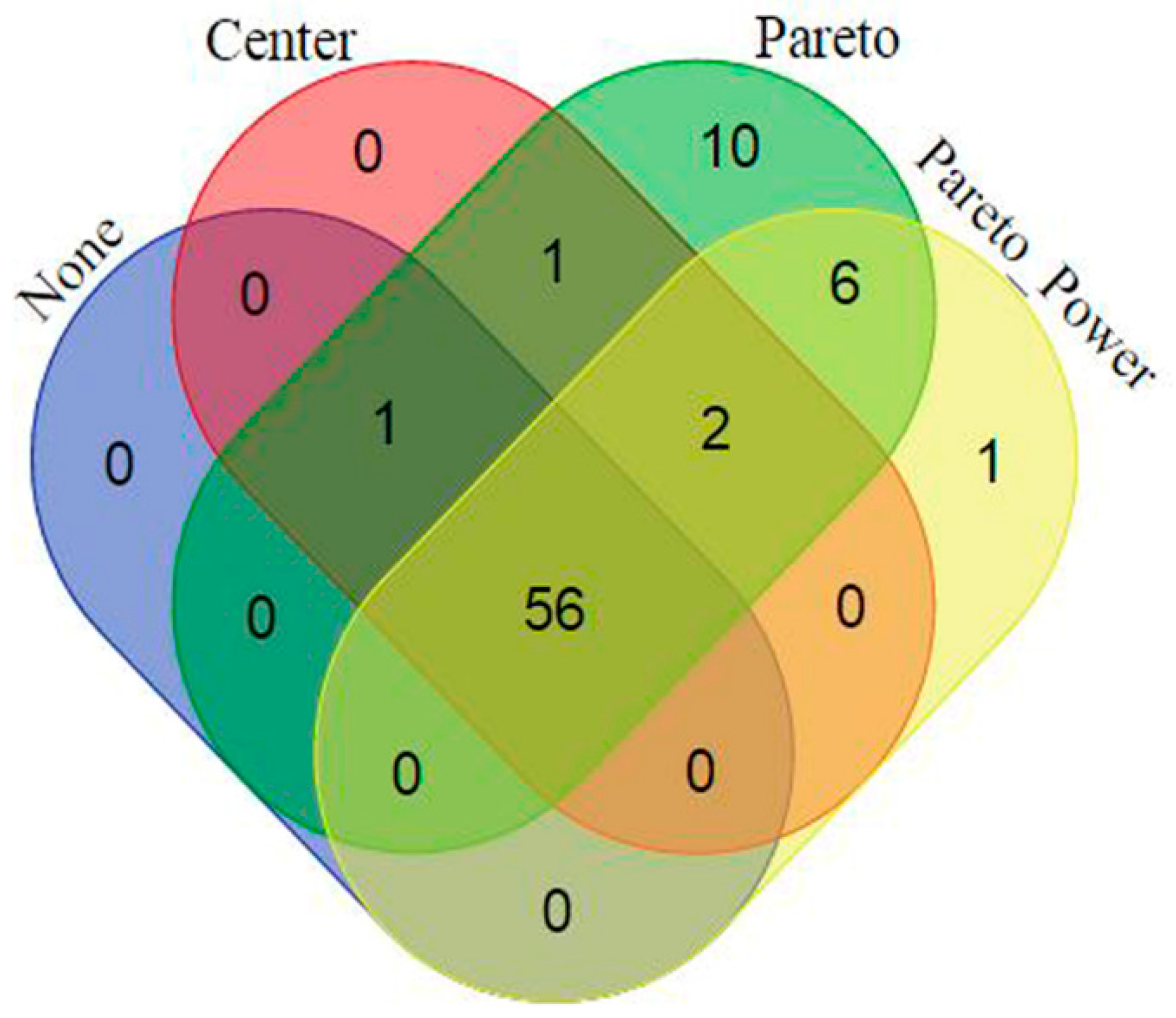
2.2. Data Scaling and Transformation Influence
3. Materials and Methods
3.1. Dataset and Raw Data Processing
3.2. Dataset Matrix Creation and Data Pre-Treatment
4. Conclusions and Perspectives
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sévin, D.C.; Kuehne, A.; Zamboni, N.; Sauer, U. Biological insights through nontargeted metabolomics. Curr. Opin. Biotechnol. 2015, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tugizimana, F.; Piater, L.A.; Dubery, I.A. Plant metabolomics: A new frontier in phytochemical analysis. S. Afr. J. Sci. 2013, 109, 18–20. [Google Scholar] [CrossRef]
- Okazaki, Y.; Saito, K. Recent advances of metabolomics in plant biotechnology. Plant Biotechnol. Rep. 2012, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bartel, J.; Krumsiek, J.; Theis, F.J. Statistical methods for the analysis of high-throughput metabolomics data. Comput. Struct. Biotechnol. J. 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Worley, B.; Powers, R. Multivariate analysis in metabolomics. Curr. Metabol. 2013, 1, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Verpoorte, R. Metabolomics: What you see is what you extract. Phytochem. Anal. 2014, 25, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Duportet, X.; Aggio, R.B.M.; Carneiro, S.; Villas-Bôas, S.G. The biological interpretation of metabolomic data can be misled by the extraction method used. Metabolomics 2012, 8, 410–421. [Google Scholar] [CrossRef]
- Yanes, O.; Tautenhahn, R.; Patti, G.J.; Siuzdak, G. Expanding coverage of the metabolome for global metabolite profiling. Anal. Chem. 2011, 83, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Mendes, P.; Dixon, R.A. Plant metabolomics: Large-scale phytochemistry in the functional genomics era. Phytochemistry 2003, 62, 817–836. [Google Scholar] [CrossRef]
- Allwood, J.W.; Ellis, D.I.; Goodacre, R. Metabolomic technologies and their application to the study of plants and plant-host interactions. Physiol. Plant. 2008, 132, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Goeddel, L.C.; Patti, G.J. Maximizing the value of metabolomic data. Bioanalysis 2012, 4, 2199–2201. [Google Scholar] [CrossRef] [PubMed]
- Boccard, J.; Rudaz, S. Harnessing the complexity of metabolomic data with chemometrics. J. Chemom. 2014, 28, 1–9. [Google Scholar] [CrossRef]
- Beisken, S.; Eiden, M.; Salek, R.M. Getting the right answers: Understanding metabolomics challenges. Expert Rev. Mol. Diagn. 2015, 15, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B.; van der Hooft, J.J.J. Updates in metabolomics tools and resources: 2014–2015. Electrophoresis 2016, 37, 86–110. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Oliver, S.G. Here is the evidence, now what is the hypothesis? The complementary roles of inductive and hypothesis-driven science in the post-genomic era. BioEssays 2004, 26, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Boccard, J.; Veuthey, J.-L.; Rudaz, S. Knowledge discovery in metabolomics: An overview of MS data handling. J. Sep. Sci. 2010, 33, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Cicek, A.E.; Roeder, K.; Ozsoyoglu, G. MIRA: Mutual information-based reporter algorithm for metabolic networks. Bioinformatics 2014, 30, i175–i184. [Google Scholar] [CrossRef] [PubMed]
- Toubiana, D.; Fernie, A.R.; Nikoloski, Z.; Fait, A. Network analysis: Tackling complex data to study plant metabolism. Trends Biotechnol. 2013, 31, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Dunn, W.B.; Ellis, D.I.; Goodacre, R.; Handl, J.; Knowles, J.D.; O’Hagan, S.; Spasić, I.; Kell, D.B. A metabolome pipeline: From concept to data to knowledge. Metabolomics 2005, 1, 39–51. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Gromski, P.S.; Xu, Y.; Hollywood, K.A.; Turner, M.L.; Goodacre, R. The influence of scaling metabolomics data on model classification accuracy. Metabolomics 2015, 11, 684–695. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, X.; Lu, X.; Lin, X.; Xu, G. A data preprocessing strategy for metabolomics to reduce the mask effect in data analysis. Front. Mol. Biosci. 2015, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Boccard, J.; Rudaz, S. Mass spectrometry metabolomic data handling for biomarker discovery. In Proteomic and Metabolomic Approaches to Biomarker Discovery; Elsevier: Amsterdam, The Netherlands, 2013; pp. 425–445. [Google Scholar]
- Trygg, J.; Holmes, E.; Lundstedt, T. Chemometrics in Metabonomics. J. Proteome Res. 2007, 6, 469–479. [Google Scholar] [CrossRef] [PubMed]
- De Livera, A.M.; Sysi-Aho, M.; Jacob, L.; Gagnon-Bartsch, J.A.; Castillo, S.; Simpson, J.A.; Speed, T.P. Statistical methods for handling unwanted variation in metabolomics data. Anal. Chem. 2015, 87, 3606–3615. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; Werf, M.J. Van Der Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R.; Broadhurst, D.; Smilde, A.K.; Kristal, B.S.; Baker, J.D.; Beger, R.; Bessant, C.; Connor, S.; Capuani, G.; Craig, A.; et al. Proposed minimum reporting standards for data analysis in metabolomics. Metabolomics 2007, 3, 231–241. [Google Scholar] [CrossRef]
- Saccenti, E.; Hoefsloot, H.C.J.; Smilde, A.K.; Westerhuis, J.A.; Hendriks, M.M.W.B. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 2013, 10, 361–374. [Google Scholar] [CrossRef]
- Buydens, L. Towards tsunami-resistant chemometrics. Anal. Sci. 2013, 813, 24–29. [Google Scholar]
- Di Guida, R.; Engel, J.; Allwood, J.W.; Weber, R.J.M.; Jones, M.R.; Sommer, U.; Viant, M.R.; Dunn, W.B. Non-targeted UHPLC-MS metabolomic data processing methods: A comparative investigation of normalisation, missing value imputation, transformation and scaling. Metabolomics 2016, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Godzien, J.; Ciborowski, M.; Angulo, S.; Barbas, C. From numbers to a biological sense: How the strategy chosen for metabolomics data treatment may affect final results. A practical example based on urine fingerprints obtained by LC-MS. Electrophoresis 2013, 34, 2812–2826. [Google Scholar] [CrossRef] [PubMed]
- Defernez, M.; Gall, G. Le strategies for data handling and statistical analysis in metabolomics studies. In Advances in Botanical Research; Elsevier Ltd.: Amsterdam, The Netherlands, 2013; Volume 67, pp. 493–555. [Google Scholar]
- Moseley, H.N.B. Error analysis and propagation in metabolomics data analysis. Comput. Struct. Biotechnol. J. 2013, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Trutschel, D.; Schmidt, S.; Grosse, I.; Neumann, S. Experiment design beyond gut feeling: Statistical tests and power to detect differential metabolites in mass spectrometry data. Metabolomics 2015, 11, 851–860. [Google Scholar] [CrossRef]
- Moco, S.; Vervoort, J.; Bino, R.; Devos, R. Metabolomics technologies and metabolite identification. TrAC Trends Anal. Chem. 2007, 26, 855–866. [Google Scholar] [CrossRef]
- Idborg, H.; Zamani, L.; Edlund, P.-O.; Schuppe-Koistinen, I.; Jacobsson, S.P. Metabolic fingerprinting of rat urine by LC/MS Part 2. Data pretreatment methods for handling of complex data. J. Chromatogr. B 2005, 828, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, C.L.; Goshawk, J. The MarkerLynx application manager: Informatics for mass spectrometric metabonomic discovery. Waters Appl. Note 2004. 720001056EN KJ-PDF. [Google Scholar]
- Veselkov, K.A.; Vingara, L.K.; Masson, P.; Robinette, S.L.; Want, E.; Li, J.V.; Barton, R.H.; Boursier-Neyret, C.; Walther, B.; Ebbels, T.M.; et al. Optimized preprocessing of ultra-performance liquid chromatography/mass spectrometry urinary metabolic profiles for improved information recovery. Anal. Chem. 2011, 83, 5864–5872. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.W.; Rutan, S.C. Chemometrics for the analysis of chromatographic data in metabolomics investigations. J. Chemom. 2014, 28, 681–687. [Google Scholar] [CrossRef]
- Peters, S.; Van Velzen, E.; Janssen, H.G. Parameter selection for peak alignment in chromatographic sample profiling: Objective quality indicators and use of control samples. Anal. Bioanal. Chem. 2009, 394, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Godzien, J.; Alonso-Herranz, V.; Barbas, C.; Armitage, E.G. Controlling the quality of metabolomics data: New strategies to get the best out of the QC sample. Metabolomics 2014, 11, 518–528. [Google Scholar] [CrossRef]
- Misra, B.B.; Assmann, S.M.; Chen, S. Plant single-cell and single-cell-type metabolomics. Trends Plant Sci. 2014, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kohli, A.; Sreenivasulu, N.; Lakshmanan, P.; Kumar, P.P. The phytohormone crosstalk paradigm takes center stage in understanding how plants respond to abiotic stresses. Plant Cell Rep. 2013, 32, 945–57. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M. A unifying view of 21st century systems biology. FEBS Lett. 2009, 583, 3891–3894. [Google Scholar] [CrossRef] [PubMed]
- Makola, M.M.; Steenkamp, P.A.; Dubery, I.A.; Kabanda, M.M.; Madala, N.E. Preferential alkali metal adduct formation by cis geometrical isomers of dicaffeoylquinic acids allows for efficient discrimination from their trans isomers during ultra-high-performance liquid chromatography/quadrupole time-of-flight mass s. Rapid Commun. Mass Spectrom. 2016, 30, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Spagou, K.; Nicholson, J.K.; Want, E.J. Technical and biological variation in UPLC-MS-based untargeted metabolic profiling of liver extracts: Application in an experimental toxicity study on galactosamine. Anal. Chem. 2011, 83, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, D.M. The Problem of overfitting. J. Chem. Inf. Comput. Sci. 2004, 44, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, D.I.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Armitage, E.G.; Godzien, J.; Alonso-Herranz, V.; López-Gonzálvez, Á.; Barbas, C. Missing value imputation strategies for metabolomics data. Electrophoresis 2015, 36, 3050–3060. [Google Scholar] [CrossRef] [PubMed]
- Ilin, A.; Raiko, T. Practical approaches to principal component analysis in the presence of missing values. J. Mach. Learn. Res. 2010, 11, 1957–2000. [Google Scholar]
- Nelson, P.R.C.; Taylor, P.A.; MacGregor, J.F. Missing data methods in PCA and PLS: Score calculations with incomplete observations. Chemom. Intell. Lab. Syst. 1996, 35, 45–65. [Google Scholar] [CrossRef]
- Wikström, C.; Albano, C.; Eriksson, L.; Fridén, H.; Johansson, E.; Nordahl, Å.; Rännar, S.; Sandberg, M.; Kettaneh-Wold, N.; Wold, S. Multivariate process and quality monitoring applied to an electrolysis process. Part I. Process supervision with multivariate control charts. Chemom. Intell. Lab. Syst. 1998, 42, 221–231. [Google Scholar] [CrossRef]
- Eriksson, L.; Trygg, J.; Wold, S. A chemometrics toolbox based on projections and latent variables. J. Chemom. 2014, 28, 332–346. [Google Scholar] [CrossRef]
- Hawkins, D.M.; Basak, S.C.; Mills, D. Assessing model fit by cross-validation. J. Chem. Inf. Comput. Sci. 2003, 43, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.; Trygg, J.; Wold, S. CV-ANOVA for significance testing of PLS and OPLS® models. J. Chemom. 2008, 22, 594–600. [Google Scholar] [CrossRef]
- Triba, M.N.; Le Moyec, L.; Amathieu, R.; Goossens, C.; Bouchemal, N.; Nahon, P.; Rutledge, D.N.; Savarin, P. PLS/OPLS models in metabolomics: The impact of permutation of dataset rows on the K-fold cross-validation quality parameters. Mol. BioSyst. 2015, 11, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Westerhuis, J.A.; Hoefsloot, H.C.J.; Smit, S.; Vis, D.J.; Smilde, A.K.; Velzen, E.J.J.; Duijnhoven, J.P.M.; Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef]
- Wiklund, S.; Johansson, E.; Sjöström, L.; Mellerowicz, E.J.; Edlund, U.; Shockcor, J.P.; Gottfries, J.; Moritz, T.; Trygg, J. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal. Chem. 2008, 80, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ambroise, C.; McLachlan, G.J. Selection bias in gene extraction on the basis of microarray gene-expression data. Proc. Natl. Acad. Sci. USA 2002, 99, 6562–6566. [Google Scholar] [CrossRef] [PubMed]
- Smilde, A.K.; Westerhuis, J.A.; Hoefsloot, H.C.J.; Bijlsma, S.; Rubingh, C.M.; Vis, D.J.; Jellema, R.H.; Pijl, H.; Roelfsema, F.; van der Greef, J. Dynamic metabolomic data analysis: A tutorial review. Metabolomics 2010, 6, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Chong, I.-G.; Jun, C.-H. Performance of some variable selection methods when multicollinearity is present. Chemom. Intell. Lab. Syst. 2005, 78, 103–112. [Google Scholar] [CrossRef]
- Mehmood, T.; Liland, K.H.; Snipen, L.; Sæbø, S. A review of variable selection methods in Partial Least Squares Regression. Chemom. Intell. Lab. Syst. 2012, 118, 62–69. [Google Scholar] [CrossRef]
- Wilkinson, L. Dot plots. Am. Stat. 1999, 53, 276–281. [Google Scholar]
- Bro, R.; Smilde, A.K. Centering and scaling in component analysis. J. Chemom. 2003, 17, 16–33. [Google Scholar] [CrossRef]
- Van Der Greef, J.; Smilde, A.K. Symbiosis of chemometrics and metabolomics: Past, present, and future. J. Chemom. 2005, 19, 376–386. [Google Scholar] [CrossRef]
- Breiman, L. Statistical modeling: The two cultures. Stat. Sci. 2001, 16, 199–215. [Google Scholar] [CrossRef]
- T’Kindt, R.; Morreel, K.; Deforce, D.; Boerjan, W.; Bocxlaer, J. Van Joint GC-MS and LC-MS platforms for comprehensive plant metabolomics: Repeatability and sample pre-treatment. J. Chromatogr. B 2009, 877, 3572–3580. [Google Scholar] [CrossRef] [PubMed]
- Tugizimana, F.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. Multi-platform metabolomic analyses of ergosterol-induced dynamic changes in nicotiana tabacum cells. PLoS ONE 2014, 9, e87846. [Google Scholar] [CrossRef] [PubMed]
- Sangster, T.; Major, H.; Plumb, R.; Wilson, A.J.; Wilson, I.D. A pragmatic and readily implemented quality control strategy for HPLC-MS and GC-MS-based metabonomic analysis. Analyst 2006, 131, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Sangster, T.P.; Wingate, J.E.; Burton, L.; Teichert, F.; Wilson, I.D. Investigation of analytical variation in metabonomic analysis using liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 2965–2970. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, H.; Hardy, N.; Beckmann, M.; Draper, J.; Smith, A.R.; Taylor, J.; Fiehn, O.; Goodacre, R.; Bino, R.J.; Hall, R.; et al. A proposed framework for the description of plant metabolomics experiments and their results. Nat. Biotechnol. 2004, 22, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Sumner, L.W.; Rhee, S.Y.; Ward, J.; Dickerson, J.; Lange, B.M.; Lane, G.; Roessner, U.; Last, R.; Nikolau, B. Minimum reporting standards for plant biology context information in metabolomic studies. Metabolomics 2007, 3, 195–201. [Google Scholar] [CrossRef]
- Salek, R.M.; Haug, K.; Conesa, P.; Hastings, J.; Williams, M.; Mahendraker, T.; Maguire, E.; Gonzalez-Beltran, A.N.; Rocca-Serra, P.; Sansone, S.-A.; et al. The MetaboLights repository: Curation challenges in metabolomics. Database 2013, 2013, bat029. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; de Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P.; et al. MetaboLights--an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013, 41, D781–D786. [Google Scholar] [CrossRef] [PubMed]
- Rocca-Serra, P.; Salek, R.M.; Arita, M.; Correa, E.; Dayalan, S.; Gonzalez-Beltran, A.; Ebbels, T.; Goodacre, R.; Hastings, J.; Haug, K.; et al. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics 2016, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gonzalez, E.; Hestilow, T.; Haskins, W.; Huang, Y. Review of peak detection algorithms in liquid-chromatography-mass spectrometry. Curr. Genom. 2009, 10, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, A.; Sleno, L. Comparison of peak-picking workflows for untargeted liquid chromatography/high-resolution mass spectrometry metabolomics data analysis. Rapid Commun. Mass Spectrom. 2015, 29, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Coble, J.B.; Fraga, C.G. Comparative evaluation of preprocessing freeware on chromatography/mass spectrometry data for signature discovery. J. Chromatogr. A 2014, 1358, 155–164. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Mass Tolerance (Da) | Intensity Threshold (counts) | X-Variable | Noise Level (%) |
|---|---|---|---|---|
| Method 1 | 0.005 | 10 | 6989 | 24 |
| Method 2 | 0.005 | 100 | 720 | 9 |
| Method 3 | 0.01 | 10 | 7309 | 23 |
| Method 4 | 0.01 | 100 | 765 | 8 |
| Data Set | Model Quality and Description | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PCA | OPLS-DA | ||||||||
| #PC | R2X (cum) | Q2 (cum) | R2X (cum) | R2Y (cum) | Q2 (cum) | CV-ANOVA p-Value | Permutation (n = 50) | ||
| R2 | Q2 | ||||||||
| Method 1 | 5 | 0.786 | 0.746 | 0.740 | 0.997 | 0.995 | 0.000 | (0.0, 0.573) | (0.0, −0.330) |
| Method 2 | 5 | 0.926 | 0.902 | 0.857 | 0.988 | 0.987 | 0.000 | (0.0, 0.0552) | (0.0, −0.212) |
| Method 3 | 6 | 0.793 | 0.744 | 0.689 | 0.989 | 0.986 | 0.000 | (0.0, 0.304) | (0.0, −0.358) |
| Method 4 | 6 | 0.934 | 0.917 | 0.894 | 0.997 | 0.997 | 0.000 | (0.0, 0.271) | (0.0, −0.340) |
| Data Treatment | Model Quality and Description | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PCA | OPLS-DA | ||||||||
| Scaling | Trans-Formation | R2X (cum) | Q2 (cum) | R2X (cum) | R2Y (cum) | Q2 (cum) | CV-ANOVA p-Value | Permutation (n = 50) | |
| R2 | Q2 | ||||||||
| None | None | 0.995 | 0.981 | 0.981 | 0.852 | 0.849 | 5.34 × 10−23 | (0.0, 0.128) | (0.0, −0.213) |
| Center | None | 0.959 | 0.923 | 0.923 | 0.991 | 0.988 | 0.000 | (0.0, 0.161) | (0.0, −0.329) |
| UV | None | 0.443 | 0.350 | 0.337 | 0.992 | 0.986 | 0.000 | (0.0, 0.650) | (0.0, −0.294) |
| Pareto | None | 0.786 | 0.746 | 0.740 | 0.997 | 0.995 | 0.000 | (0.0, 0.573) | (0.0, −0.330) |
| UV | Log | 0.641 | 0.517 | 0.548 | 0.998 | 0.996 | 0.000 | (0.0, 0.665) | (0.0, −0.222) |
| Pareto | Log | 0.667 | 0.517 | 0.548 | 0.998 | 0.996 | 0.000 | (0.0, 0.633) | (0.0, −0.184) |
| UV | Power | 0.435 | 0.336 | 0.307 | 0.994 | 0.988 | 0.000 | (0.0, 0.649) | (0.0, −0.311) |
| Pareto | Power | 0.948 | 0.900 | 0.922 | 0.993 | 0.990 | 0.000 | (0.0, 0.267) | (0.0, −0.480) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tugizimana, F.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. A Conversation on Data Mining Strategies in LC-MS Untargeted Metabolomics: Pre-Processing and Pre-Treatment Steps. Metabolites 2016, 6, 40. https://doi.org/10.3390/metabo6040040
Tugizimana F, Steenkamp PA, Piater LA, Dubery IA. A Conversation on Data Mining Strategies in LC-MS Untargeted Metabolomics: Pre-Processing and Pre-Treatment Steps. Metabolites. 2016; 6(4):40. https://doi.org/10.3390/metabo6040040
Chicago/Turabian StyleTugizimana, Fidele, Paul A. Steenkamp, Lizelle A. Piater, and Ian A. Dubery. 2016. "A Conversation on Data Mining Strategies in LC-MS Untargeted Metabolomics: Pre-Processing and Pre-Treatment Steps" Metabolites 6, no. 4: 40. https://doi.org/10.3390/metabo6040040
APA StyleTugizimana, F., Steenkamp, P. A., Piater, L. A., & Dubery, I. A. (2016). A Conversation on Data Mining Strategies in LC-MS Untargeted Metabolomics: Pre-Processing and Pre-Treatment Steps. Metabolites, 6(4), 40. https://doi.org/10.3390/metabo6040040