
MetMatch: A Semi-Automated Software Tool for the Comparison and Alignment of LC-HRMS Data from Different Metabolomics Experiments

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
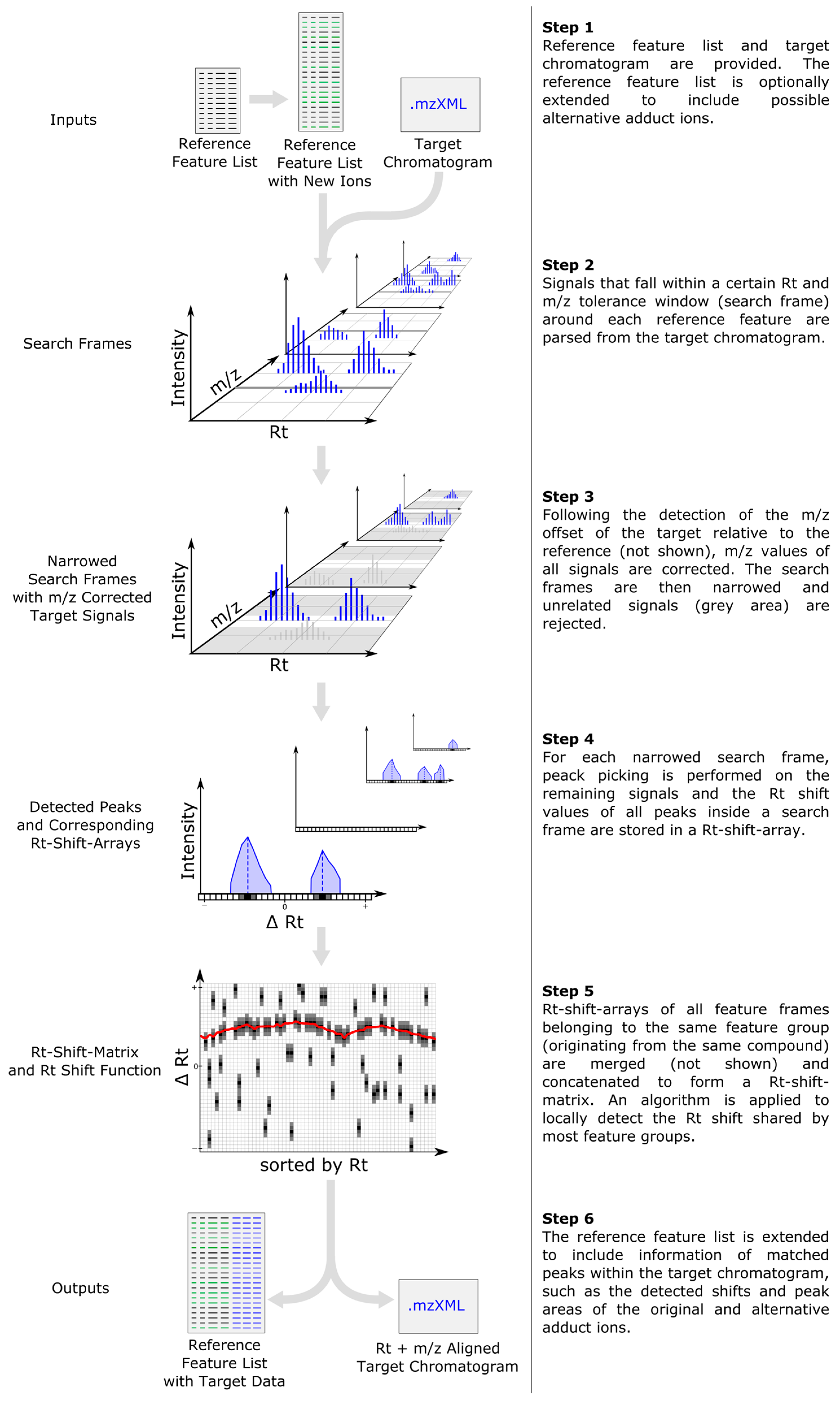
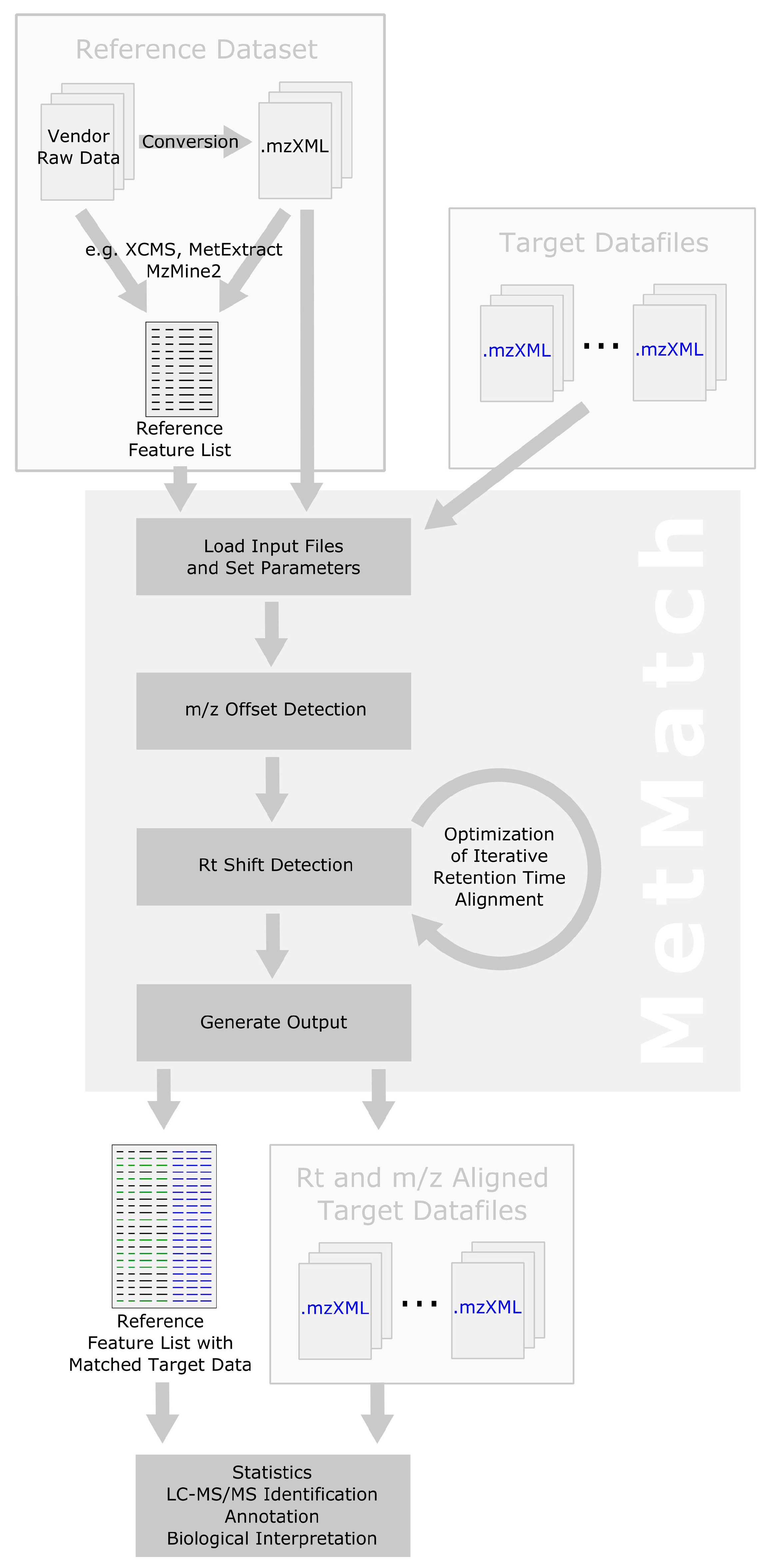
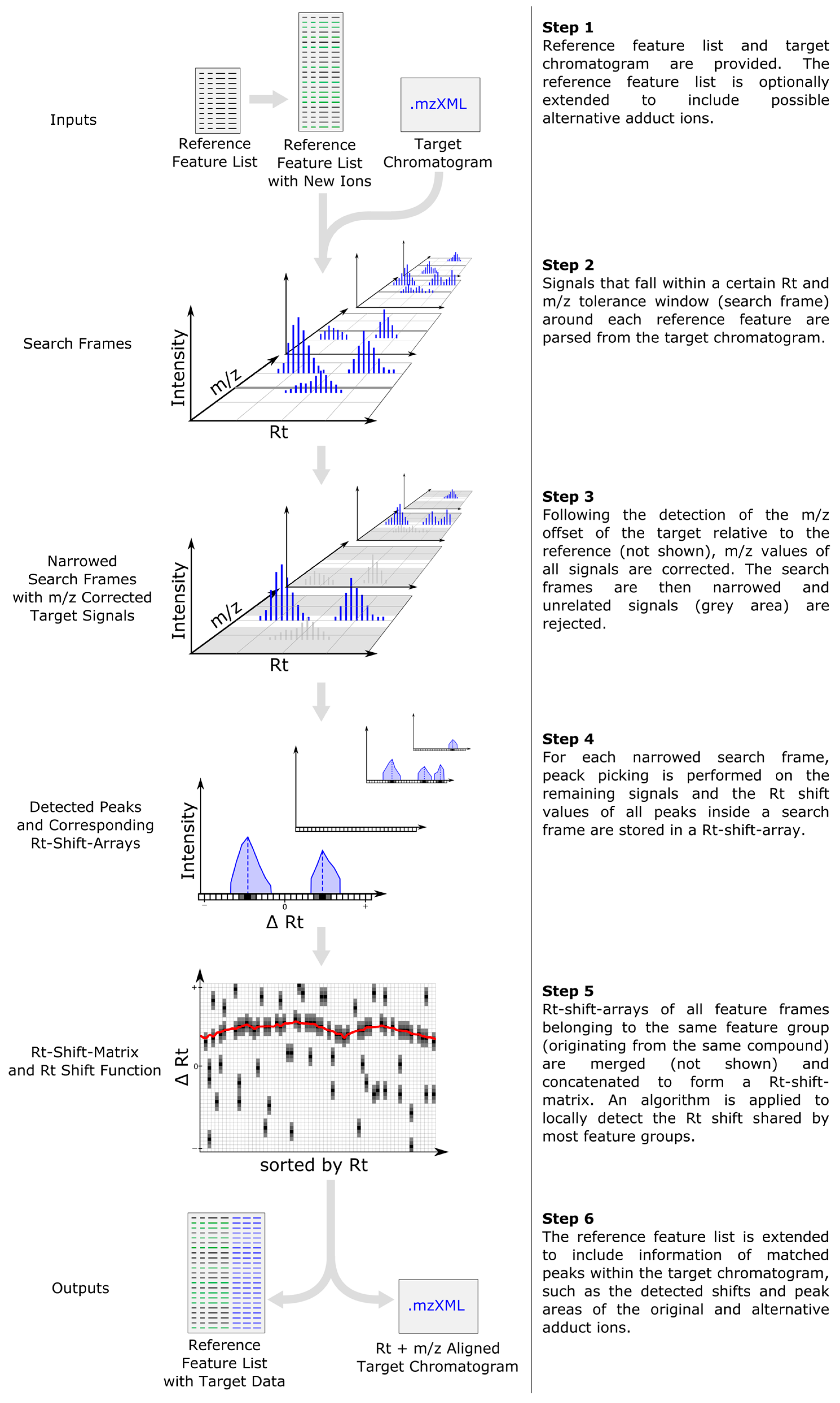
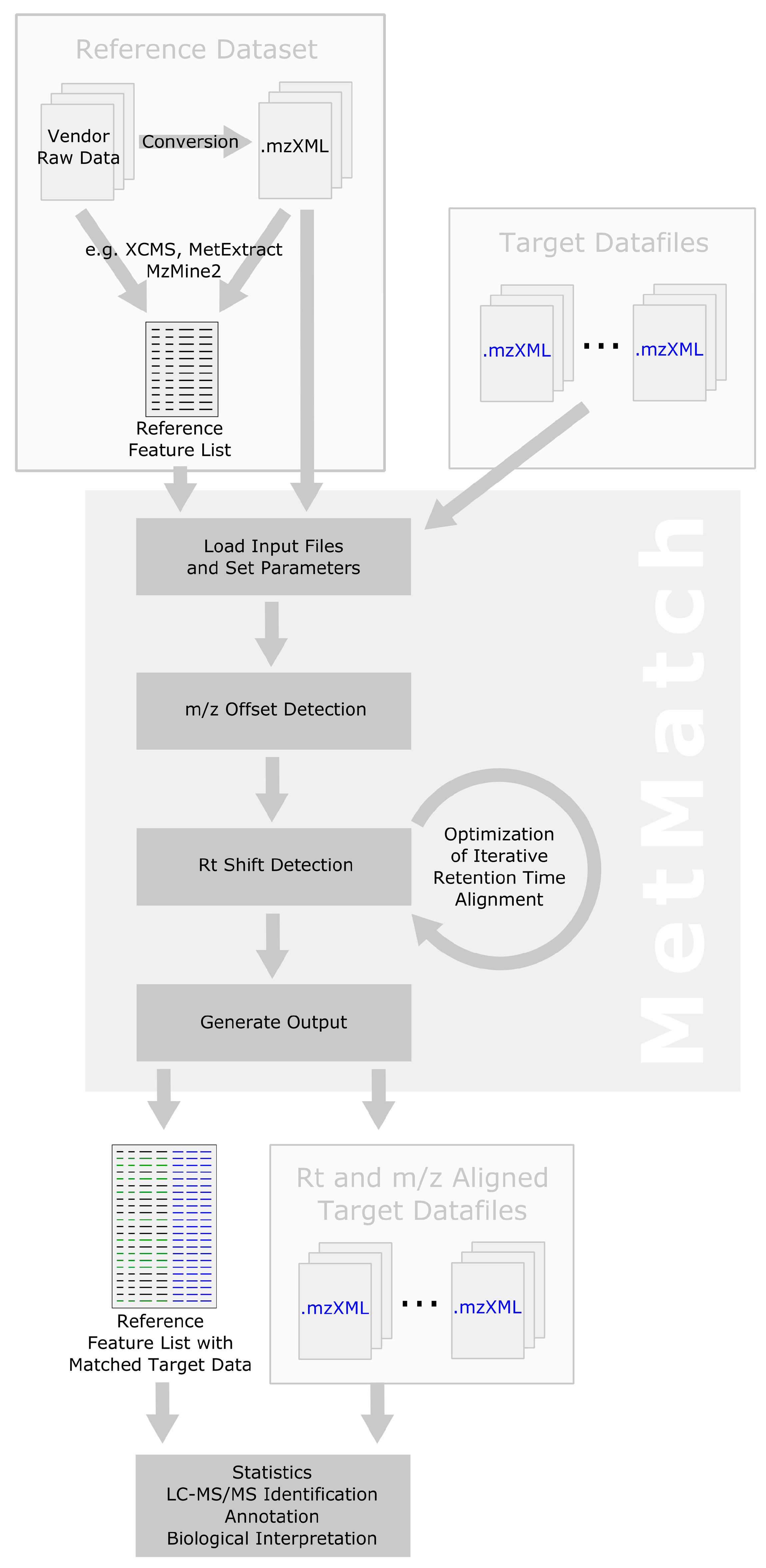
2.1. Computational Workflow
2.2. Inclusion of Alternative Ion Forms (Step 1)
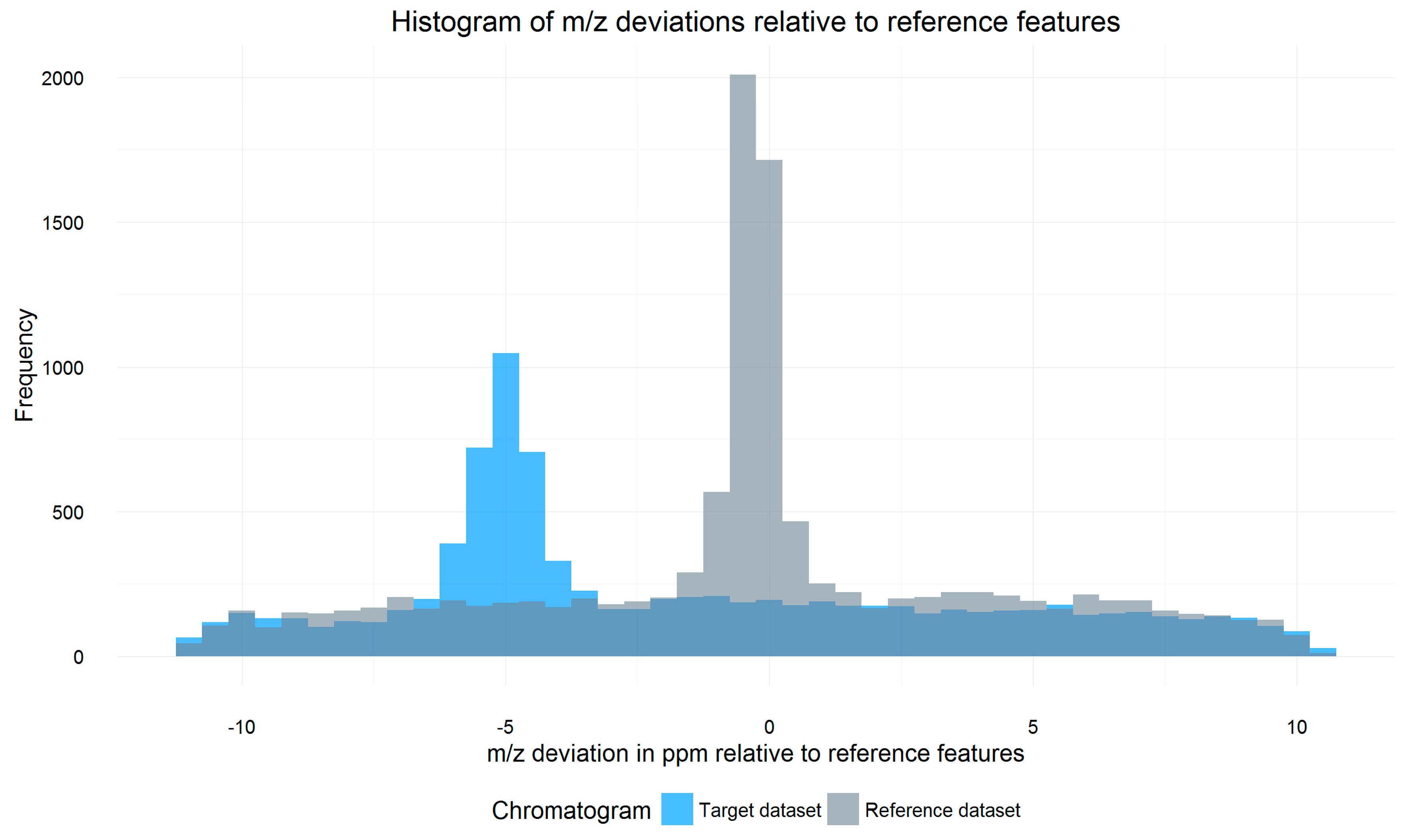
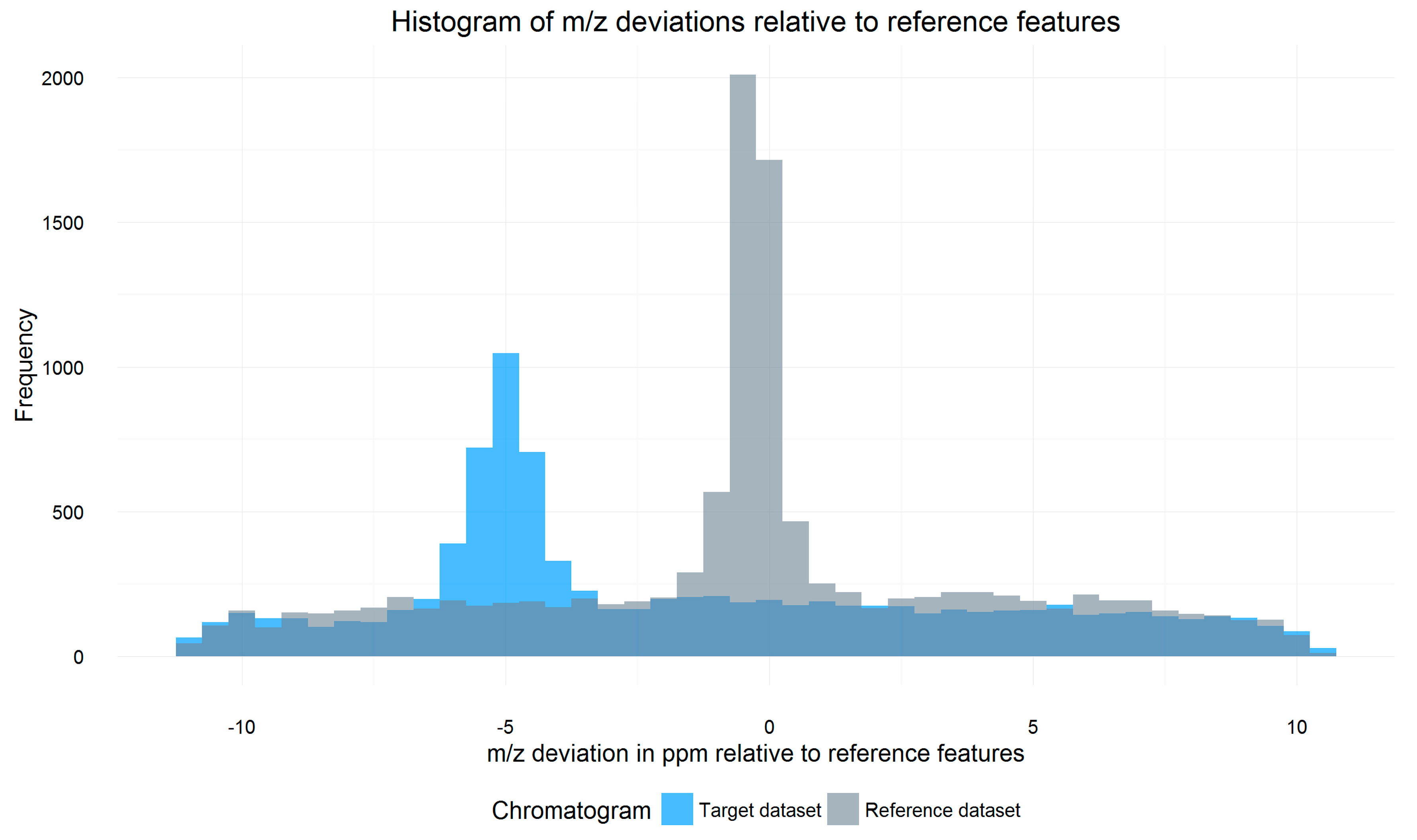
2.3. m/z Offset Detection (Steps 2 and 3)
2.4. Peak Picking (Step 4)
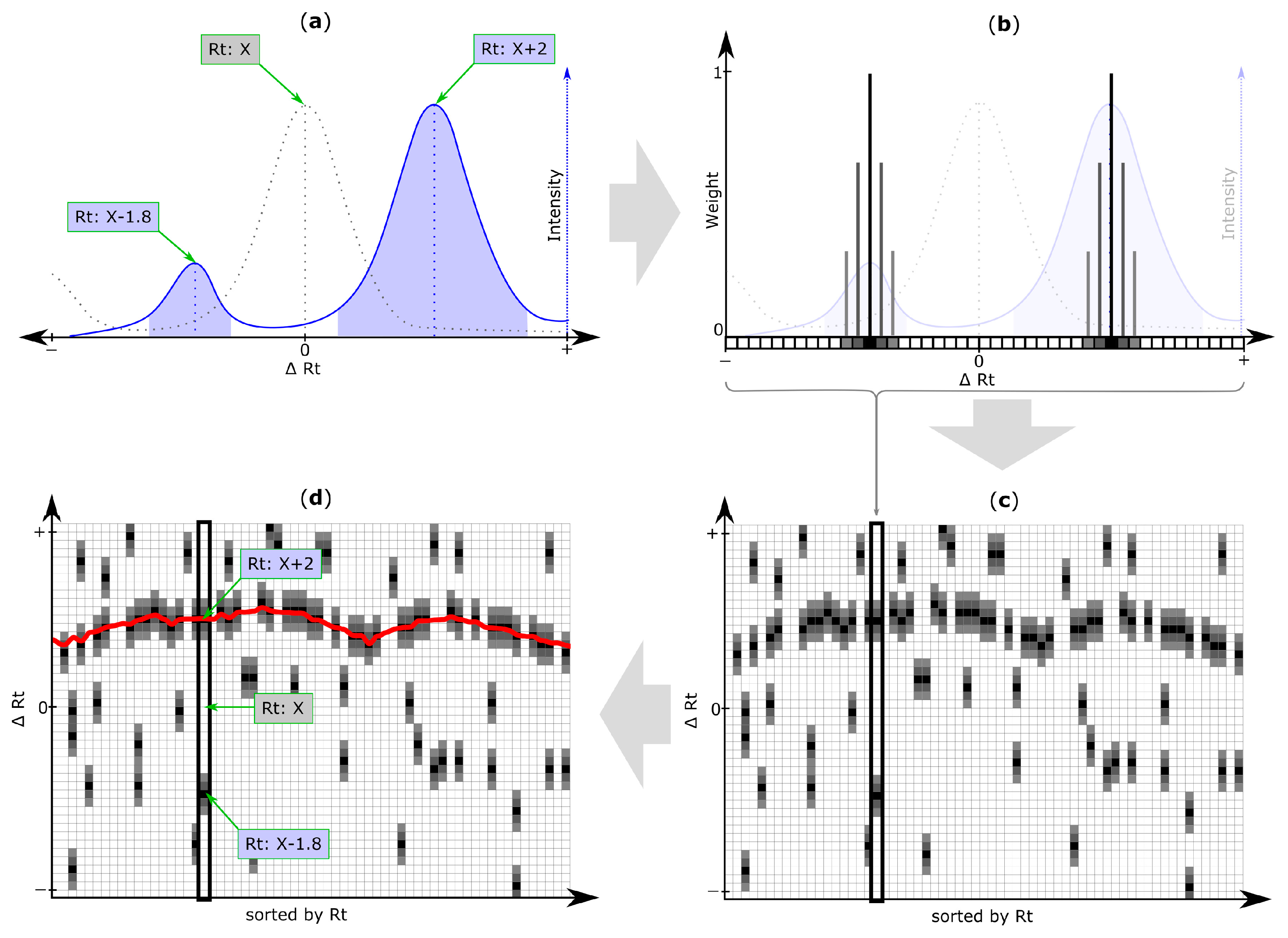
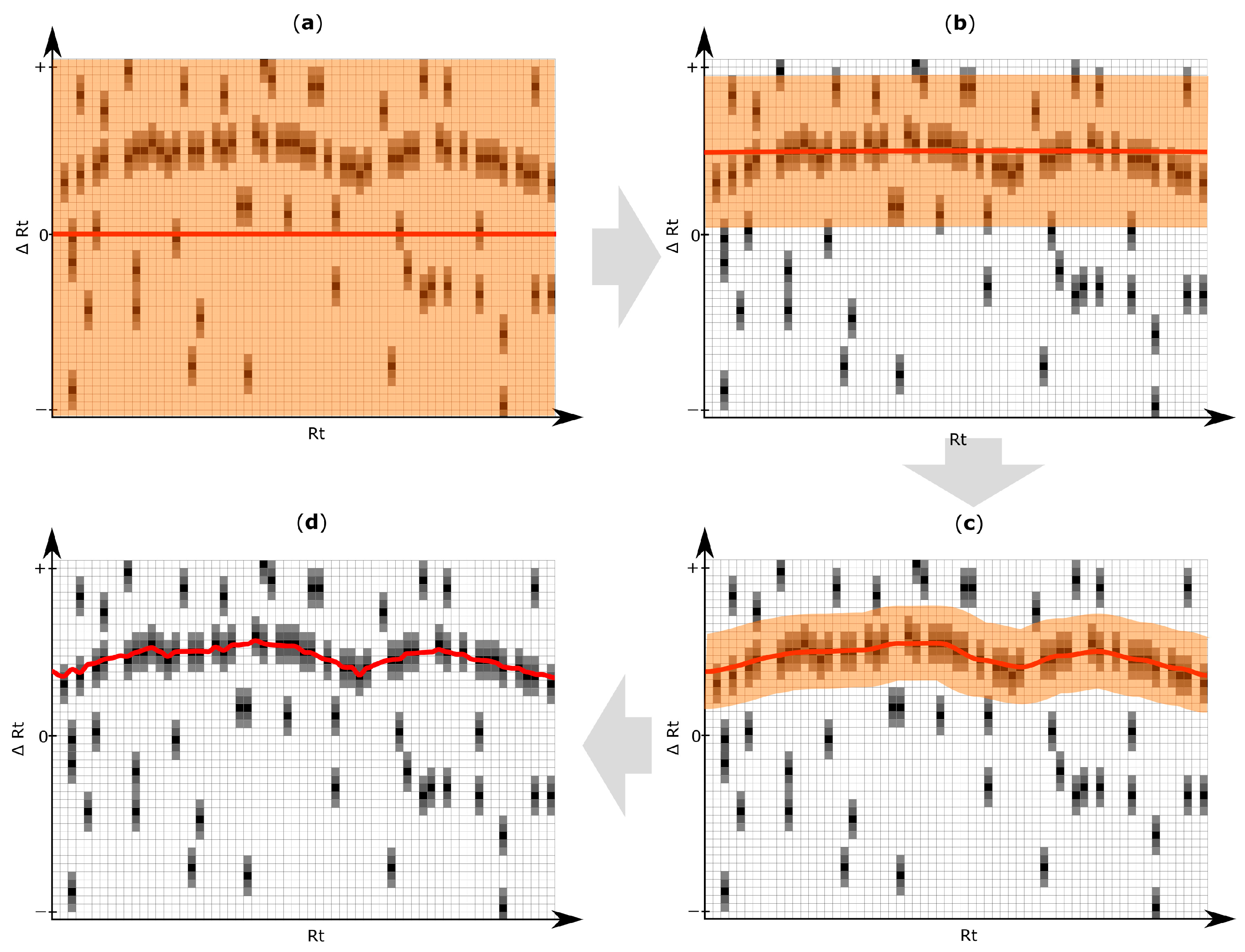
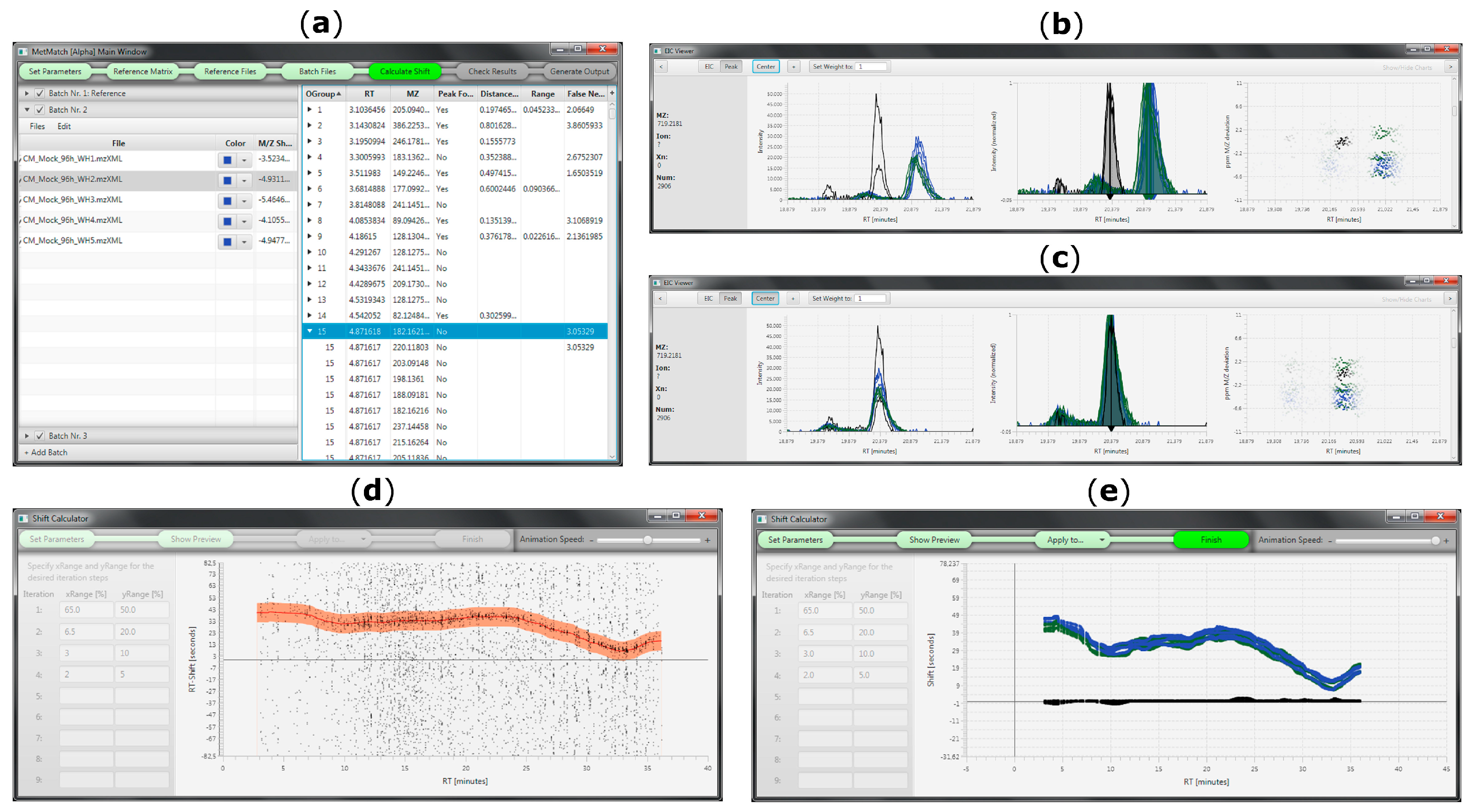
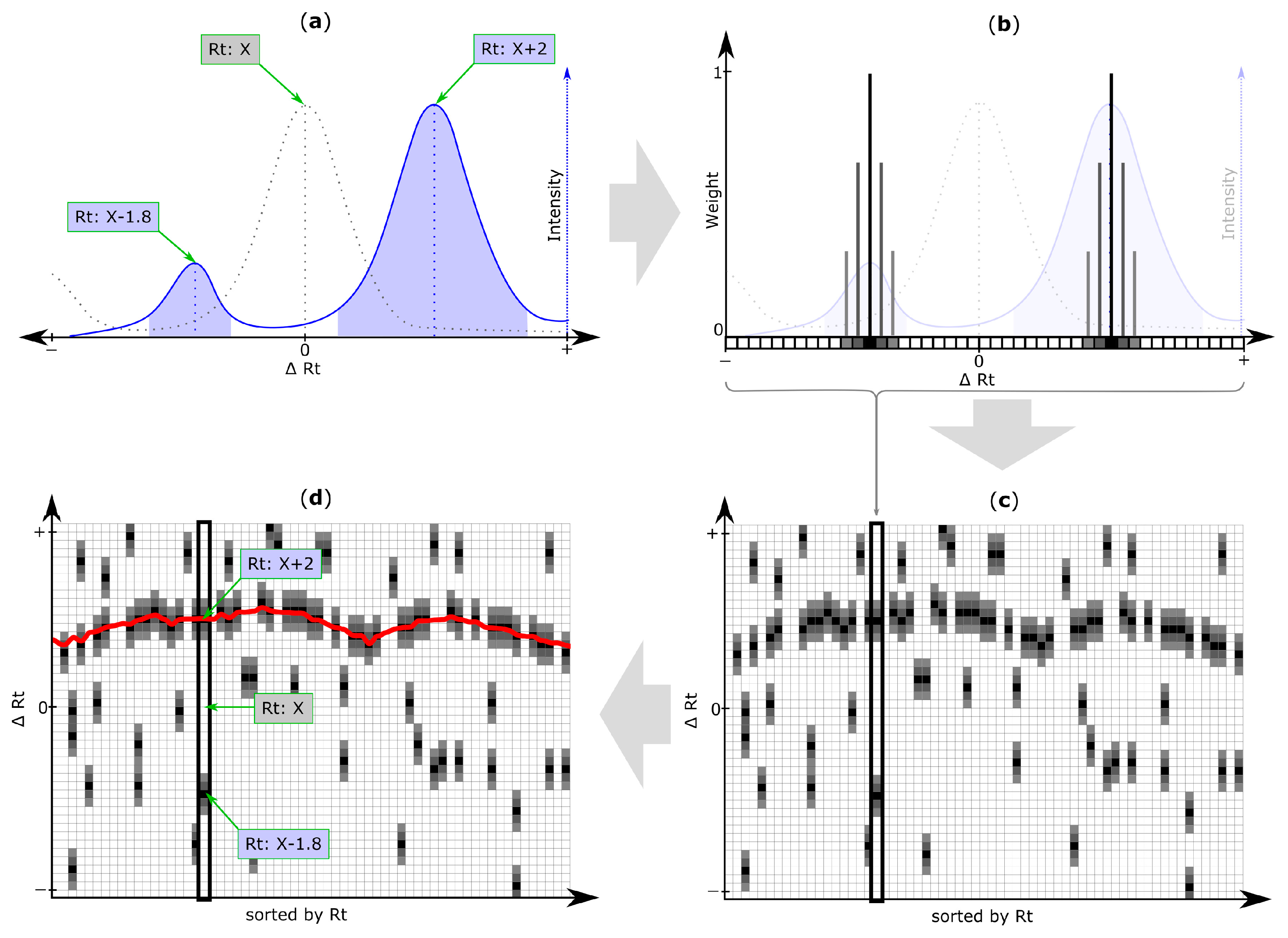
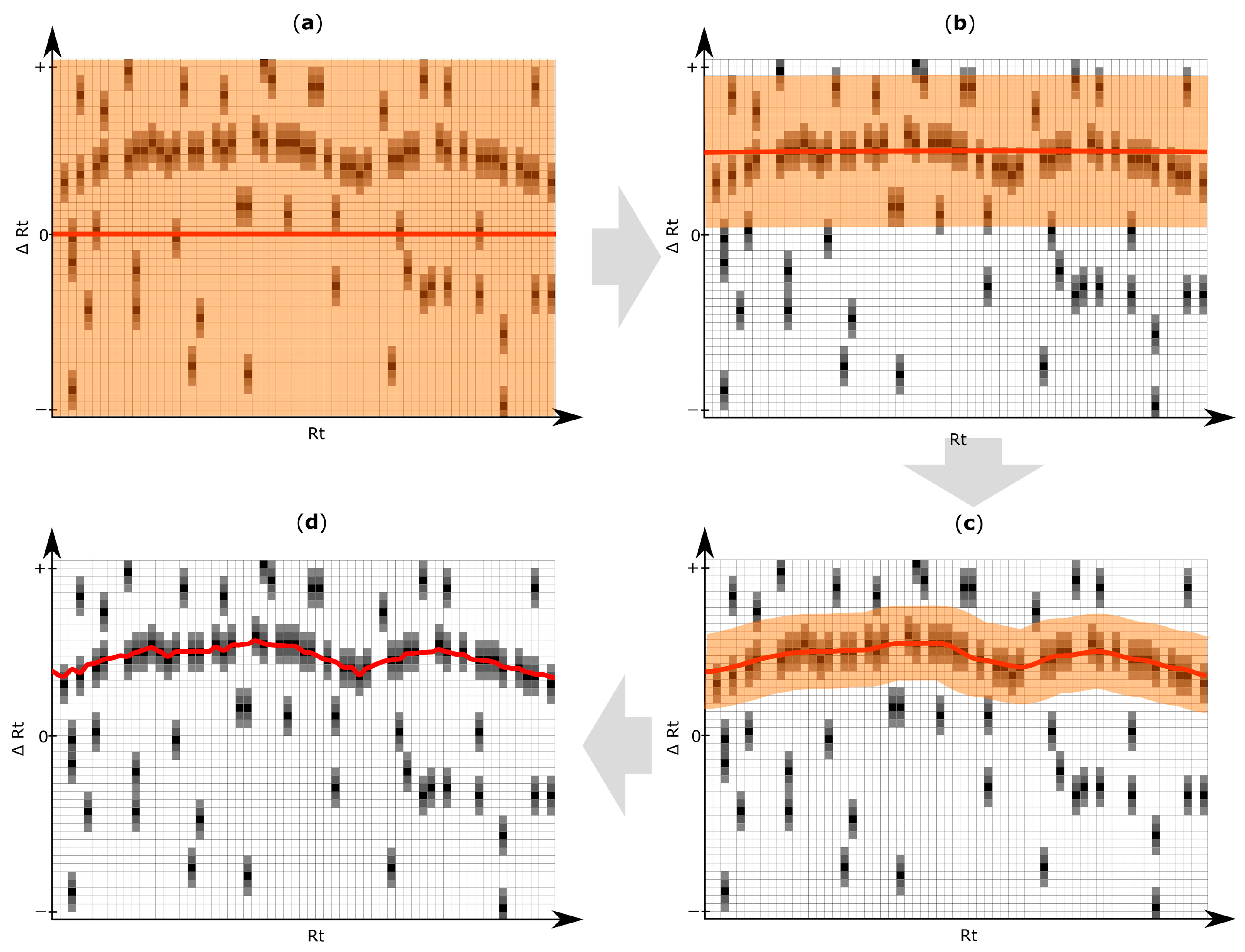
2.5. Rt Shift Detection (Step 5)
2.6. Output of Generated Results (Step 6)
2.7. Phe-Derived Biological Data
2.8. T-2 Toxin/HT-2 Toxin-Derived Biological Data
3. Results and Discussion
3.1. Accepted Input Files
3.2. Detection of Differently Formed Ions
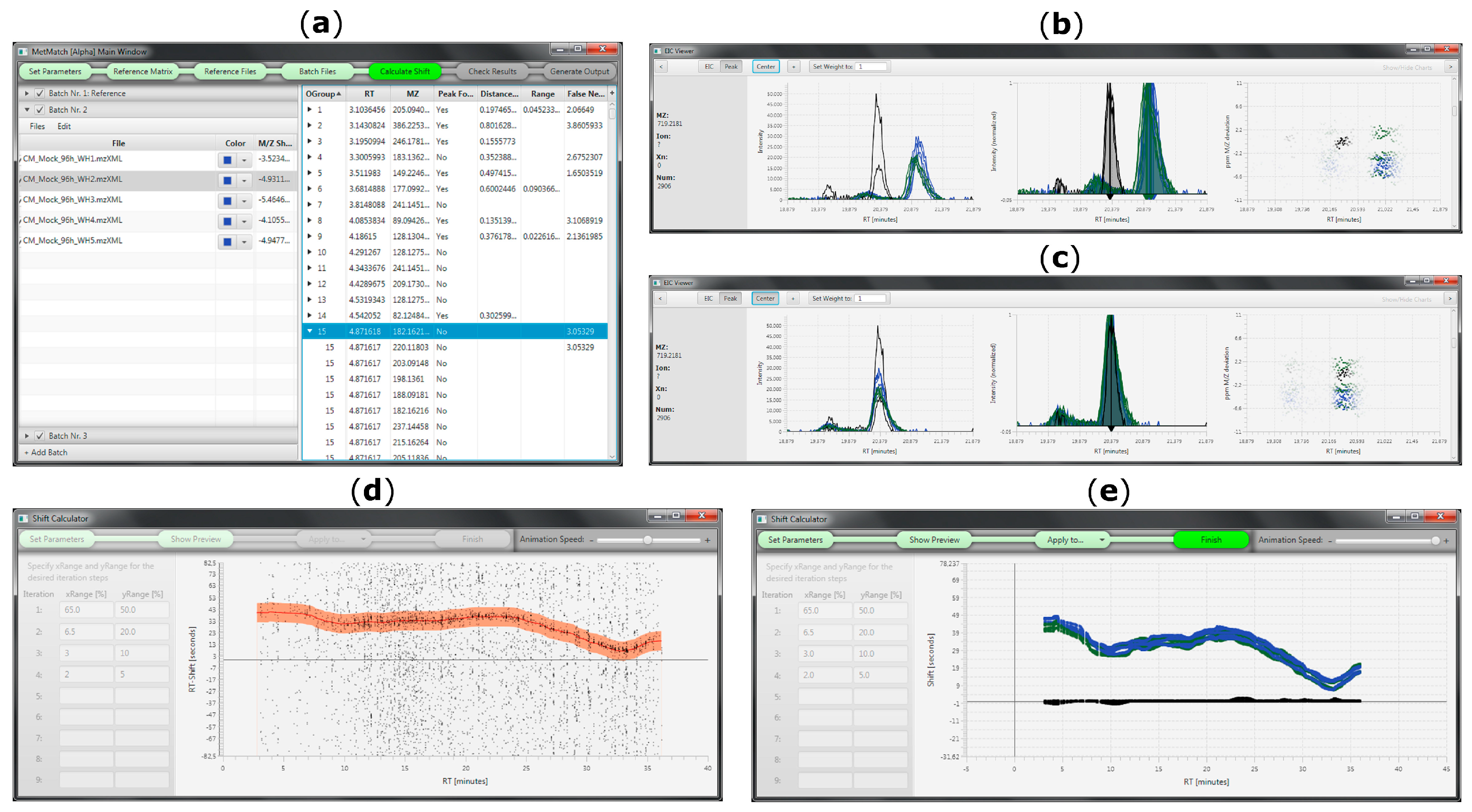
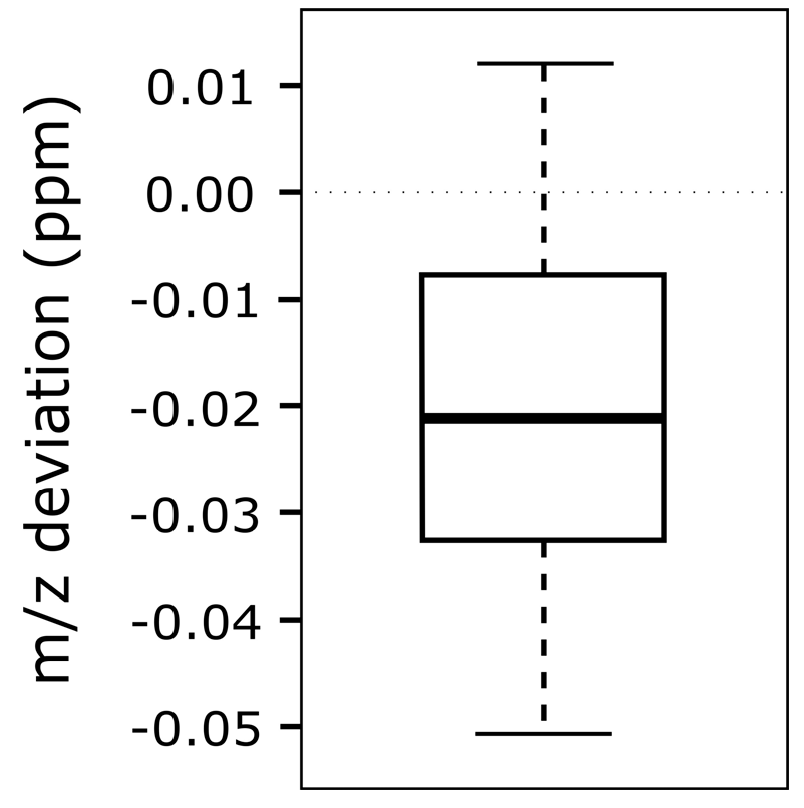
3.3. m/z Shift Detection
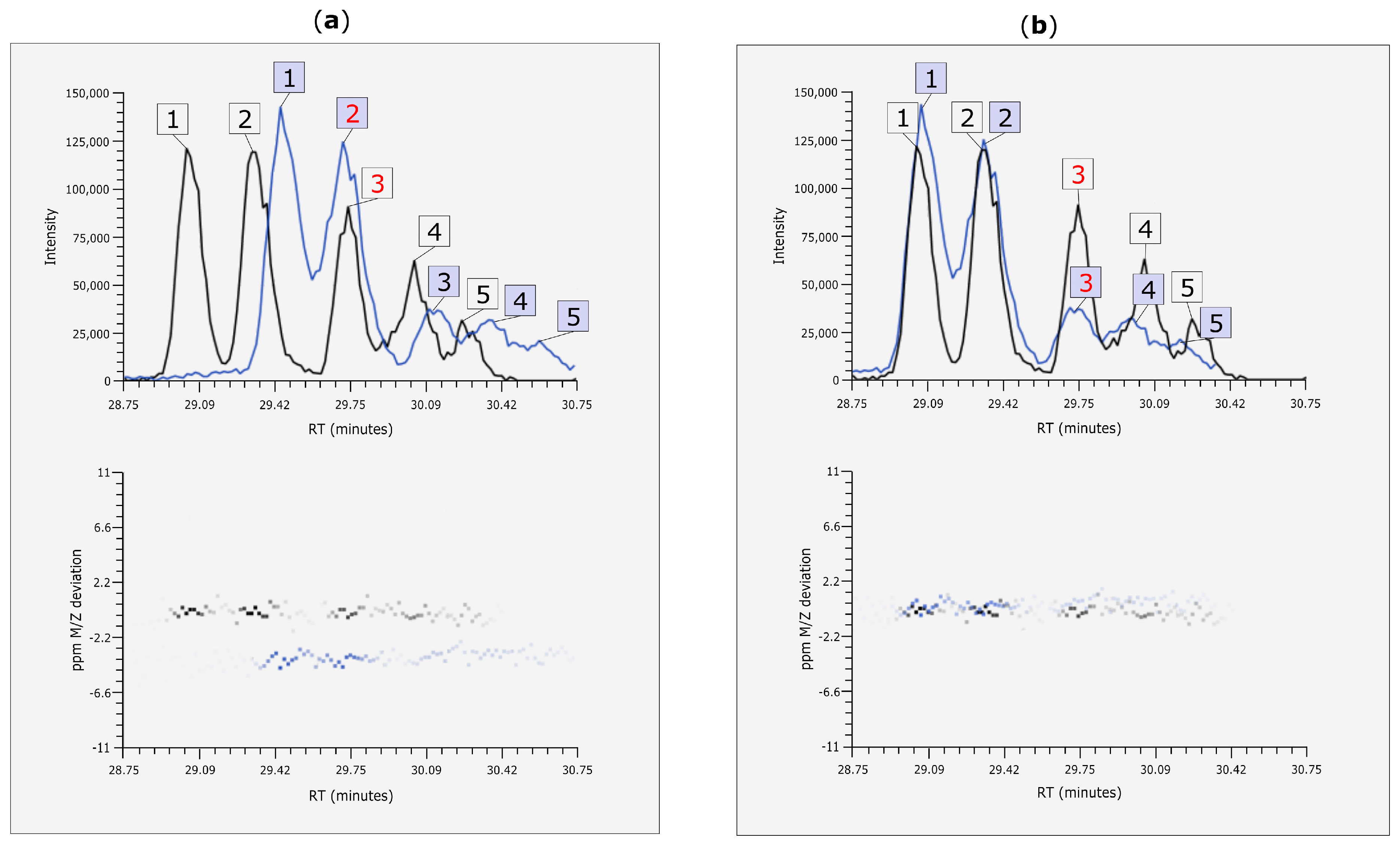
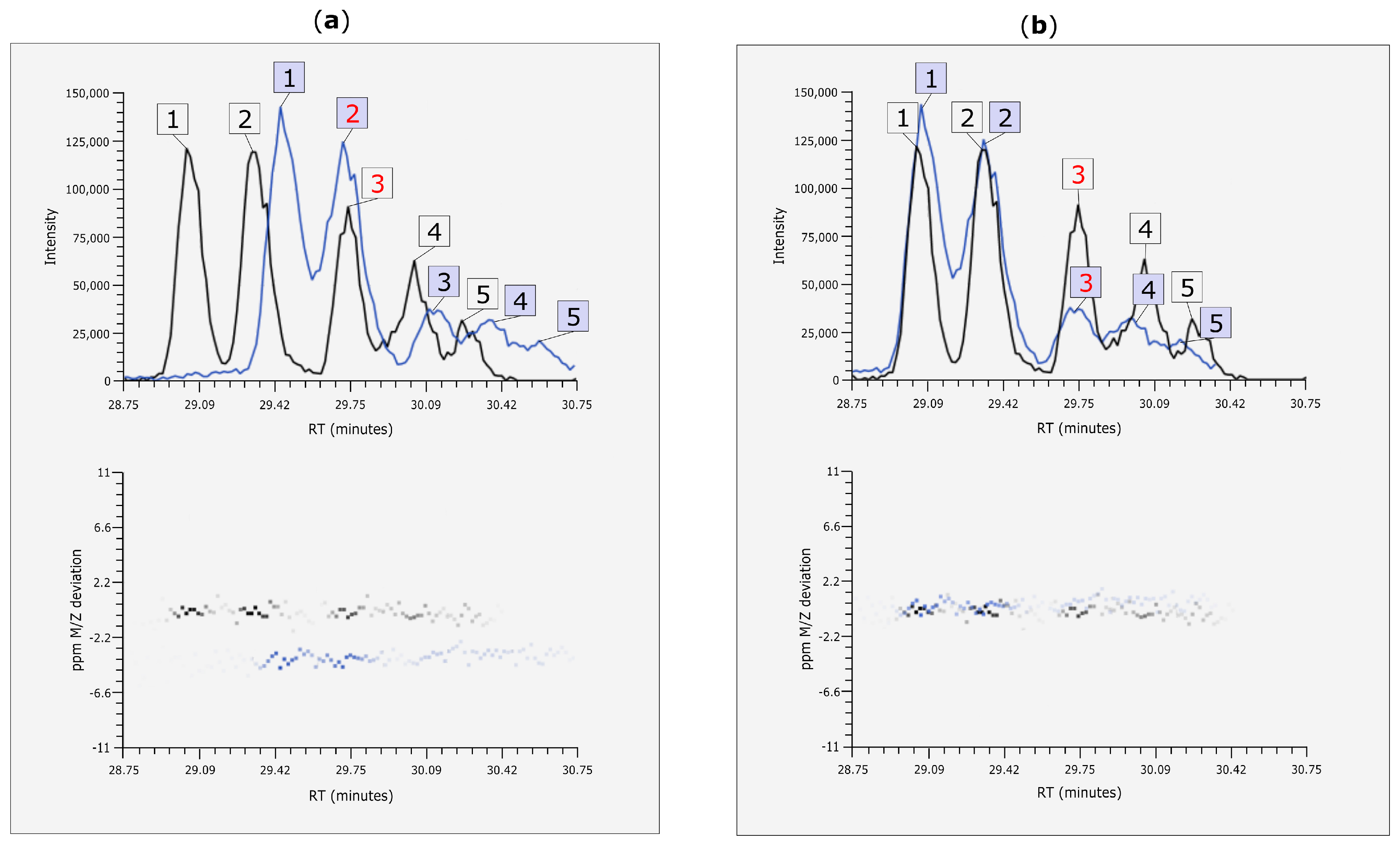
3.4. Rt Shift Detection
3.5. Manual Corrections of Automatically Generated Results
3.6. Output Format
3.7. Runtimes
3.8. Evaluation of the Phe-Derived Biological Metabolites
3.9. Evaluation of the T-2 Toxin/HT-2 Toxin-Derived Biological Data
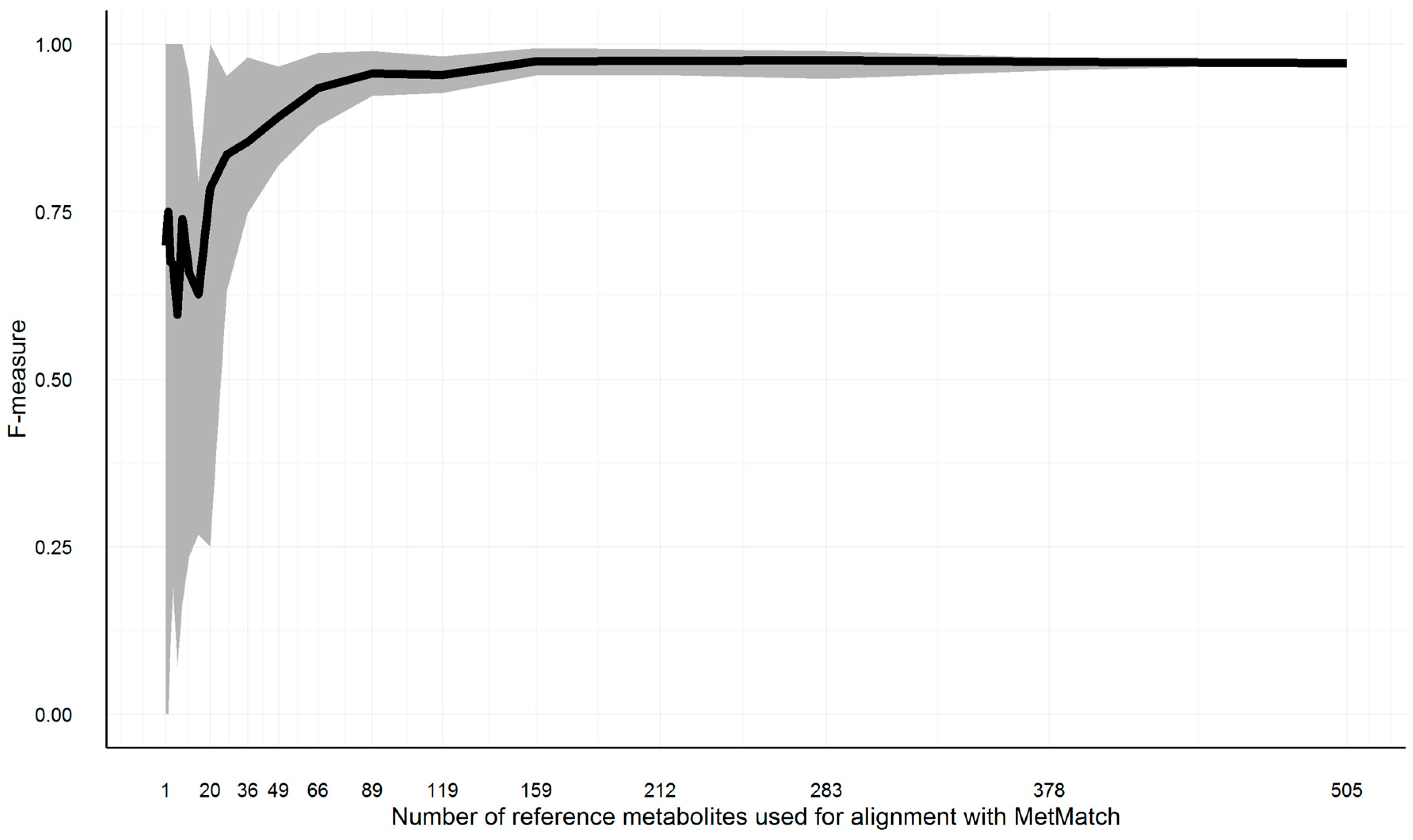
3.10. Evaluation of Artificial Data
3.10.1. Formation of Different Ion Species
3.10.2. m/z Shift Detection
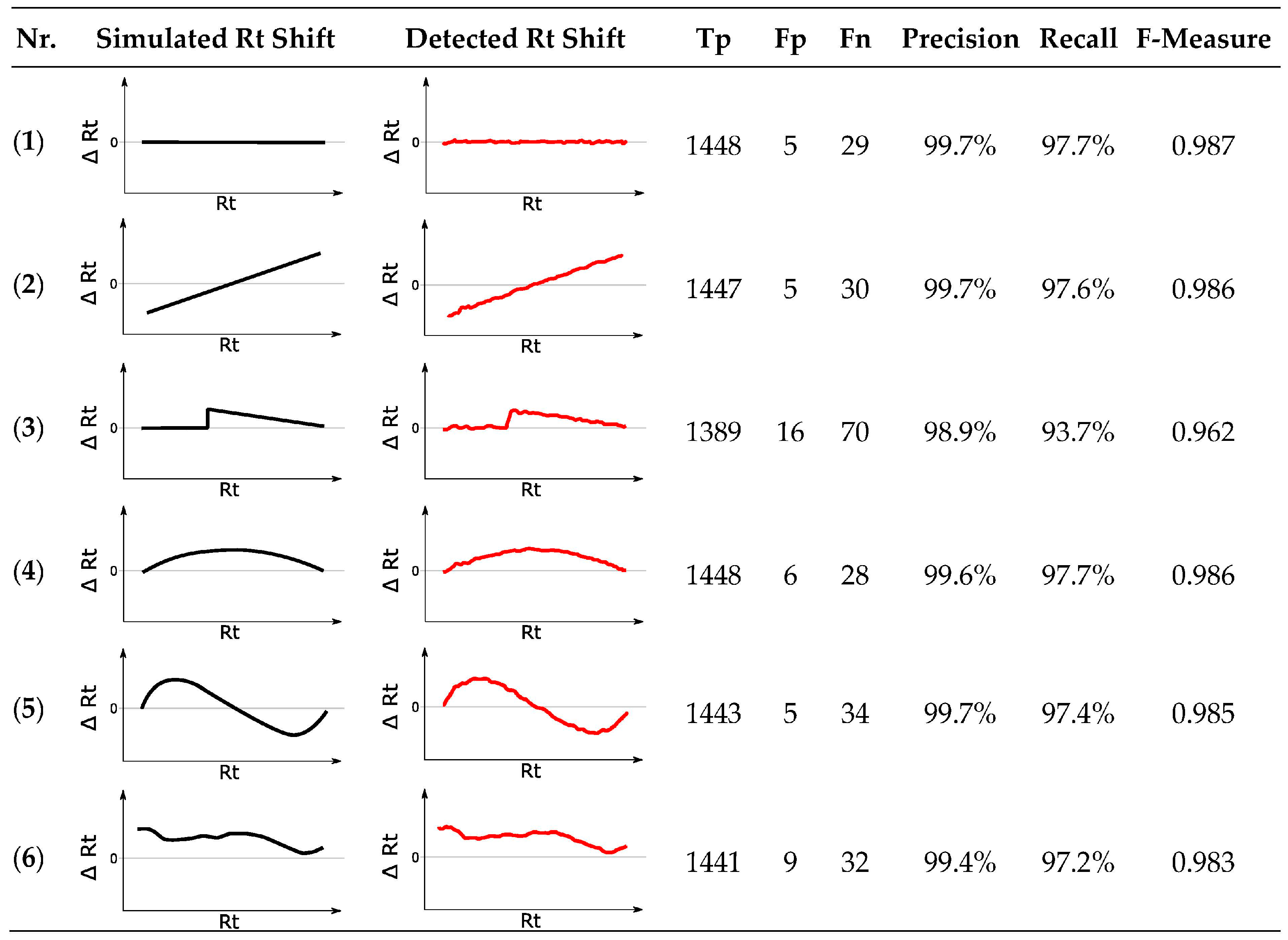
3.10.3. Rt Shift Detection
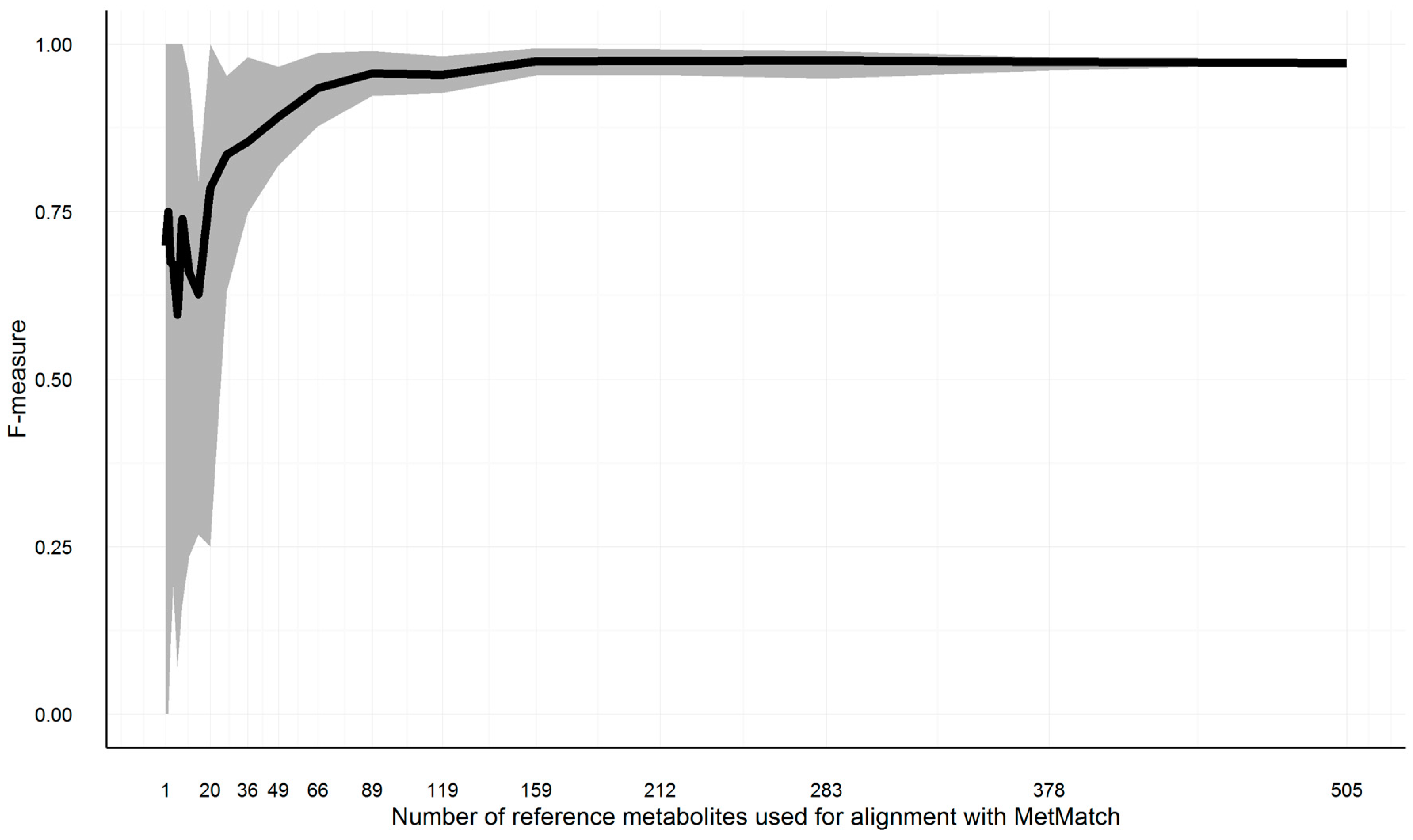
3.10.4. Minimum Required Common Feature Groups
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations and Terminology
| Binning | A range (e.g., m/z or Rt shift values) is divided into a series of equally long intervals (bins). |
| Dataset | A list of LC-HRMS datafiles belonging to one analytical batch, originating from the same experiment. |
| EIC | Extracted ion chromatogram. An EIC represents the abundance/intensity of a certain m/z value in each mass spectrum of a chromatogram. The mass spectra are sorted according to their acquisition time relative to injecting the sample into the HPLC instrument. Usually a small deviation from the selected m/z value (± ppm; depending on the used HRMS instrument) is allowed to account for minor scan-to-scan m/z inconsistencies observed during acquisition of a chromatogram. |
| Feature | A feature represents a particular metabolite ion of interest. It is characterized by its m/z value (experimental and theoretical) and retention time (as calculated by the used peak-picking algorithm). Features are usually the result of an untargeted metabolomics data processing approach and typically comprise multiple chromatographic peaks, one per LC-HRMS chromatogram. |
| Feature group | All features originating from the same chemical substance and hence sharing the same Rt and peak shape. |
| F-measure | Harmonic mean of precision and recall. |
| Fn | False negatives. Features that were not matched. |
| Fp | False positives. If a matched feature does not meet the criteria to be considered as Tp, it is an Fp. |
| LC-HRMS | Liquid chromatography coupled to high resolution mass spectrometry. |
| m/z | Mass-to-charge ratio. |
| Peak/Chromatographic peak | A chromatographic peak representing a particular feature in a chromatogram. Other than a feature, a peak has a defined m/z value, a retention time (as defined by the used peak-picking algorithm) as well as a peak area. Peaks are detected by the means of a peak-picking algorithm. |
| Peak apex | The time at which the most abundant signal of a respective chromatographic peak is detected after injection of the sample into the HPLC instrument. |
| Peak center | The time at which a chromatographic peak has its center of gravity. (Time) measurement is started with injecting the sample into the HPLC instrument. |
| Precision | Percentage of matched reference features that were correctly matched. Calculation: Number of Tp divided by the number of Tp plus Fp. |
| Recall | Percentage of all reference features that were correctly matched. Calculation: Number of Tp divided by the number of all reference features. |
| Reference dataset | A single or multiple LC-HRMS datafiles, which are used to generate the reference feature list. |
| Reference feature list | List of features to which target datasets are matched. |
| Rt | Retention time. |
| Rt shift function | A function that holds the calculated retention time shift between a target chromatogram relative to the reference feature list. It is used to map the retention times of detected chromatographic peaks in the target chromatogram to the retention time of the features in the reference list. |
| Search frame | Signals of a chromatogram (single datafile) that fall within a certain Rt and m/z tolerance window around a reference feature. These values have to be slightly larger than the maximum expected shift. (e.g., 1.5 min Rt shift and 10 parts per million (ppm) m/z shift for a typical LC-HRMS chromatogram). |
| Rt shift array | An array containing the Rt shift of all peaks detected within a search frame relative to the retention time of the respective reference feature. First, a search frame is discretized into smaller bins (number of bins is set by the user of MetMatch) each representing a certain Rt offset relative to the retention time of the respective reference feature. Then, those Rt bins for which chromatographic peaks of the respective reference feature were detected in the target chromatogram, are assigned weight values greater than zero (either automatically or user-guided). Any Rt offset bin for which no chromatographic peak was detected, is assigned zero value and thus do not contribute to the Rt shift detection algorithm. The number of entries in each Rt shift array is set by the user during the iterative Rt alignment step. Each entry in the Rt shift array corresponds to a certain Rt deviation between the retention times of the reference features and their corresponding chromatographic peaks in the target chromatograms. |
| Rt shift matrix | The individual Rt shift arrays of all reference feature groups are concatenated column-wise and sorted by the retention time of their reference feature group. Each column represents a feature group, while rows represent discretized Rt shift values. |
| Target dataset | A list of LC-HRMS datafiles to be matched to a reference feature list. |
| Tp | True positives. Matched features are considered Tp if their m/z deviation is ≤5 ppm and their Rt deviation is ≤8.4 s (manually validated threshold value for the distinction of Tp and Fp in this datafile) relative to the reference feature after shift correction. The formation of different ion species is taken into account when comparing the m/z. |
References
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Ventura, D.; Prince, J.T. LC-MS alignment in theory and practice: A comprehensive algorithmic review. Brief. Bioinform. 2015, 16, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Aberg, K.M.; Alm, E.; Torgrip, R.J. The correspondence problem for metabonomics datasets. Anal. Bioanal. Chem. 2009, 394, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Kassidas, A.; MacGregor, J.F.; Taylor, P.A. Synchronization of batch trajectories using dynamic time warping. AIChE J. 1998, 44, 864–875. [Google Scholar] [CrossRef]
- Nielsen, N.-P.V.; Carstensen, J.M.; Smedsgaard, J. Aligning of single and multiple wavelength chromatographic profiles for chemometric data analysis using correlation optimised warping. J. Chromatogr. A 1998, 805, 17–35. [Google Scholar] [CrossRef]
- Eilers, P.H. Parametric time warping. Anal. Chem. 2004, 76, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Suits, F.; Lepre, J.; Du, P.; Bischoff, R.; Horvatovich, P. Two-dimensional method for time aligning liquid chromatography-mass spectrometry data. Anal. Chem. 2008, 80, 3095–3104. [Google Scholar] [CrossRef] [PubMed]
- Prince, J.T.; Marcotte, E.M. Chromatographic alignment of ESI-LC-MS proteomics data sets by ordered bijective interpolated warping. Anal. Chem. 2006, 78, 6140–6152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Asara, J.M.; Adamec, J.; Ouzzani, M.; Elmagarmid, A.K. Data pre-processing in liquid chromatography-mass spectrometry-based proteomics. Bioinformatics 2005, 21, 4054–4059. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. Xcms: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Katajamaa, M.; Miettinen, J.; Oresic, M. Mzmine: Toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics 2006, 22, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. Mzmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Bertsch, A.; Gropl, C.; Reinert, K.; Kohlbacher, O. Openms and TOPP: Open source software for LC-MS data analysis. Methods Mol. Biol. 2011, 696, 353–367. [Google Scholar] [PubMed]
- Bloemberg, T.G.; Gerretzen, J.; Lunshof, A.; Wehrens, R.; Buydens, L.M. Warping methods for spectroscopic and chromatographic signal alignment: A tutorial. Anal. Chim. Acta 2013, 781, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Christin, C.; Hoefsloot, H.C.; Smilde, A.K.; Suits, F.; Bischoff, R.; Horvatovich, P.L. Time alignment algorithms based on selected mass traces for complex LC-MS data. J. Proteome Res. 2010, 9, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Christin, C.; Smilde, A.K.; Hoefsloot, H.C.; Suits, F.; Bischoff, R.; Horvatovich, P.L. Optimized time alignment algorithm for LC-MS data: Correlation optimized warping using component detection algorithm-selected mass chromatograms. Anal. Chem. 2008, 80, 7012–7021. [Google Scholar] [CrossRef] [PubMed]
- Bueschl, C.; Kluger, B.; Berthiller, F.; Lirk, G.; Winkler, S.; Krska, R.; Schuhmacher, R. Metextract: A new software tool for the automated comprehensive extraction of metabolite-derived LC/MS signals in metabolomics research. Bioinformatics 2012, 28, 736–738. [Google Scholar] [CrossRef] [PubMed]
- MetMatch. Available online: https://metabolomics-ifa.boku.ac.at/MetMatch/index.html (accessed on 31 October 2016).
- Kluger, B.; Bueschl, C.; Neumann, N.; Stuckler, R.; Doppler, M.; Chassy, A.W.; Waterhouse, A.L.; Rechthaler, J.; Kampleitner, N.; Thallinger, G.G.; et al. Untargeted profiling of tracer-derived metabolites using stable isotopic labeling and fast polarity-switching lc-esi-hrms. Anal. Chem. 2014, 86, 11533–11537. [Google Scholar] [CrossRef] [PubMed]
- Bueschl, C.; Kluger, B.; Lemmens, M.; Adam, G.; Wiesenberger, G.; Maschietto, V.; Marocco, A.; Strauss, J.; Bodi, S.; Thallinger, G.G.; et al. A novel stable isotope labelling assisted workflow for improved untargeted LC-HRMS based metabolomics research. Metabolomics 2014, 10, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Parich, A.; Bueschl, C.; Schoefbeck, D.; Neumann, N.K.; Kluger, B.; Schuster, K.; Krska, R.; Adam, G.; Lemmens, M.; et al. GC-MS based targeted metabolic profiling identifies changes in the wheat metabolome following deoxynivalenol treatment. Metabolomics 2015, 11, 722–738. [Google Scholar] [CrossRef] [PubMed]
- Meng-Reiterer, J.; Varga, E.; Nathanail, A.V.; Bueschl, C.; Rechthaler, J.; McCormick, S.P.; Michlmayr, H.; Malachova, A.; Fruhmann, P.; Adam, G.; et al. Tracing the metabolism of ht-2 toxin and t-2 toxin in barley by isotope-assisted untargeted screening and quantitative LC-HRMS analysis. Anal. Bioanal. Chem. 2015, 407, 8019–8033. [Google Scholar] [CrossRef] [PubMed]
- Fourment, M.; Gillings, M.R. A comparison of common programming languages used in bioinformatics. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Algorithm1. Available online: https://metabolomics-ifa.boku.ac.at/metmatch/figures/Algorithm1.gif (accessed on 31 October 2016).
- Pedrioli, P.G.; Eng, J.K.; Hubley, R.; Vogelzang, M.; Deutsch, E.W.; Raught, B.; Pratt, B.; Nilsson, E.; Angeletti, R.H.; Apweiler, R.; et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004, 22, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. Proteowizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Podwojski, K.; Fritsch, A.; Chamrad, D.C.; Paul, W.; Sitek, B.; Stuhler, K.; Mutzel, P.; Stephan, C.; Meyer, H.E.; Urfer, W.; et al. Retention time alignment algorithms for LC/MS data must consider non-linear shifts. Bioinformatics 2009, 25, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Mitra, V.; Smilde, A.; Hoefsloot, H.; Suits, F.; Bischoff, R.; Horvatovich, P. Inversion of peak elution order prevents uniform time alignment of complex liquid-chromatography coupled to mass spectrometry datasets. J. Chromatogr. A 2014, 1373, 61–72. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion ID a | Group ID a | Rt a | m/z a | Ion Form b | Uncharged Ion Mass b | Ion Charge b | Ionisation Mode b |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 2.78 | 160.075 | [M + H]+ | 159.068 | 1 | + |
| 2 | 1 | 2.78 | 177.102 | [M + NH4]+ | 159.068 | 1 | + |
| 3 | 2 | 3.19 | 482.296 | [M + NH4]+ | 464.262 | 1 | + |
| 4 | 3 | 3.27 | 330.458 | [M + NH4]+ | 312.424 | 1 | + |
| Ion ID a | Group ID a | Rt a | m/z a | Ion Form a | Uncharged Ion Mass a | Reference Peak Area a | File Peak Area b | File Peak Rt b | File Peak m/z b |
|---|---|---|---|---|---|---|---|---|---|
| 1 a | 1 | 2.78 | 160.075 | [M + H]+ | 159.068 | 103,038 | - | - | - |
| 2 a | 1 | 2.78 | 177.102 | [M + NH4]+ | 159.068 | 6,234,458 | - | - | - |
| 901 b | 1 | 2.78 | 182.057 | [M + Na]+ | 159.068 | - | 3,423,472 | 2.83 | 182.057 |
| 3 a | 2 | 3.19 | 482.296 | [M + NH4]+ | 464.262 | 523,238 | - | - | - |
| 902 b | 2 | 3.19 | 487.251 | [M + Na]+ | 464.262 | - | 2,137,423 | 3.26 | 487.251 |
| 4 a | 3 | 3.27 | 330.458 | [M + NH4]+ | 312.424 | 13,459,147 | - | - | - |
| 903 b | 3 | 3.27 | 487.251 | [M + Na]+ | 312.424 | - | 15,358,291 | 3.38 | 487.251 |
| Total Number of Features | Matched Features | Tp | Fp | Fn | Precision | Recall | F-Measure |
|---|---|---|---|---|---|---|---|
| 1482 | 1446 | 1446 | 0 | 36 | 100% | 97.6% | 0.988 |
| Total Number of Features | Matched Features | Tp | Fp | Fn | Precision | Recall | F-Measure |
|---|---|---|---|---|---|---|---|
| 1482 | 1446 | 1446 | 0 | 36 | 100% | 97.6% | 0.988 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koch, S.; Bueschl, C.; Doppler, M.; Simader, A.; Meng-Reiterer, J.; Lemmens, M.; Schuhmacher, R. MetMatch: A Semi-Automated Software Tool for the Comparison and Alignment of LC-HRMS Data from Different Metabolomics Experiments. Metabolites 2016, 6, 39. https://doi.org/10.3390/metabo6040039
Koch S, Bueschl C, Doppler M, Simader A, Meng-Reiterer J, Lemmens M, Schuhmacher R. MetMatch: A Semi-Automated Software Tool for the Comparison and Alignment of LC-HRMS Data from Different Metabolomics Experiments. Metabolites. 2016; 6(4):39. https://doi.org/10.3390/metabo6040039
Chicago/Turabian StyleKoch, Stefan, Christoph Bueschl, Maria Doppler, Alexandra Simader, Jacqueline Meng-Reiterer, Marc Lemmens, and Rainer Schuhmacher. 2016. "MetMatch: A Semi-Automated Software Tool for the Comparison and Alignment of LC-HRMS Data from Different Metabolomics Experiments" Metabolites 6, no. 4: 39. https://doi.org/10.3390/metabo6040039
APA StyleKoch, S., Bueschl, C., Doppler, M., Simader, A., Meng-Reiterer, J., Lemmens, M., & Schuhmacher, R. (2016). MetMatch: A Semi-Automated Software Tool for the Comparison and Alignment of LC-HRMS Data from Different Metabolomics Experiments. Metabolites, 6(4), 39. https://doi.org/10.3390/metabo6040039