
Neural Tube Defects: From a Proteomic Standpoint

Abstract
:1. Introduction—Inborn Errors of Development: Neural Tube Defects
1.1. Clinical Features of NTDs
1.2. Standard Procedures for the Diagnosis NTDs
- —prenatal myelomeningocele repair and prognosis
- —postnatal myelomeningocele surgical repair and prognosis
- —pregnancy termination with autopsy [12].
1.3. Determining the Causative Factors or Contributing Factors of NTDs
1.3.1. Importance of Vitamins and Vitamin-Related Genes
1.3.2. Importance of Methylation-Related Genes
1.3.3. Importance of Metabolites
1.4. Use of Animal Models for the Study of Different Types of NTDs
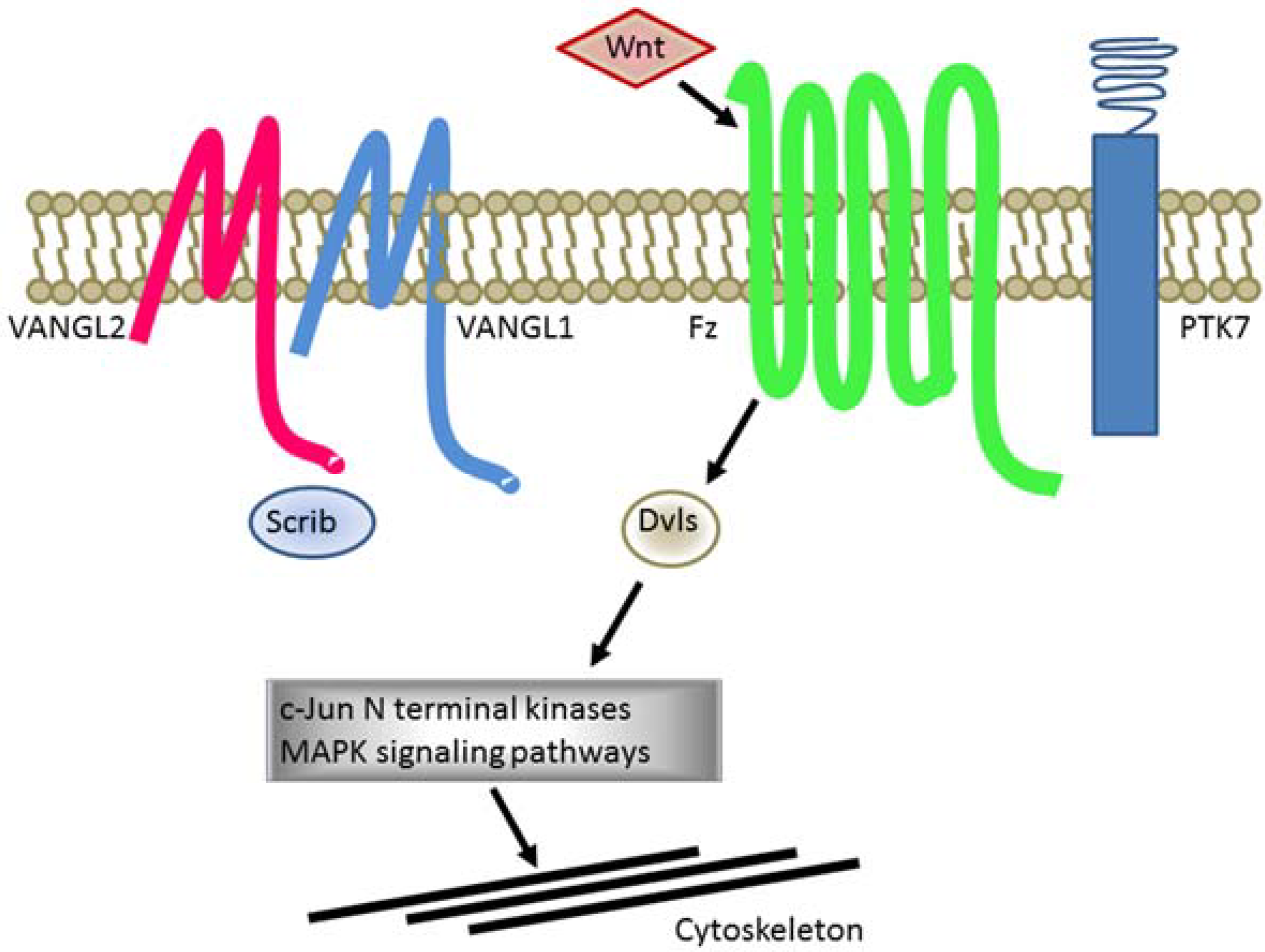
1.5. Defects of Planar Cell Polarity Play a Causative Role in NTDs
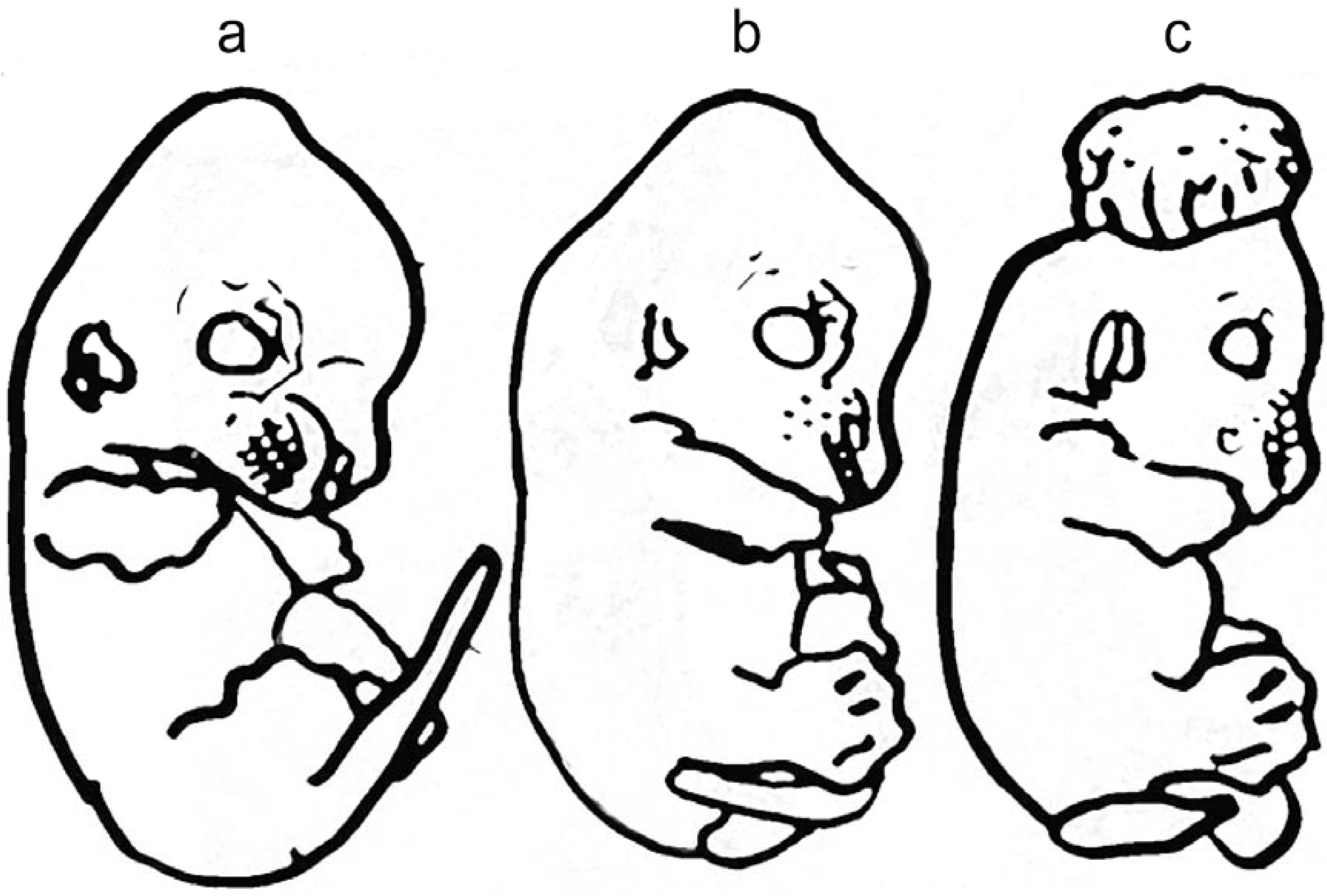

{kind=link}
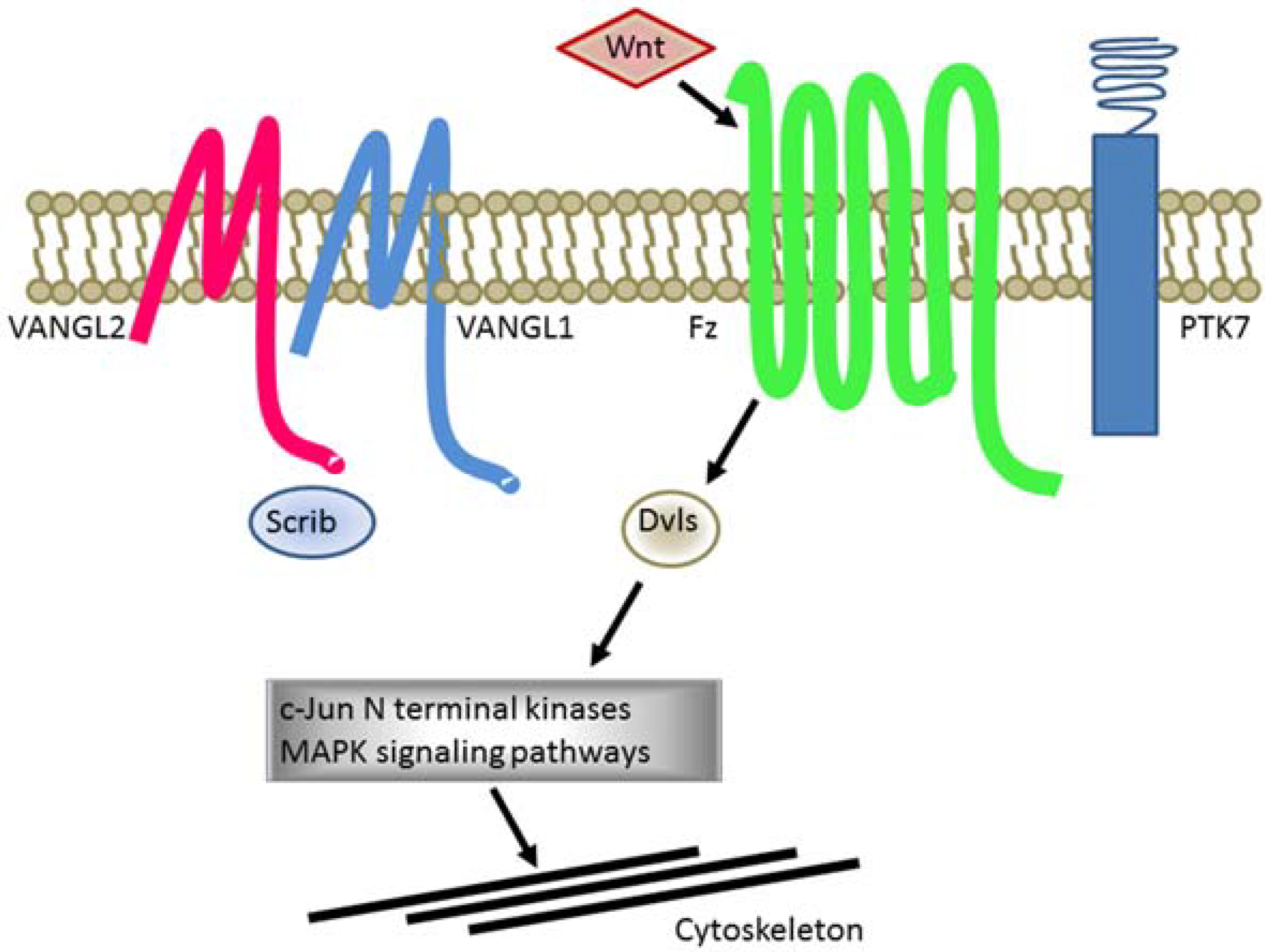{kind=link}
| PCP Genes | Phenotypes in Homozygous Mutant Mice |
|---|---|
| Celsr | Craniorachischisis |
| Dvl1/2 or DVL2/3 | Craniorachischisis |
| Fz3/6 | Craniorachischisis |
| Pk1 | None |
| Ptk7 (CCK-4) | Craniorachischisis |
| Scrib | Craniorachischisis |
| SEC24b | Craniorachischisis |
| Vangl1 | None |
| Vangl2 | Craniorachischisis |
1.6. Proteomics to Understand NTDs
1.6.1. Identifying Putative NTD Genetic Modifier Genes Using Proteomic Studies
1.6.2. Comparative Proteomic Analyses in Rat/Animal Spina Bifida Model
1.6.3. Analysis of Metabolic Factors from Human Biological Fluids
1.6.4. Analysis of Amniotic Fluids
1.6.5. Detection of Human Polymorphisms in Clinical Samples Using Proteomics Studies
1.6.6. Using Affinity Based Strategies to Analyze Protein Networks
1.6.7. Experimental and Computational Strategies Used for the Analysis of Diseases
1.7. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dolk, H.; Loane, M.; Garne, E. The prevalence of congenital anomalies in Europe. Adv. Exp. Med. Biol. 2010, 686, 349–364. [Google Scholar] [PubMed]
- Botto, L.D.; Moore, C.A.; Khoury, M. J.; Erickson, J.D. Neural-tube defects. N. Engl. J. Med. 1999, 341, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.A.; Li, S.; Li, Z.; Hong, S.X.; Gu, H.Q.; Berry, R.J.; Mulinare, J.; Erickson, J.D. Elevated rates of severe neural tube defects in a high-prevalence area in northern China. Am. J. Med. Genet. 1997, 73, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Frey, L.; Hauser, W.A. Epidemiology of neural tube defects. Epilepsia 2003, 44, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Leck, I. Causation of neural tube defects: Clues from epidemiology. Br. Med. Bull. 1974, 30, 158–163. [Google Scholar] [PubMed]
- Padmanabhan, R. Etiology, pathogenesis and prevention of neural tube defects. Congenit. Anom. 2006, 46, 55–67. [Google Scholar] [CrossRef]
- Copp, A.J.; Greene, N.D. Neural tube defects—Disorders of neurulation and related embryonic processes. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Rifat, Y.; Parekh, V.; Wilanowski, T.; Hislop, N.R.; Auden, A.; Ting, S.B.; Cunningham, J.M.; Jane, S.M. Regional neural tube closure defined by the Grainy head-like transcription factors. Dev. Biol. 2010, 345, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Obladen, M. Cats, frogs, and snakes: Early concepts of neural tube defects. J. Child Neurol. 2011, 26, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J.; Stanier, P.; Greene, N.D. Neural tube defects: Recent advances, unsolved questions, and controversies. Lancet Neurol. 2013, 12, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Ghi, T.; Contro, E.; de Musso, F. Neural Tube Defects: Craniorachischisis and Spina Bifida. In Obstetric Imaging; Copel, J.A., Ed.; Elsevier Saunders: Philadelphia, PA, USA, 2012. [Google Scholar]
- Wilson, R.D.; Audibert, F.; Brock, J.A.; Campagnolo, C.; Carroll, J.; Cartier, L.; Chitayat, D.; Gagnon, A.; Johnson, J.A.; Langlois, S.; et al. Prenatal screening, diagnosis, and pregnancy management of fetal neural tube defects. J. Obstet. Gynaecol. Can. 2014, 36, 927–939. [Google Scholar] [PubMed]
- Norem, C.T.; Schoen, E.J.; Walton, D.L.; Krieger, R.C.; O'Keefe, J.; To, T.T.; Ray, G.T. Routine ultrasonography compared with maternal serum alpha-fetoprotein for neural tube defect screening. Obstet. Gynecol. 2005, 106, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Cargill, Y.; Morin, L.; Bly, S.; Butt, K.; Denis, N.; Gagnon, R.; Hietala-Coyle, M.A.; Lim, K.; Ouellet, A.; Racicot, M.H.; Salem, S.; et al. Content of a complete routine second trimester obstetrical ultrasound examination and report. J. Obstet. Gynaecol. Can. 2009, 31, 272–275, 276–280. [Google Scholar] [PubMed]
- Loncar, J.; Barnabei, V.M.; Larsen, J.W., Jr. Advent of maternal serum markers for Down syndrome screening. Obstet. Gynecol. Surv. 1995, 50, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.C.; Miller, K.E.; Sellers, A.D. Maternal serum triple analyte screening in pregnancy. Am. Fam. Physician. 2002, 65, 915–920. [Google Scholar] [PubMed]
- Yi, Y.; Lindemann, M.; Colligs, A.; Snowball, C. Economic burden of neural tube defects and impact of prevention with folic acid: A literature review. Eur. J. Pediatr. 2011, 170, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Mahalik, S.K.; Vaze, D.; Kanojia, R.P.; Narasimhan, K.L.; Rao, K.L. Multiple neural tube defects may not be very rare. Childs Nerv. Syst. 2013, 29, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Smithells, R.W.; Sheppard, S.; Schorah, C.J. Vitamin dificiencies and neural tube defects. Arch. Dis. Child. 1976, 51, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Kirke, P.N.; Molloy, A.M.; Daly, L.E.; Burke, H.; Weir, D.G.; Scott, J.M. Maternal plasma folate and vitamin B12 are independent risk factors for neural tube defects. Q. J. Med. 1993, 86, 703–708. [Google Scholar] [PubMed]
- Carter, M.; Ulrich, S.; Oofuji, Y.; Williams, D.A.; Ross, M.E. Crooked tail (Cd) models human folate-responsive neural tube defects. Hum. Mol. Genet. 1999, 8, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Copp, A.J. Embryonic folate metabolism and mouse neural tube defects. Science 1998, 280, 2107–2109. [Google Scholar] [CrossRef] [PubMed]
- Barbera, J.P.; Rodriguez, T.A.; Greene, N.D.; Weninger, W.J.; Simeone, A.; Copp, A.J.; Beddington, R.S.; Dunwoodie, S. Folic acid prevents exencephaly in Cited2 deficient mice. Hum. Mol. Genet. 2002, 11, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, B.J.; Tang, L.S.; Triplett, A.; Aleman, F.; Finnell, R.H. Spontaneous neural tube defects in splotch mice supplemented with selected micronutrients. Toxicol. Appl. Pharmacol. 2006, 213, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Kuizon, S.; Junaid, M.A. Folic acid supplementation in pregnancy and implications in health and disease. J. Biomed. Sci. 2014, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Wong, P.W.; Susmano, A.; Sora, J.; Norusis, M.; Ruggie, N. Thermolabile methylenetetrahydrofolate reductase: An inherited risk factor for coronary artery disease. Am. J. Hum. Genet. 1991, 48, 536–545. [Google Scholar] [PubMed]
- Refsum, H.; Ueland, P.M.; Nygård, O.; Vollset, S.E. Homocysteine and cardiovascular disease. Annu. Rev. Med. 1998, 49, 31–62. [Google Scholar] [CrossRef] [PubMed]
- Van der Put, N.M.; Gabreëls, F.; Stevens, E.M.; Smeitink, J.A.; Trijbels, F.J.; Eskes, T.K.; van den Heuvel, L.P.; Blom, H.J. A second common mutation in the methylenetetrahydrofolate reductase gene: An additional risk factor for neural-tube defects? Am. J. Hum. Genet. 1998, 62, 1044–1051. [Google Scholar]
- Yan, L.; Zhao, L.; Long, Y.; Zou, P.; Ji, G.; Gu, A.; Zhao, P. Association of the maternal MTHFR C677T polymorphism with susceptibility to neural tube defects in offsprings: Evidence from 25 case-control studies. PLoS One 2012, 7, e41689. [Google Scholar] [CrossRef] [PubMed]
- Dalal, A.; Pradhan, M.; Tiwari, D.; Behari, S.; Singh, U.; Mallik, G.K.; Das, V.; Agarwal, S. MTHFR 677C-->T and 1298A-->C polymorphisms: Evaluation of maternal genotypic risk and association with level of neural tube defect. Gynecol. Obstet. Investig. 2007, 63, 146–150. [Google Scholar] [CrossRef]
- Wilcken, B.; Wilcken, B.; Bamforth, F.; Li, Z.; Zhu, H.; Ritvanen, A.; Renlund, M.; Stoll, C.; Alembik, Y.; Dott, B.; Czeizel, A.E. Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): Findings from over 7000 newborns from 16 areas world wide. J. Med. Genet. 2003, 40, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Barford, D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004, 14, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J.; Juriloff, D.M. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Van Straaten, H.W.; Copp, A.J. Curly tail: A 50-year history of the mouse spina bifida model. Anat. Embryol. 2001, 203, 225–237. [Google Scholar]
- Embury, S.; Seller, M.J.; Adinolfi, M.; Polani, P.E. Neural tube defects in curly-tail mice. I. Incidence, expression and similarity to the human condition. Proc. R. Soc. Lond. B Biol. Sci. 1979, 206, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Pena de Castro, S.C. Proteomic and Molecular Analysis of Neural Tube Defects in the Mouse Embryo. In Institute of Child Health; University College London: London, UK, 2011. [Google Scholar]
- Gustavsson, P.; Greene, N.D.; Lad, D.; Pauws, E.; de Castro, S.C.; Stanier, P.; Copp, A.J. Increased expression of Grainyhead-like-3 rescues spina bifida in a folate-resistant mouse model. Hum. Mol. Genet. 2007, 16, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Ting, S.B.; Wilanowski, T.; Auden, A.; Hall, M.; Voss, A.K.; Thomas, T.; Parekh, V.; Cunningham, J.M.; Jane, S.M. Inositol- and folate-resistant neural tube defects in mice lacking the epithelial-specific factor Grhl-3. Nat. Med. 2003, 9, 1513–1519. [Google Scholar]
- Yu, Z.; Lin, K.K.; Bhandari, A.; Spencer, J.A.; Xu, X.; Wang, N.; Lu, Z.; Gill, G.N.; Roop, D.R.; Wertz, P.; Andersen, B. The Grainyhead-like epithelial transactivator Get-1/Grhl3 regulates epidermal terminal differentiation and interacts functionally with LMO4. Dev. Biol. 2006, 299, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, E.I.; Sugihara, T.M.; Wang, N.; Lasso, R.J.; Gudnason, J.F.; Lipkin, S.M.; Andersen, B. Identification and characterization of Grainyhead-like epithelial transactivator (GET-1), a novel mammalian Grainyhead-like factor. Dev. Dyn. 2003, 226, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, S.; Jane, S.M.; Darido, C. The planar cell polarity pathway in vertebrate epidermal development, homeostasis and repair. Organogenesis 2011, 7, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Jessen, J.R. Analyzing planar cell polarity during zebrafish gastrulation. Methods Mol. Biol. 2012, 839, 69–78. [Google Scholar] [PubMed]
- Copp, A.J.; Greene, N.D.; Murdoch, J.N. Dishevelled: Linking convergent extension with neural tube closure. Trends Neurosci. 2003, 26, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.J.; Mlodzik, M. Planar cell polarization: An emerging model points in the right direction. Annu. Rev. Cell Dev. Biol. 2005, 21, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Ciruna, B.; Solnica-Krezel, L. Convergence and extension movements during vertebrate gastrulation. Curr. Top. Dev. Biol. 2009, 89, 163–192. [Google Scholar] [PubMed]
- Juriloff, D.M.; Harris, M.J. A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Zohn, I.E.; Chesnutt, C.R.; Niswander, L. Cell polarity pathways converge and extend to regulate neural tube closure. Trends Cell Biol. 2003, 13, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Klingensmith, J.; Nusse, R.; Perrimon, N. The Drosophila segment polarity gene dishevelled encodes a novel protein required for response to the wingless signal. Genes Dev. 1994, 8, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Theisen, H.; Purcell, J.; Bennett, M.; Kansagara, D.; Syed, A.; Marsh, J.L. Dishevelled is required during wingless signaling to establish both cell polarity and cell identity. Development 1994, 120, 347–360. [Google Scholar] [PubMed]
- Chae, J.; Kim, M.J.; Goo, J.H.; Collier, S.; Gubb, D.; Charlton, J.; Adler, P.N.; Park, W.J. The Drosophila tissue polarity gene starry night encodes a member of the protocadherin family. Development 1999, 126, 5421–5429. [Google Scholar] [PubMed]
- Gubb, D.; Green, C.; Huen, D.; Coulson, D.; Johnson, G.; Tree, D.; Collier, S.; Roote, J. The balance between isoforms of the prickle LIM domain protein is critical for planar polarity in Drosophila imaginal discs. Genes Dev. 1999, 13, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Shima, Y.; Shimada, Y.; Hirano, S.; Burgess, R.W.; Schwarz, T.L.; Takeichi, M.; Uemura, T. Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell 1999, 98, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Bosoi, C.M.; Kooistra, M.; Salem, S.; Finnell, R.H.; De Marco, P.; Merello, E.; Bassuk, A.G.; Capra, V.; Gros, P. Novel mutations in VANGL1 in neural tube defects. Hum. Mutat. 2009, 30, E706–E715. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Torban, E.; McDearmid, J.R.; Reynolds, A.; Berghout, J.; Mathieu, M.; Kirillova, I.; De Marco, P.; Merello, E.; Hayes, J.M. Mutations in VANGL1 associated with neural-tube defects. N. Engl. J. Med. 2007, 356, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.P.; Zhang, T.; Li, H.; Wu, B.L.; Jin, L.; Wang, H.Y. VANGL2 mutations in human cranial neural-tube defects. N. Engl. J. Med. 2010, 362, 2232–2235. [Google Scholar] [CrossRef] [PubMed]
- Merello, E.; Mascelli, S.; Raso, A.; Piatelli, G.; Consales, A.; Cama, A.; Kibar, Z.; Capra, V.; Marco, P.D. Expanding the mutational spectrum associated to neural tube defects: Literature revision and description of novel VANGL1 mutations. Birth Defects Res. A Clin. Mol. Teratol. 2015, 103, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Suriben, R.; Kivimäe, S.; Fisher, D.A.; Moon, R.T.; Cheyette, B.N. Posterior malformations in Dact1 mutant mice arise through misregulated Vangl2 at the primitive streak. Nat. Genet. 2009, 41, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ding, Y.; Lei, Y.P.; Yang, X.Y.; Xie, G.M.; Wen, J.; Cai, C.Q.; Li, H.; Chen, Y.; Zhang, T.; et al. Identification of novel rare mutations of DACT1 in human neural tube defects. Hum. Mutat. 2012, 33, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.A.; Chernoff, G.F. Intermittent pattern of neural tube closure in two strains of mice. Teratology 1993, 47, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J.; Greene, N.D.; Murdoch, J.N. The genetic basis of mammalian neurulation. Nat. Rev. Genet. 2003, 4, 784–793. [Google Scholar] [CrossRef] [PubMed]
- De Castro, S.C.; Leung, K.Y.; Savery, D.; Burren, K.; Rozen, R.; Copp, A.J.; Greene, N.D. Neural tube defects induced by folate deficiency in mutant curly tail (Grhl3) embryos are associated with alteration in folate one-carbon metabolism but are unlikely to result from diminished methylation. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 612–618. [Google Scholar]
- Wilkins, M.R.; Pasquali, C.; Appel, R.D.; Ou, K.; Golaz, O.; Sanchez, J.C.; Yan, J.X.; Gooley, A.A.; Hughes, G.; Humphery-Smith, I.; et al. From proteins to proteomes: Large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology 1996, 14, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Thelen, J.J. Introduction to Proteomics: A brief historical perspective on contemporary approaches. In Plant Proteomics; Šamaj, J., Ed.; Springer: Berlin, Germany, 2007. [Google Scholar]
- Neumann, P.E.; Frankel, W.N.; Letts, V.A.; Coffin, J.M.; Copp, A.J.; Bernfield, M. Multifactorial inheritance of neural tube defects: Localization of the major gene and recognition of modifiers in ct mutant mice. Nat. Genet. 1994, 6, 357–362. [Google Scholar] [CrossRef] [PubMed]
- De Castro, S.C.; Malhas, A.; Leung, K.Y.; Gustavsson, P.; Vaux, D.J.; Copp, A.J.; Greene, N.D. Lamin b1 polymorphism influences morphology of the nuclear envelope, cell cycle progression, and risk of neural tube defects in mice. PLoS Genet. 2012, 8, e1003059. [Google Scholar]
- Fan, Y.; Wang, L.; Zhou, F.; Zhang, Y.; Li, H.; Shan, L.; Yuan, Z. Comparative proteomics of spinal cords of rat fetuses with spina bifida aperta. J. Proteomics 2011, 75, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Correia-Pinto, J.; Reis, J.L.; Hutchins, G.M.; Baptista, M.J.; Estevão-Costa, J.; Flake, A.W.; Leite-Moreira, A.F. In utero meconium exposure increases spinal cord necrosis in a rat model of myelomeningocele. J. Pediatr. Surg. 2002, 37, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Drewek, M.J.; Bruner, J.P.; Whetsell, W.O.; Tulipan, N. Quantitative analysis of the toxicity of human amniotic fluid to cultured rat spinal cord. Pediatr. Neurosurg. 1997, 27, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Luo, G.A.; Liang, Q.L.; Wang, Y.; Yang, H.H.; Wang, Y.M.; Zheng, X.Y.; Song, X.M.; Chen, G.; Zhang, T.; Wu, J.X. Neural tube defects and disturbed maternal folate- and homocysteine-mediated one-carbon metabolism. Exp. Neurol. 2008, 212, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.P.; Li, H.; Hao, L.Z.; Zhao, Z.T. The effectiveness of prenatal serum biomarker screening for neural tube defects in second trimester pregnant women: A meta-analysis. Prenat. Diagn. 2009, 29, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yuan, Z.; Zhao, Q. SELDI-TOF-MS proteomic profiling of serum, urine, and amniotic fluid in neural tube defects. PLoS One 2014, 9, e103276. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Su, M.; Pei, L.; Zhang, T.; Ma, X.; Qiu, Y.; Xia, H.; Wang, F.; Zheng, X.; Gu, X.; et al. Metabolic signature of pregnant women with neural tube defects in offspring. J. Proteome Res. 2011, 10, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Pei, L.; Chen, G.; Song, X.; Zhao, A.; Chen, T.; Su, M.; Zhang, Y.; Liu, J.; Ren, A. Metabonomic profiling of human placentas reveals different metabolic patterns among subtypes of neural tube defects. J. Proteome Res. 2014, 13, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Turner, D.; Singh, R.; Ruest, L.B.; Pierce, W.M., Jr.; Knudsen, T.B. Fetal alcohol syndrome (FAS) in C57BL/6 mice detected through proteomics screening of the amniotic fluid. Birth Defects Res. A Clin. Mol. Teratol. 2008, 82, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Thadikkaran, L.; Crettaz, D.; Siegenthaler, M.A.; Gallot, D.; Sapin, V.; Iozzo, R.V.; Queloz, P.A.; Schneider, P.; Tissot, J.D. The role of proteomics in the assessment of premature rupture of fetal membranes. Clin. Chim. Acta 2005, 360, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Tsangaris, G.T.; Karamessinis, P.; Kolialexi, A.; Garbis, S.D.; Antsaklis, A.; Mavrou, A.; Fountoulakis, M. Proteomic analysis of amniotic fluid in pregnancies with Down syndrome. Proteomics 2006, 6, 4410–4419. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Fan, Y.; Li, H.; Liu, W.; Gu, H.; Zhou, F.; Yuan, Z. Proteomic analysis of amniotic fluid of pregnant rats with spina bifida aperta. J. Proteomics 2012, 75, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Kelleher, P.C.; Bélanger, L.; Dallaire, L. Reactivity of amniotic fluid alpha-fetoprotein with concanavallin A in diagnosis of neural tube defects. Br. Med. J. 1979, 1, 920–921. [Google Scholar] [CrossRef] [PubMed]
- Taketa, K. Characterization of sugar chain structures of human alpha-fetoprotein by lectin affinity electrophoresis. Electrophoresis 1998, 19, 2595–2602. [Google Scholar] [CrossRef] [PubMed]
- Lemire, J.M.; Fausto, N. Multiple alpha-fetoprotein RNAs in adult rat liver: Cell type-specific expression and differential regulation. Cancer Res. 1991, 51, 4656–4664. [Google Scholar] [PubMed]
- Tsurubuchi, T.; Ichi, S.; Shim, K.W.; Norkett, W.; Allender, E.; Mania-Farnell, B.; Tomita, T.; McLone, D.G.; Ginsberg, N.; Mayanil, C.S. Amniotic fluid and serum biomarkers from women with neural tube defect-affected pregnancies: A case study for myelomeningocele and anencephaly: Clinical article. J. Neurosurg. Pediatr. 2013, 12, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.; Chiriacò, F.; Del Boccio, P.; Tagliaferro, L.; Acierno, R.; Menegazzi, P.; Pinca, E.; Pignatelli, F.; Storelli, C.; Federici, G.; et al. A proteomic approach for the characterization of C677T mutation of the human gene methylenetetrahydrofolate reductase. Proteomics 2006, 6, 5350–5361. [Google Scholar] [CrossRef] [PubMed]
- Svasti, J.; Kurosky, A.; Bennett, A.; Bowman, B.H. Molecular basis for the three major forms of human serum vitamin D binding protein (group-specific component). Biochemistry 1979, 18, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Bichlmaier, R.; Cleve, H. Molecular analysis of the gene for the human vitamin-D-binding protein (group-specific component): Allelic differences of the common genetic GC types. Hum. Genet. 1992, 89, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Miller, J.J.; Corbit, K.C.; Giles, R.H.; Brauer, M.J.; Otto, E.A.; Baye, L.M.; Wen, X.; Scales, S.J.; Kwong, M.; et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011, 145, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peränen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.Z.; Miller, J.J.; Jackson, P.K. High-throughput generation of tagged stable cell lines for proteomic analysis. Proteomics 2009, 9, 2888–2891. [Google Scholar] [CrossRef] [PubMed]
- Montcouquiol, M.; Crenshaw, E.B., 3rd; Kelley, M.W. Noncanonical Wnt signaling and neural polarity. Annu. Rev. Neurosci. 2006, 29, 363–386. [Google Scholar] [CrossRef] [PubMed]
- Nourry, C.; Grant, S.G.; Borg, J.P. PDZ domain proteins: Plug and play! Sci. STKE 2003. [Google Scholar] [CrossRef]
- Belotti, E.; Polanowska, J.; Daulat, A.M.; Audebert, S.; Thome, V.; Lissitzky, J.C.; Lembo, F.; Biblek, K.; Omi, S.; Lenfant, N.; et al. The human PDZome: A gateway to PSD95-Disc large-zonula occludens (PDZ)-mediated functions. Mol. Cell. Proteomics 2013, 12, 2587–2603. [Google Scholar] [CrossRef] [PubMed]
- Montcouquiol, M.; Sans, N.; Huss, D.; Kach, J.; Dickman, J.D.; Forge, A.; Rachel, R.A.; Copeland, N.G.; Jenkins, N.A.; Bogani, D.; et al. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 2003, 423, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Biechele, T.L.; Robitaille, M.; Muster, J.; Allison, K.H.; Angers, S.; Moon, R.T. A protein complex of SCRIB, NOS1AP and VANGL1 regulates cell polarity and migration, and is associated with breast cancer progression. Oncogene 2012, 31, 3696–3708. [Google Scholar] [CrossRef] [PubMed]
- Belotti, E.; Puvirajesinghe, T.M.; Audebert, S.; Baudelet, E.; Camoin, L.; Pierres, M.; Lasvaux, L.; Ferracci, G.; Montcouquiol, M.; Borg, J.P. Molecular characterisation of endogenous Vangl2/Vangl1 heteromeric protein complexes. PLoS One 2012, 7, e46213. [Google Scholar] [CrossRef] [PubMed]
- Duran, M.C.; Mas, S.; Martin-Ventura, J.L.; Meilhac, O.; Michel, J.B.; Gallego-Delgado, J.; Lázaro, A.; Tuñon, J.; Egido, J.; Vivanco, F. Proteomic analysis of human vessels: Application to atherosclerotic plaques. Proteomics 2003, 3, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, C.; Thumar, J.; Mayya, V.; Hwang, S.I.; Zebroski, H.; Claffey, K.P.; Haudenschild, C.; Eng, J.K.; Lundgren, D.H.; Han, D.K. Proteomics analysis of human coronary atherosclerotic plaque: A feasibility study of direct tissue proteomics by liquid chromatography and tandem mass spectrometry. Mol. Cell. Proteomics 2007, 6, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, E.; Maiti, S.; Brahmachari, S.K.; Sengupta, S. Predicting protein homocysteinylation targets based on dihedral strain energy and pKa of cysteines. Proteins 2008, 71, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Lardelli, M.; Williams, R.; Mitsiadis, T.; Lendahl, U. Expression of the Notch 3 intracellular domain in mouse central nervous system progenitor cells is lethal and leads to disturbed neural tube development. Mech. Dev. 1996, 59, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Voyiaziakis, E.; Markenson, D.F.; Sokol, K.A.; Hayek, T.; Breslow, J.L. Apo B gene knockout in mice results in embryonic lethality in homozygotes and neural tube defects, male infertility, and reduced HDL cholesterol ester and apo A-I transport rates in heterozygotes. J. Clin. Investig. 1995, 96, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Kaucka, M.; Plevova, K.; Pavlova, S.; Janovska, P.; Mishra, A.; Verner, J.; Prochazkova, J.; Krejci, P.; Kotaskova, J.; Ovesna, P.; et al. The planar cell polarity pathway drives pathogenesis of chronic lymphocytic leukemia by the regulation of B-lymphocyte migration. Cancer Res. 2013, 73, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Lhoumeau, A.C.; Arnoulet, C.; Aulas, A.; Marchetto, S.; Audebert, S.; Puppo, F.; Chabannon, C.; Sainty, D.; Santoni, M.J.; et al. The cell polarity PTK7 receptor acts as a modulator of the chemotherapeutic response in acute myeloid leukemia and impairs clinical outcome. Blood 2010, 116, 2315–2323. [Google Scholar]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puvirajesinghe, T.M.; Borg, J.-P. Neural Tube Defects: From a Proteomic Standpoint. Metabolites 2015, 5, 164-183. https://doi.org/10.3390/metabo5010164
Puvirajesinghe TM, Borg J-P. Neural Tube Defects: From a Proteomic Standpoint. Metabolites. 2015; 5(1):164-183. https://doi.org/10.3390/metabo5010164
Chicago/Turabian StylePuvirajesinghe, Tania M., and Jean-Paul Borg. 2015. "Neural Tube Defects: From a Proteomic Standpoint" Metabolites 5, no. 1: 164-183. https://doi.org/10.3390/metabo5010164
APA StylePuvirajesinghe, T. M., & Borg, J.-P. (2015). Neural Tube Defects: From a Proteomic Standpoint. Metabolites, 5(1), 164-183. https://doi.org/10.3390/metabo5010164