Computational Analyses of Spectral Trees from Electrospray Multi-Stage Mass Spectrometry to Aid Metabolite Identification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Functionalities Implemented in the Iontree Package
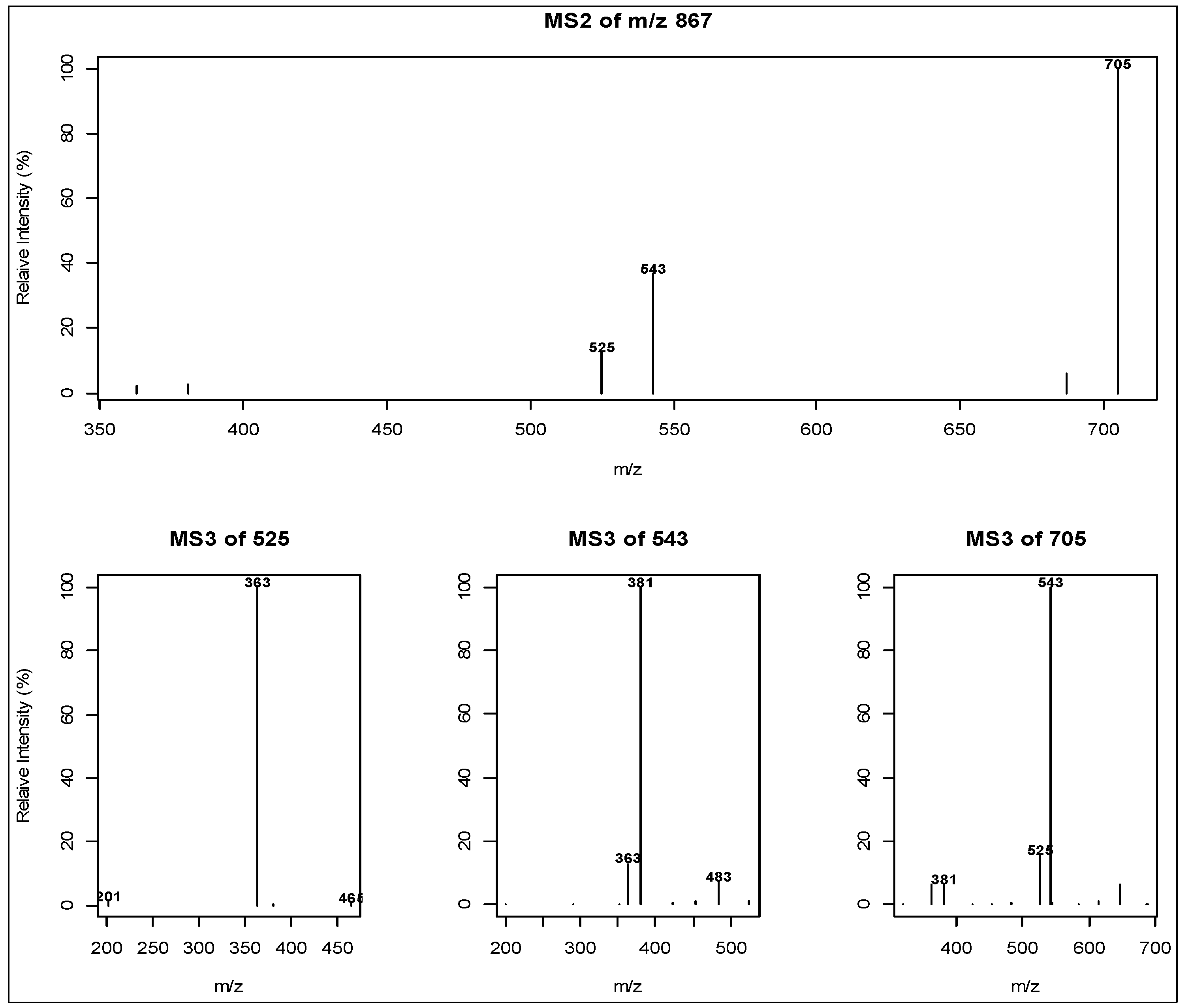
2.2. Direct infusion Low Resolution Ion Trap Mass Spectrometry
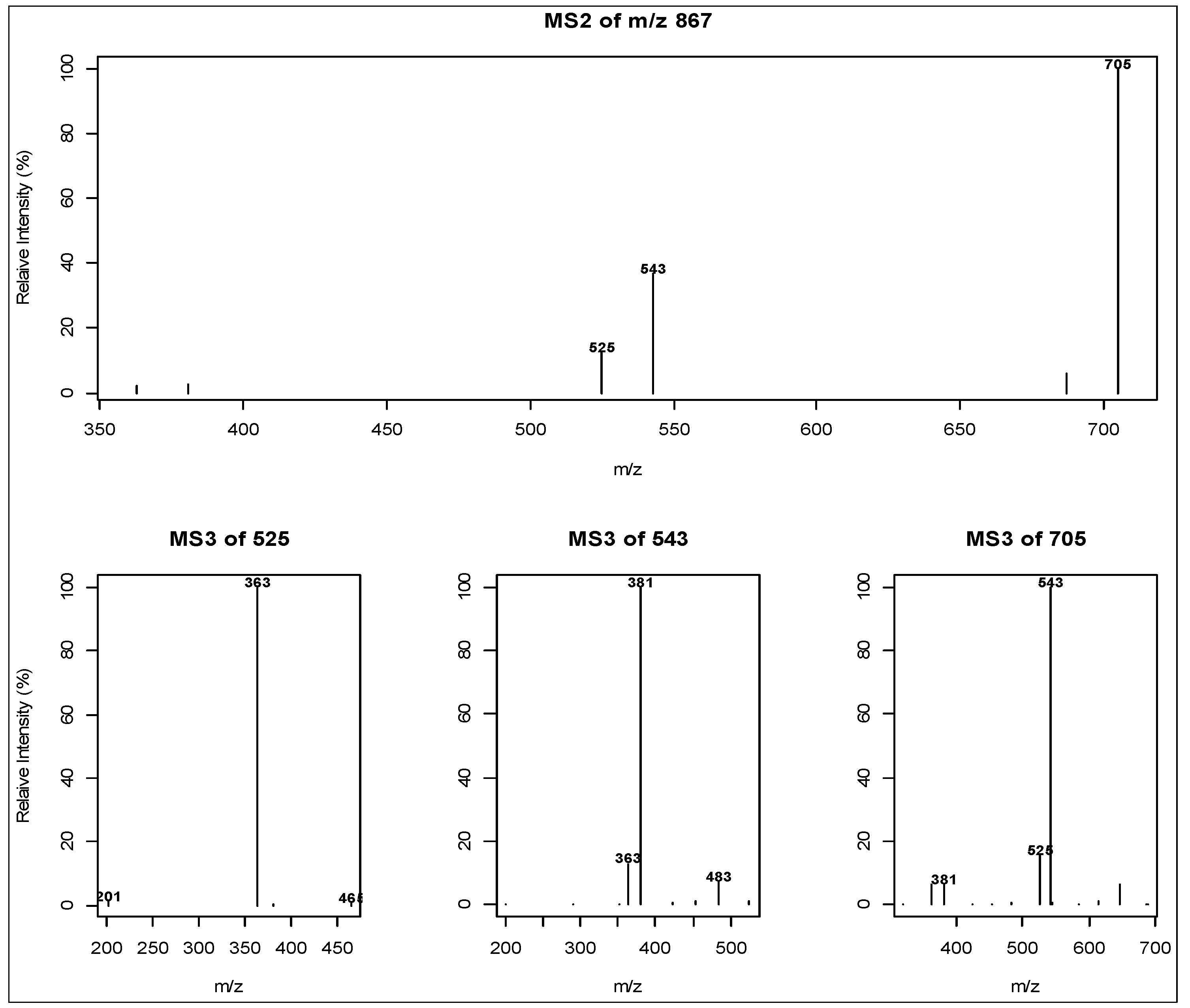
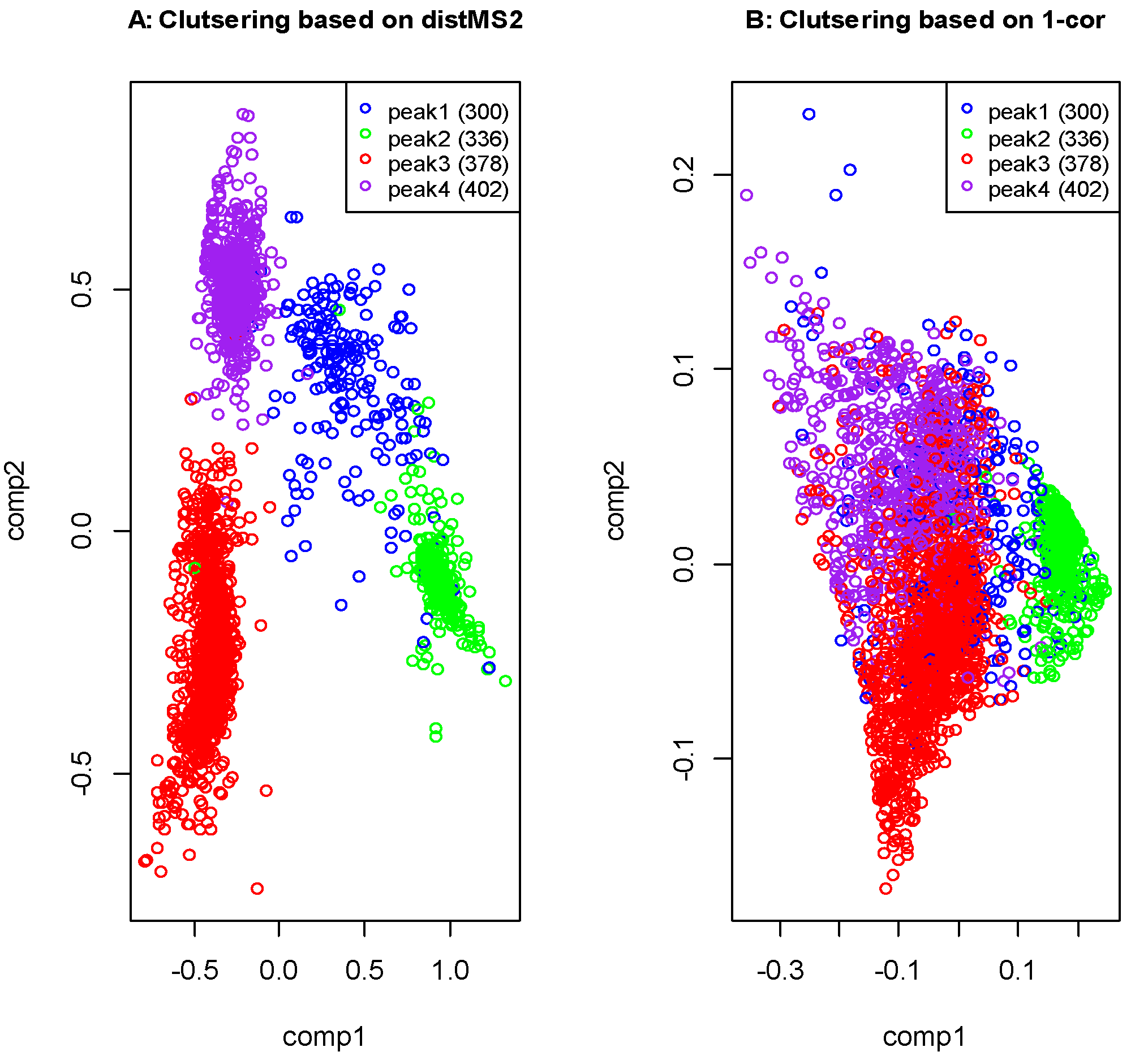
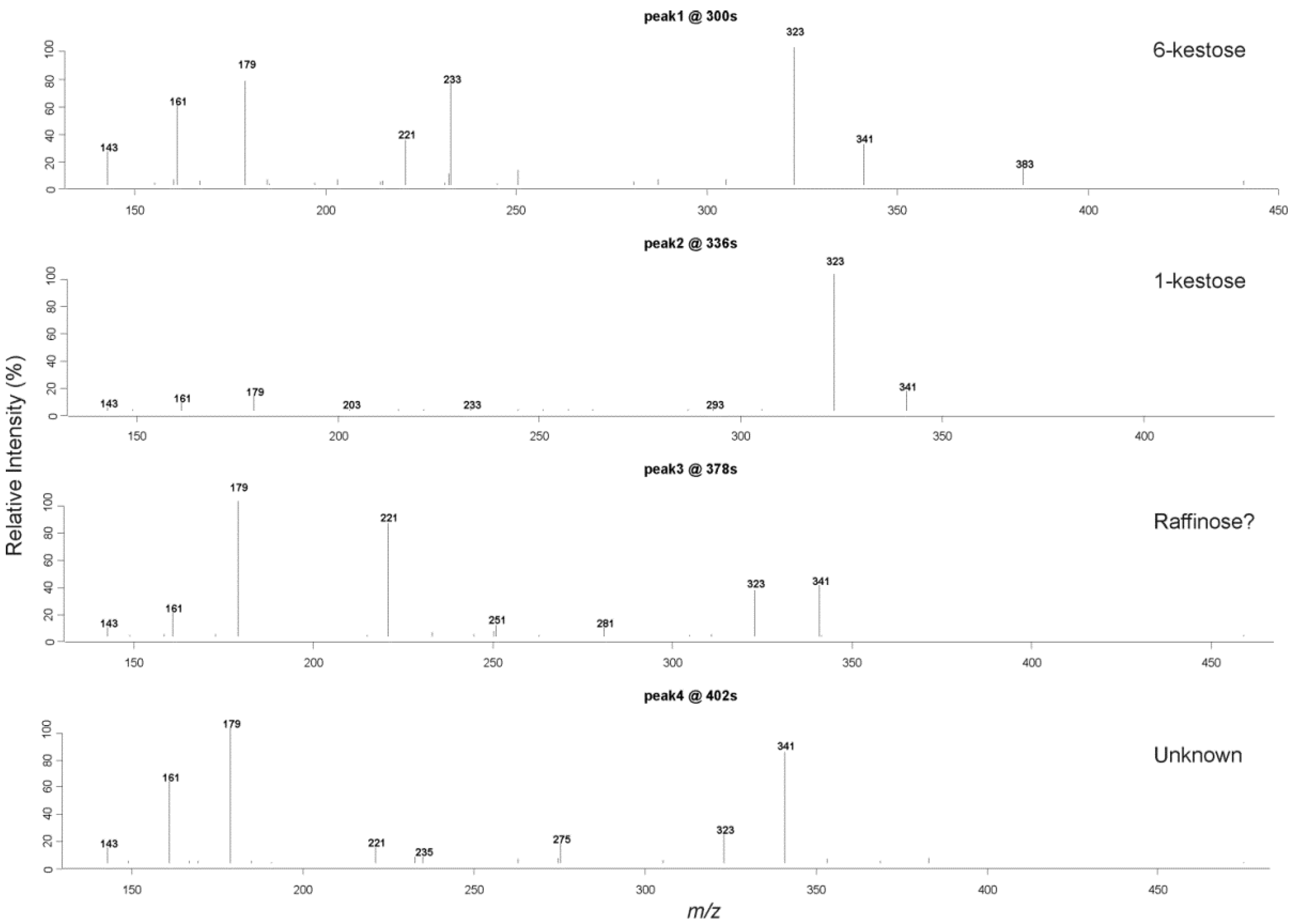
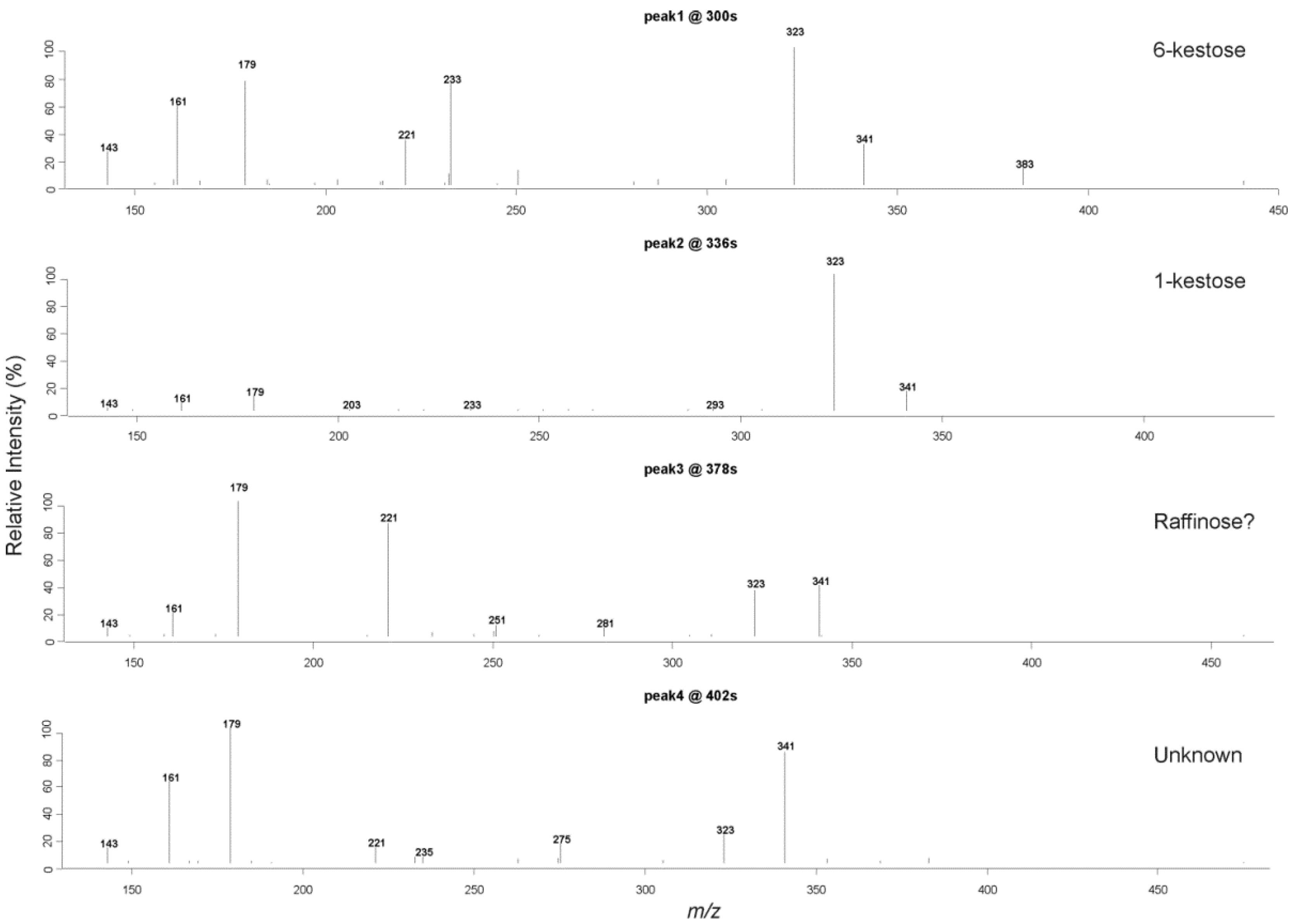
2.3. Liquid Chromatography Low Resolution Mass Spectrometry
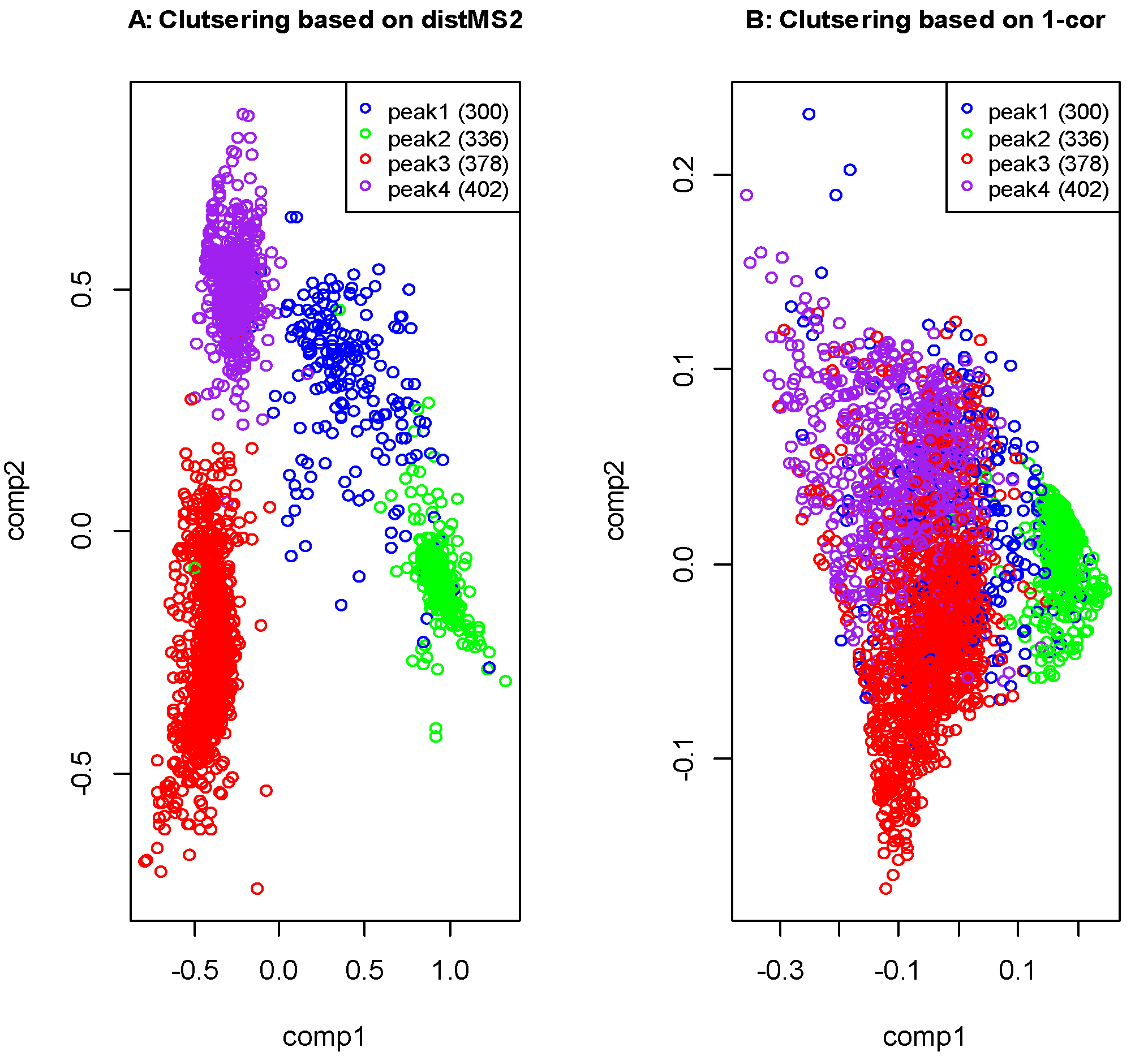
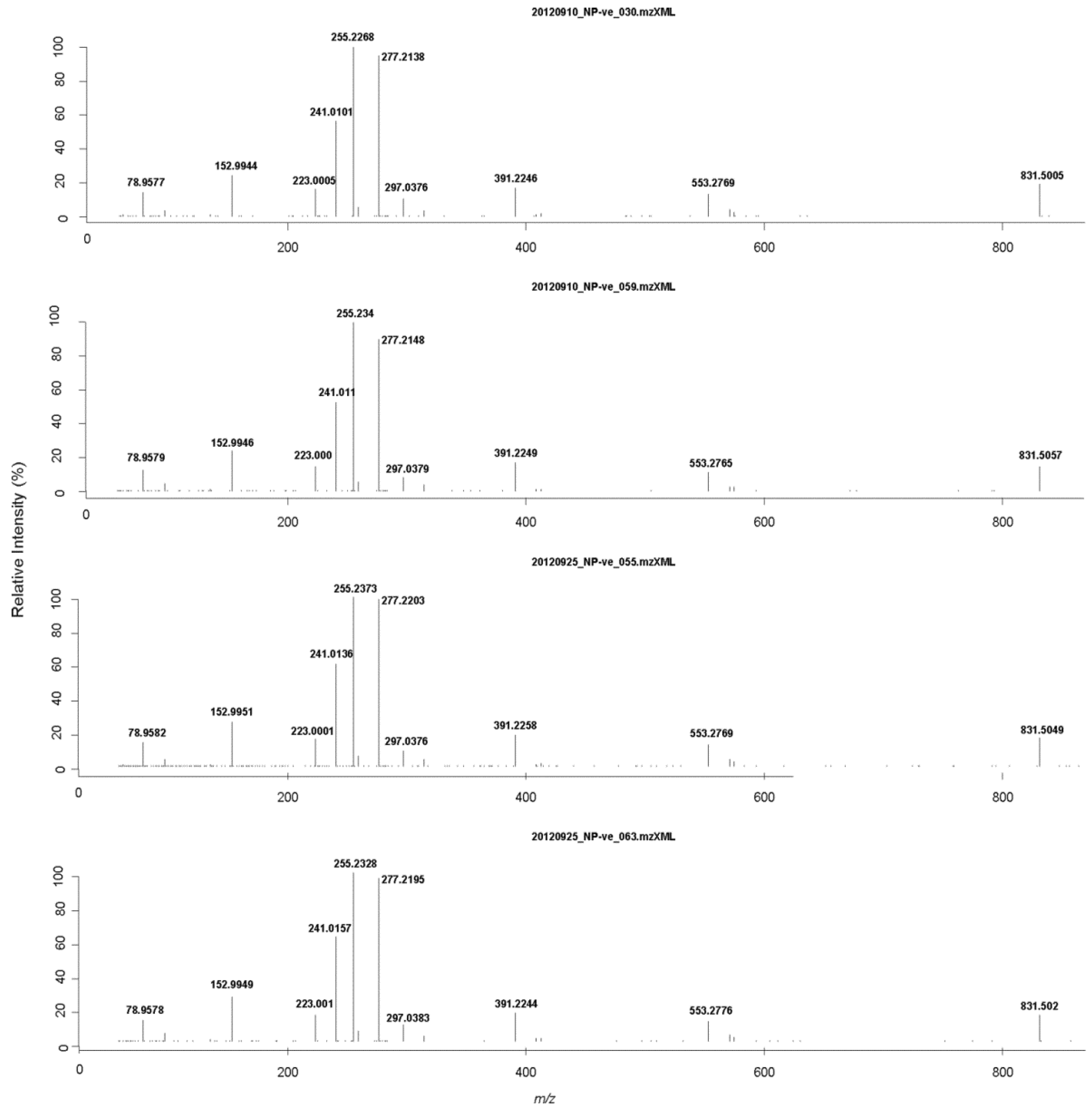
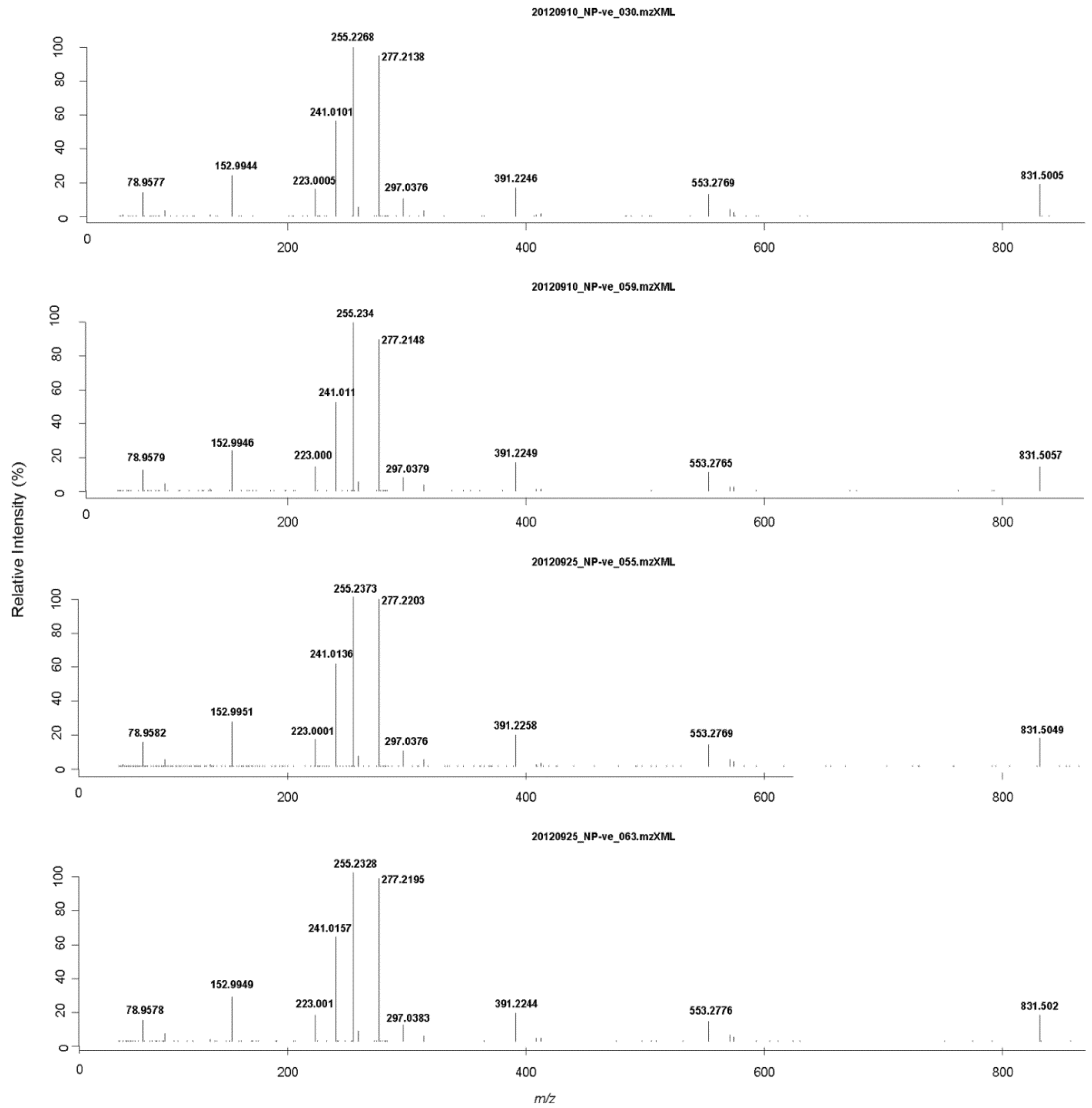
2.4. Liquid Chromatography High Resolution Mass Spectrometry
3. Experimental Section
3.1. Plant Material and Analytical Methods
3.2. Software Tools and Database Resources
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kind, T.; Fiehn, O. Advances in structure elucidation of small molecules using mass spectrometry. Bioanal. Rev. 2010, 2, 23–60. [Google Scholar] [CrossRef]
- Dunn, W.; Erban, A.; Weber, R.; Creek, D.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2013, 9, S44–S66. [Google Scholar]
- Draper, J.; Enot, D.; Parker, D.; Beckmann, M.; Snowdon, S.; Lin, W.; Zubair, H. Metabolite signal identification in accurate mass metabolomics data with MZedDB, an interactive m/z annotation tool utilising predicted ionisation behaviour ‘rules’. BMC Bioinformatics 2009, 10. [Google Scholar] [CrossRef]
- Wishart, D.S. Advances in metabolite identification. Bioanalysis 2011, 3, 1769–1782. [Google Scholar] [CrossRef]
- Scheubert, K.; Hufsky, F.; Bocker, S. Computational mass spectrometry for small molecules. J. Cheminform. 2013, 5. [Google Scholar] [CrossRef]
- Sumner, L.; Amberg, A.; Barrett, D.; Beale, M.; Beger, R.; Daykin, C.; Fan, T.M.; Fiehn, O.; Goodacre, R.; Griffin, J.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Zhu, Z.-J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics 2007, 8. [Google Scholar] [CrossRef]
- Olsen, J.V.; Mann, M. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc. Natl. Acad. Sci. USA 2004, 101, 13417–13422. [Google Scholar] [CrossRef]
- Scheubert, K.; Hufsky, F.; Rasche, F.; Bocker, S. Computing fragmentation trees from metabolite multiple mass mpectrometry data. J. Comput. Biol. 2011, 18, 1387–1397. [Google Scholar]
- Sadygov, R.G.; Cociorva, D.; Yates, J.R. Large-scale database searching using tandem mass spectra: Looking up the answer in the back of the book. Nat. Methods 2004, 1, 195–202. [Google Scholar] [CrossRef]
- Würtinger, P.; Oberacher, H. Evaluation of the performance of a tandem mass spectral library with mass spectral data extracted from literature. Drug Test. Anal. 2012, 4, 235–241. [Google Scholar] [CrossRef]
- Benton, H.P.; Wong, D.M.; Trauger, S.A.; Siuzdak, G. XCMS2: Processing tandem mass spectrometry data for metabolite identification and structural characterization. Anal. Chem. 2008, 80, 6382–6389. [Google Scholar] [CrossRef]
- Gatto, L.; Lilley, K.S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2012, 28, 288–289. [Google Scholar] [CrossRef]
- Van der Hooft, J.; Vervoort, J.; Bino, R.; de Vos, R. Spectral trees as a robust annotation tool in LC-MS based metabolomics. Metabolomics 2012, 8, 691–703. [Google Scholar] [CrossRef]
- Ridder, L.; van der Hooft, J.J.J.; Verhoeven, S.; de Vos, R.C.H.; van Schaik, R.; Vervoort, J. Substructure-based annotation of high-resolution multistage MSn spectral trees. Rapid Commun. Mass Spectrom. 2012, 26, 2461–2471. [Google Scholar] [CrossRef]
- Rojas-Cherto, M.; Peironcely, J.E.; Kasper, P.T.; van der Hooft, J.J.J.; de Vos, R.C.H.; Vreeken, R.; Hankemeier, T.; Reijmers, T. Metabolite identification using automated comparison of high-resolution multistage mass spectral trees. Anal. Chem. 2012, 84, 5524–5534. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Cho, K.; Uritboonthai, W.; Zhu, Z.; Patti, G.J.; Siuzdak, G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat. Biotechnol. 2012, 30, 826–828. [Google Scholar]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Wolf, S.; Schmidt, S.; Muller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics 2010, 11. [Google Scholar] [CrossRef]
- Cao, M.; Koulman, A.; Johnson, L.J.; Lane, G.A.; Rasmussen, S. Advanced data-mining strategies for the analysis of direct-infusion ion trap mass spectrometry data from the association of perennial ryegrass with Its endophytic fungus, Neotyphodium lolii. Plant Physiol. 2008, 146, 1501–1514. [Google Scholar] [CrossRef]
- Koulman, A.; Cao, M.; Faville, M.; Lane, G.; Mace, W.; Rasmussen, S. Semi-quantitative and structural metabolic phenotyping by direct infusion ion trap mass spectrometry and its application in genetical metabolomics. Rapid Commun. Mass Spectrom. 2009, 23, 2253–2263. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- R package iontree. Available online: http://www.bioconductor.org/packages/release/bioc/html/iontree.html (accessed on 6 August 2013).
- Pedrioli, P.G.A.; Eng, J.K.; Hubley, R.; Vogelzang, M.; Deutsch, E.W.; Raught, B.; Pratt, B.; Nilsson, E.; Angeletti, R.H.; Apweiler, R.; et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004, 22, 1459–1466. [Google Scholar] [CrossRef]
- Hsieh, E.J.; Hoopmann, M.R.; MacLean, B.; MacCoss, M.J. Comparison of database search strategies for high precursor mass accuracy MS/MS data. J. Proteome Res. 2009, 9, 1138–1143. [Google Scholar]
- Deutsch, E.W.; Shteynberg, D.; Lam, H.; Sun, Z.; Eng, J.K.; Carapito, C.; von Haller, P.D.; Tasman, N.; Mendoza, L.; Farrah, T.; et al. Trans-Proteomic Pipeline supports and improves analysis of electron transfer dissociation data sets. Proteomics 2010, 10, 1190–1195. [Google Scholar] [CrossRef]
- Jain, A.K.; Murty, M.N.; Flynn, P.J. Data clustering: A review. ACM Comput. Surv. 1999, 31, 264–323. [Google Scholar] [CrossRef]
- Li, Q.; Xia, Q.; Wang, T.; Meila, M.; Hackett, M. Analysis of the stochastic variation in LTQ single scan mass spectra. Rapid Commun. Mass Spectrom. 2006, 20, 1551–1557. [Google Scholar] [CrossRef]
- SQLite. Available online: http://www.sqlite.org (accessed on 6 August 2013).
- Koulman, A.; Tapper, B.A.; Fraser, K.; Cao, M.; Lane, G.A.; Rasmussen, S. High-throughput direct-infusion ion trap mass spectrometry: A new method for metabolomics. Rapid Commun. Mass Spectrom. 2007, 21, 421–428. [Google Scholar] [CrossRef]
- Beckmann, M.; Parker, D.; Enot, D.P.; Duval, E.; Draper, J. High-throughput, nontargeted metabolite fingerprinting using nominal mass flow injection electrospray mass spectrometry. Nat. Protoc. 2008, 3, 486–504. [Google Scholar]
- Weissberg, A.; Dagan, S. Interpretation of ESI(+)-MS-MS spectra—Towards the identification of “unknowns”. Int. J. Mass Spectrom. 2011, 299, 158–168. [Google Scholar] [CrossRef]
- Harrison, S.J.; Fraser, K.; Lane, G.A.; Villas-Boas, S.; Rasmussen, S. A reverse-phase liquid chromatography/mass spectrometry method for the analysis of high-molecular-weight fructooligosaccharides. Anal. Biochem. 2009, 395, 113–115. [Google Scholar] [CrossRef]
- Du, P.; Kibbe, W.A.; Lin, S.M. Improved peak detection in mass spectrum by incorporating continuous wavelet transform-based pattern matching. Bioinformatics 2006, 22, 2059–2065. [Google Scholar] [CrossRef]
- Harrison, S.; Xue, H.; Lane, G.; Villas-Boas, S.; Rasmussen, S. Linear ion trap MSn of enzymatically synthesized 13C-labeled fructans revealing differentiating fragmentation patterns of β (1–2) and β (1–6) fructans and providing a tool for oligosaccharide identification in complex mixtures. Anal. Chem. 2011, 84, 1540–1548. [Google Scholar]
- Lange, E.; Tautenhahn, R.; Neumann, S.; Gropl, C. Critical assessment of alignment procedures for LC-MS proteomics and metabolomics measurements. BMC Bioinformatics 2008, 9. [Google Scholar] [CrossRef]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef]
- Bellew, M.; Coram, M.; Fitzgibbon, M.; Igra, M.; Randolph, T.; Wang, P.; May, D.; Eng, J.; Fang, R.; Lin, C.; et al. A suite of algorithms for the comprehensive analysis of complex protein mixtures using high-resolution LC-MS. Bioinformatics 2006, 22, 1902–1909. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- MassBank. Available online: http://www.massbank.jp (accessed on 6 August 2013).
- METLIN. Available online: http://metlin.scripps.edu (accessed on 6 August 2013).
- LipidMaps. Available online: http://www.lipidmaps.org (accessed on 6 August 2013).
- HMDB. Available online: http://www.hmdb.ca (accessed on 6 August 2013).
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cao, M.; Fraser, K.; Rasmussen, S. Computational Analyses of Spectral Trees from Electrospray Multi-Stage Mass Spectrometry to Aid Metabolite Identification. Metabolites 2013, 3, 1036-1050. https://doi.org/10.3390/metabo3041036
Cao M, Fraser K, Rasmussen S. Computational Analyses of Spectral Trees from Electrospray Multi-Stage Mass Spectrometry to Aid Metabolite Identification. Metabolites. 2013; 3(4):1036-1050. https://doi.org/10.3390/metabo3041036
Chicago/Turabian StyleCao, Mingshu, Karl Fraser, and Susanne Rasmussen. 2013. "Computational Analyses of Spectral Trees from Electrospray Multi-Stage Mass Spectrometry to Aid Metabolite Identification" Metabolites 3, no. 4: 1036-1050. https://doi.org/10.3390/metabo3041036
APA StyleCao, M., Fraser, K., & Rasmussen, S. (2013). Computational Analyses of Spectral Trees from Electrospray Multi-Stage Mass Spectrometry to Aid Metabolite Identification. Metabolites, 3(4), 1036-1050. https://doi.org/10.3390/metabo3041036