Small Molecule Identification with MOLGEN and Mass Spectrometry



Abstract
1. Introduction
2. Methods
2.1. Category 1: Best Molecular Formula with LC–MS/MS
- –
- Challenges 1, 4–6, 10, 12–15, 17: el = CHNOPS, ppm = 5, acc = 10, oei, exist, ion = +H.
- –
- Challenges 2–3: el = CHNOPS, ppm = 5, acc = 10, oei, exist, ion = −H.
- –
- Challenge 11: el = CHNOPS, ppm = 5, acc = 10, oei, exist, m = 232.088, ion = −e. Here, m = 232.088 set the mass for M+, and ion mode ‘−e’ corresponds with M+.
- –
- Challenge 16: el = CHNOPS, ppm = 5, acc = 10, oei, exist, m = 359.1481 ion = +e. Here, m = 359.1481 was used to set an M+ ion mass, with ion mode +e to obtain the corresponding formulas.
2.2. Category 2: Best Structure with LC–MS/MS

2.3. Category 3: Best Molecular Formula with GC–MS

- –
- Challenge 1: C ≥ 7, O ≥ 2, H unlimited, F, Cl, Br, I, N, P, S, Si = 0. RDB = 6–7.
- –
- Challenge 2: C ≥ 7, O ≥ 2, N ≥1, H unlimited, F, Cl, Br, I, P, S, Si = 0. RDB = 7–8.
- –
- Challenge 3: O, Cl = 1, C, H unlimited. F, Br, I, N, P, S, Si = 0. RDB = 4.
- –
- Challenge 4: O, Cl = 1, C, H unlimited. F, Br, I, N, P, S, Si = 0. RDB = 4.
- –
- Challenge 5: Cl = 2, C, H unlimited. F, Br, I, N, P, S, O, Si = 0. RDB = 4.
- –
- Challenge 6: C, H unlimited. F, Cl, Br, I, N, P, S, O, Si = 0. RDB = 8. MW = 154. Note: the two highest peaks in the CSV file were removed to improve results; likely impurities.
- –
- Challenge 7: O = 1, Cl = 2, C, H unlimited. F, Br, I, N, P, S, Si = 0. RDB = 5.
- –
- Challenge 8: C, H unlimited. F, Cl, Br, I, N, P, S, O, Si = 0. RDB = 9.
- –
- Challenge 9: O = 1–2, Cl = 1, C, H unlimited. F, Br, I, N, P, S, Si = 0. RDB = 5.
- –
- Challenge 10: O = 1, Cl = 3, C, H unlimited. F, Br, I, N, P, S, Si = 0. RDB = 4.
- –
- Challenge 11: MW = 257; N = 1, O, S = 2–20, P = 1–20, C, H ≥ 4. F, Br, I, Si = 0.
- –
- Challenge 12: MW = 288; C ≥ 6, H ≥ 1, Cl ≥ 5. F, Br, I, N, P, S, O, Si = 0. RDB = 1.
- –
- Challenge 13: S = 1–20, C, H unlimited. F, Cl, Br, I, N, P, S, Si = 0.
- –
- Challenge 14: C ≥ 1, H ≥ 3, O= 3, P, S ≥ 1. F, Cl, Br, I, N, Si = 0.
- –
- Challenge 15: C ≥ 12, H, N ≥ 0, O ≥ 1. F, Cl, Br, I, P, S, Si = 0. RDB = 9.
- –
- Challenge 16: C, P, S ≥ 1, H ≥ 3, O = 2, N unlimited. F, Cl, Br, I, Si = 0.
2.4. Category 4: Best Structure with GC–MS


2.5. Evaluation Measures and Ranking

3. Results and Discussion
3.1. Category 1: Best Molecular Formula with LC–MS/MS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
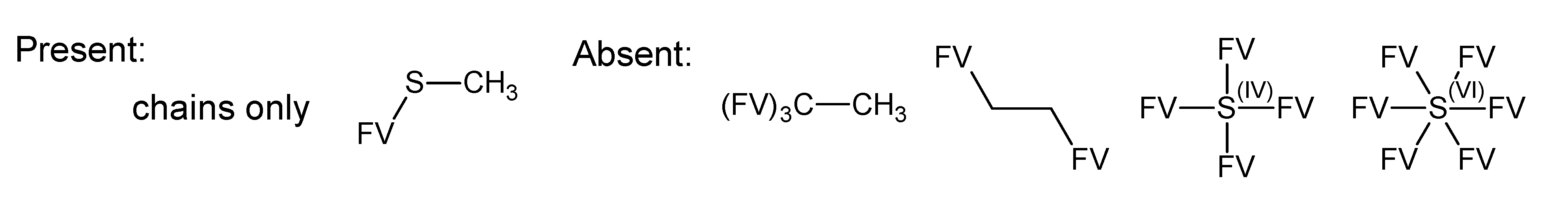{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Challenge Number | Molecular Formula | PubChem CID | Formulas Entered | Rank with MS MV | Rank with MS/MS MV | Rank with combMV | Rank of Winner |
|---|---|---|---|---|---|---|---|
| 1 | C18H36N4O11 | 6032 | 54 | 12 | 1 (38) | 11 | 1 |
| 2 | C28H32O14 | 5280665 | 249 | 1 | 1 (239) | 1 | 1 |
| 3 | C14H27NO9S3 | 46173875 | 90 | 1 | 27 (25) | 14 | 1 |
| 4 | C19H17NO4 | 12304178 | 15 | 1 | 1 (10) | 1 | 1 |
| 5 | C19H23NO4 | 10233 | 10 | 1 | 1 (13) | 1 | 1 |
| 6 | C21H21NO6 | 197775 | 31 | 1 | 1 (19) | 1 | 2 |
| 10 | C14H9NO2 | 6710 | 3 | 1 | 1 | 1 | 1 |
| 11 | C17H12O | 104977 | 6 | 1 (6) | 1 (4) | 1 (4) | NA |
| 12 | C17H16N4O4 | 221491 | 18 | 1 (18) | 1 (4) | 1 (4) | NA |
| 13 | C19H17OP | 76293 | 10 | 1 | 1 (3) | 1 | 1 |
| 14 | C12H9N | 98617 | 2 | 1 | 1 | 1 | 1 |
| 15 | C12H13NO2 | 2145522 | 2 | 1 | 1 (2) | 1 | 1 |
| 16 | C18H21N3O5 | 18091616 | 20 | NA | 2 (3) | 2 (3) | NA |
| 17 | C13H13N3 | 68380 | 3 | 1 | 1 | 1 | 1 |
3.2. Category 2: Best structure with LC–MS/MS
| Challenge Number | Molecular Formula | PubChem CID | Structures Possible(%) | Structures Entered | MOLGEN Rank | MOLGEN RRP | Rank of Winner |
|---|---|---|---|---|---|---|---|
| 10 | C14H9NO2 | 6710 | >1E8 (<1%) | 171 | 63 | 0.365 | NA |
| 11 | C17H12O | 104977 | >1E8 (<1%) | 8 | 3 | 0.286 | NA |
| 13 | C19H17OP | 76293 | >1E8 (<1%) | 4 | 3 | 0.667 | 1 |
| 14 | C12H9N | 98617 | >1E8 (18%) | 41 | 22 | 0.525 | 12 |
| 15 | C12H13NO2 | 2145522 | >1E8 (1.5%) | 32 | 26 | 0.806 | 1 |
| 17 | C13H13N3 | 68380 | >1E8 (<1%) | 1295 | 58 | 0.044 | NA |
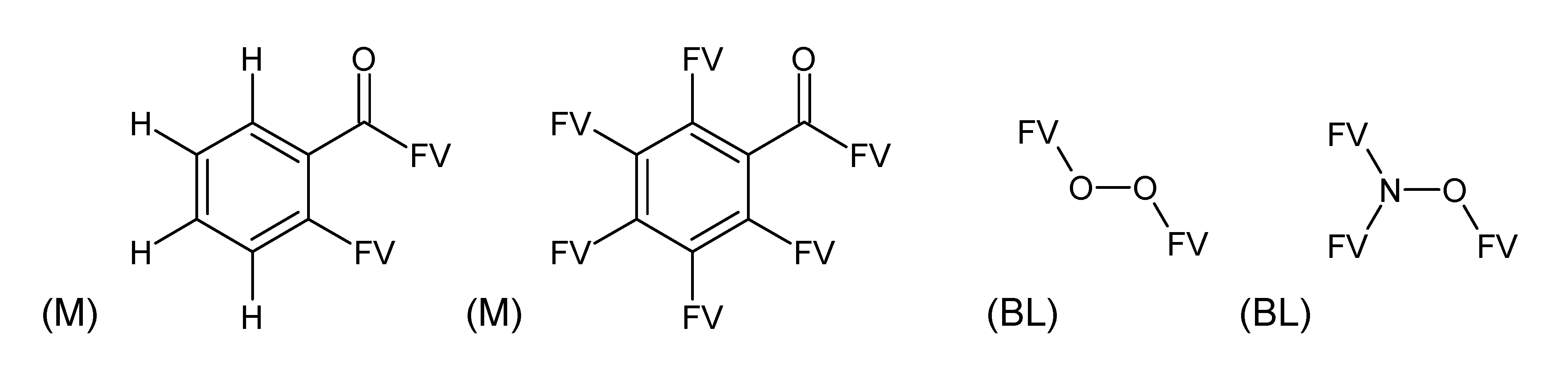
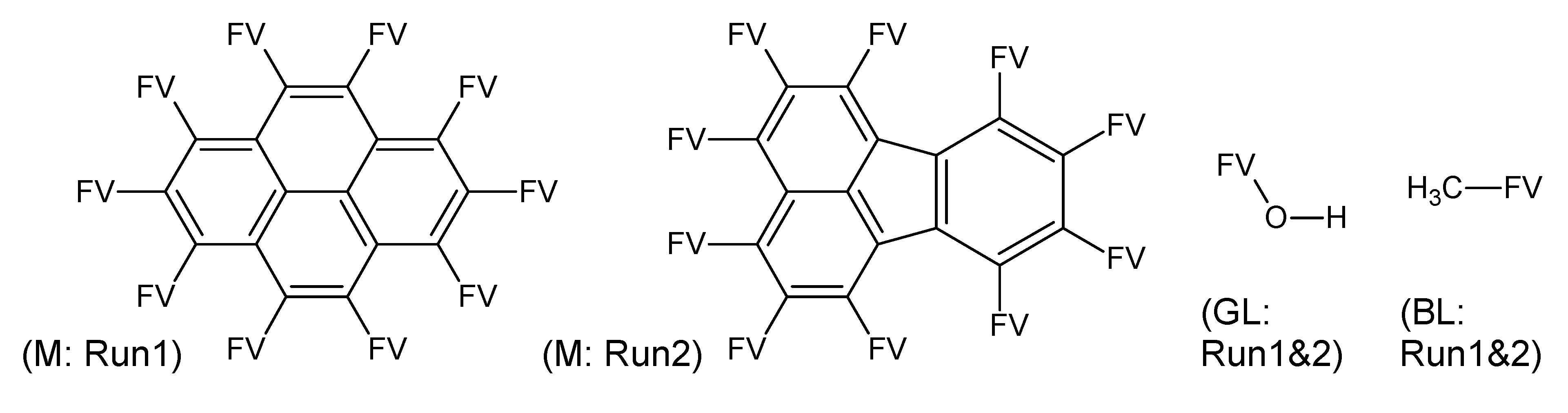
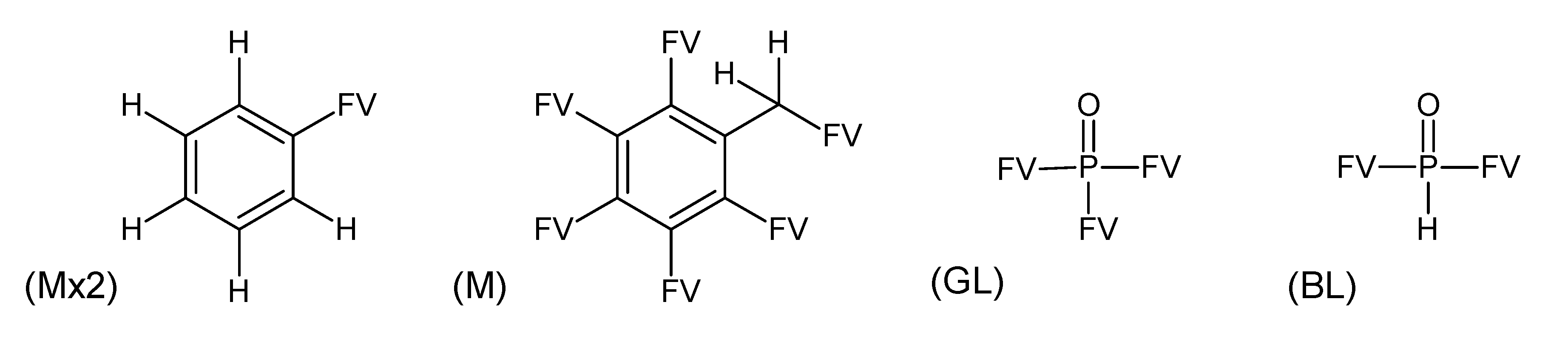
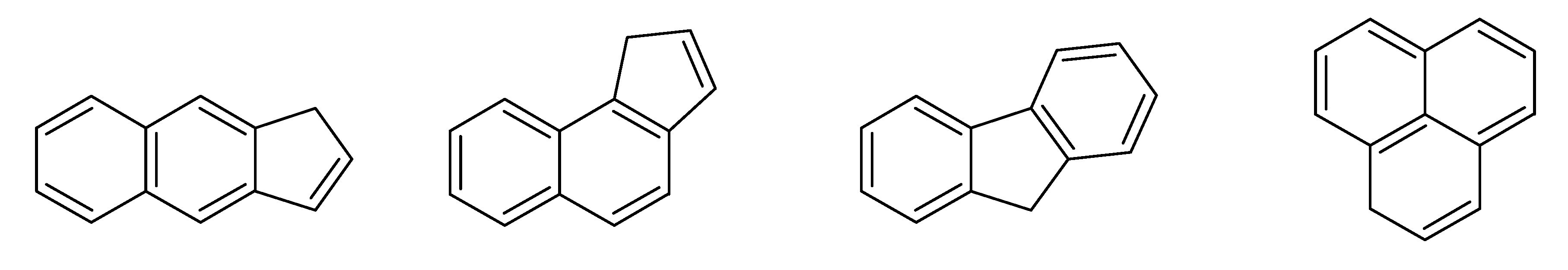
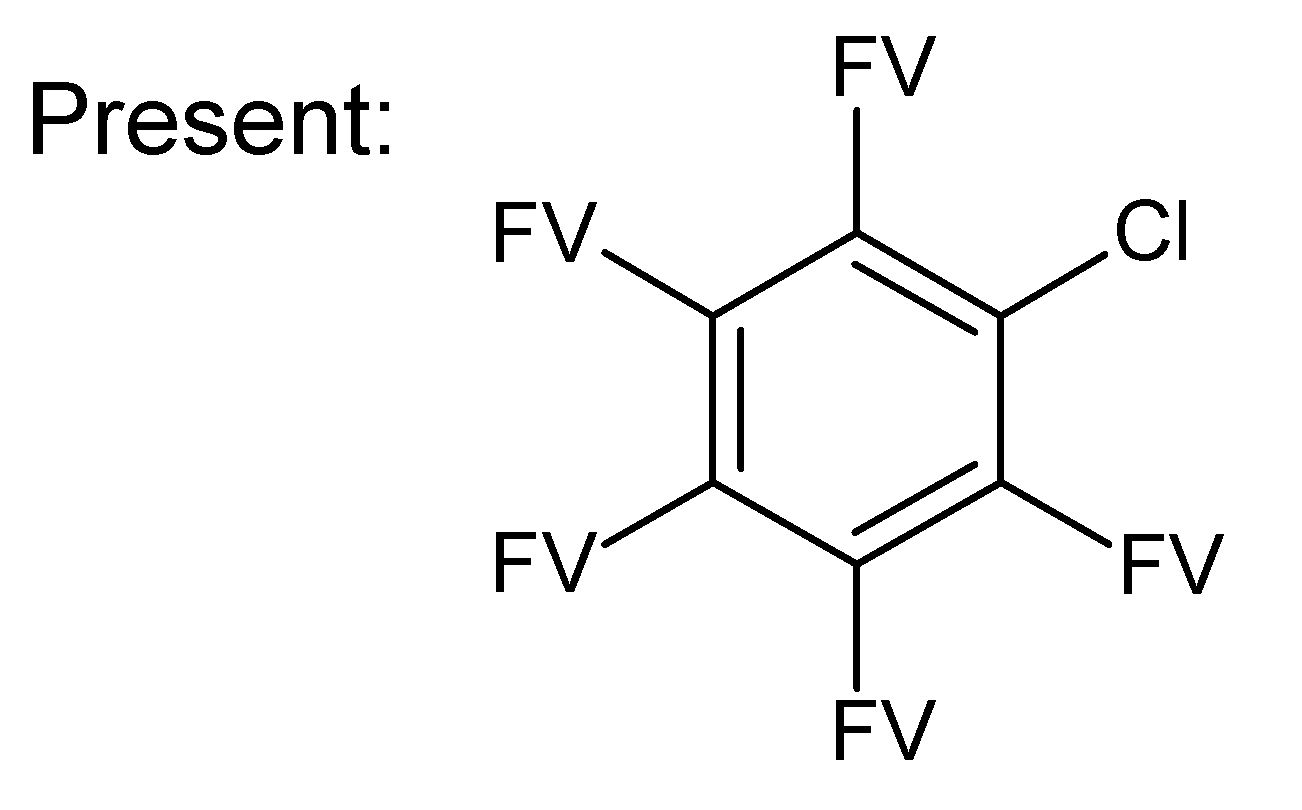
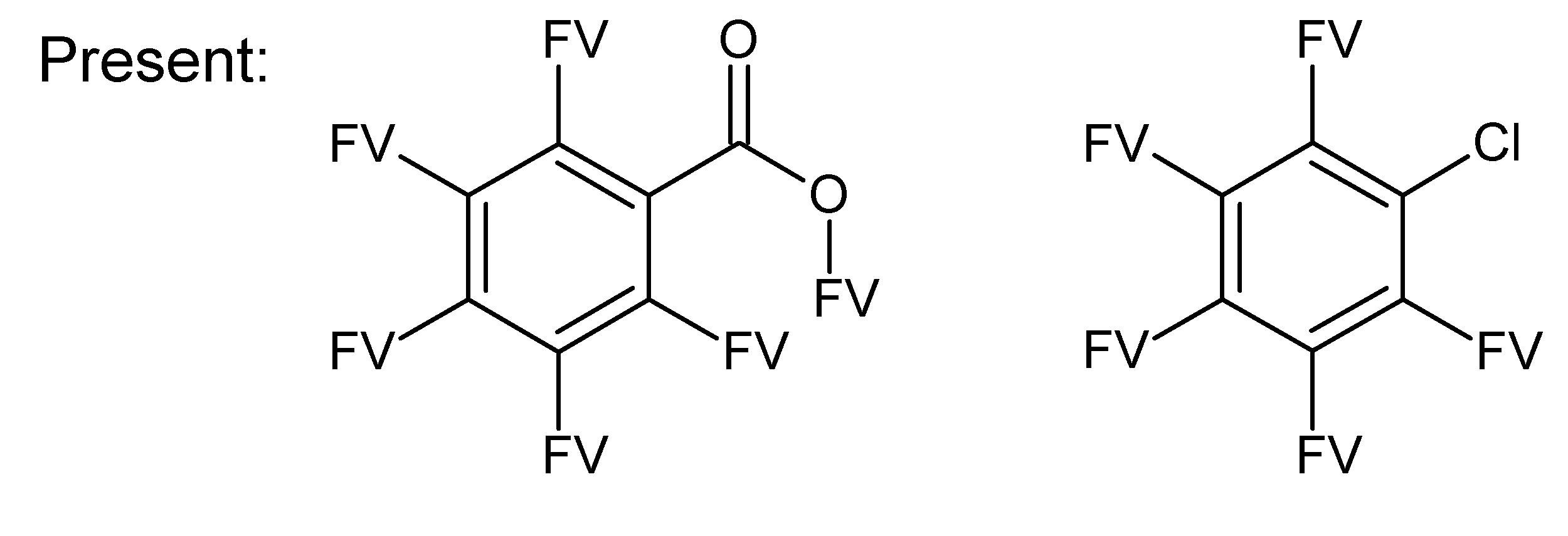
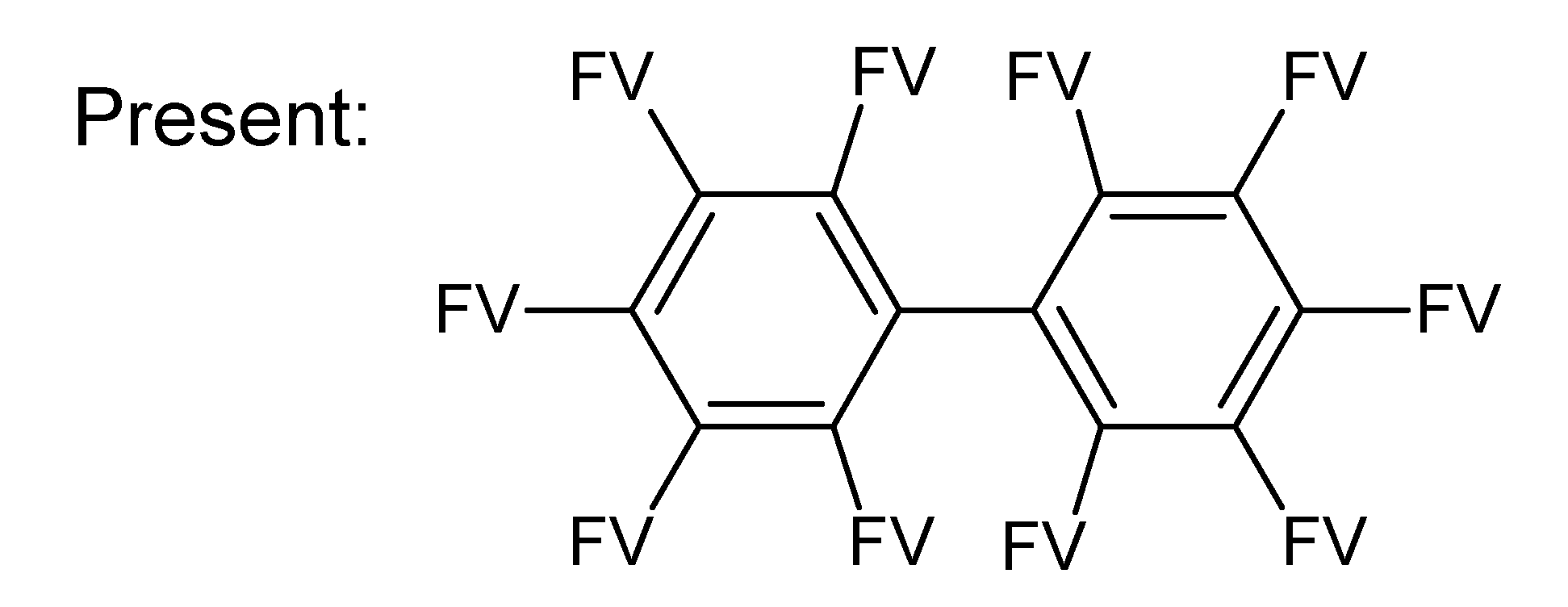
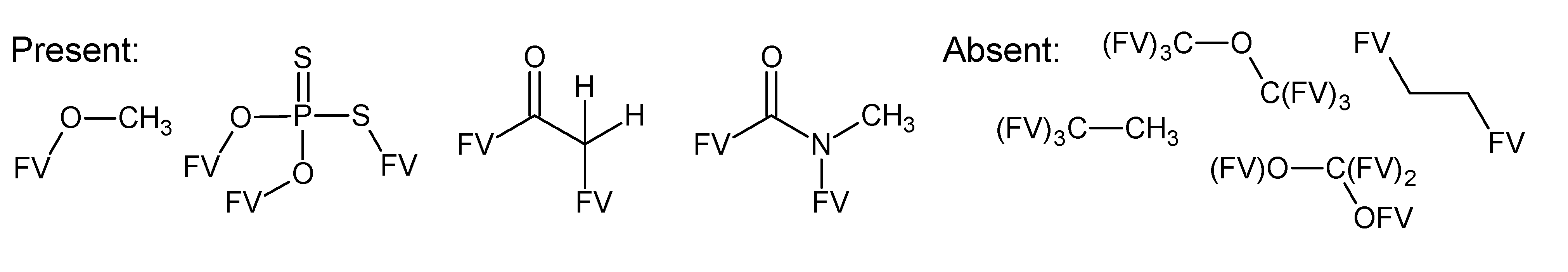
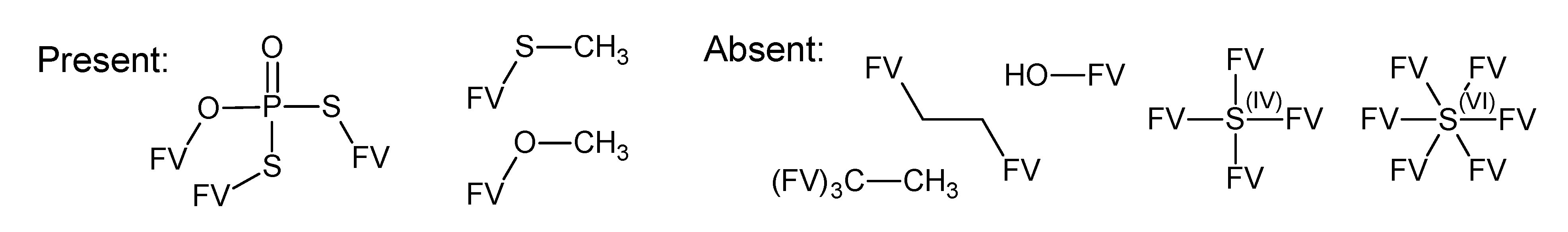
- –
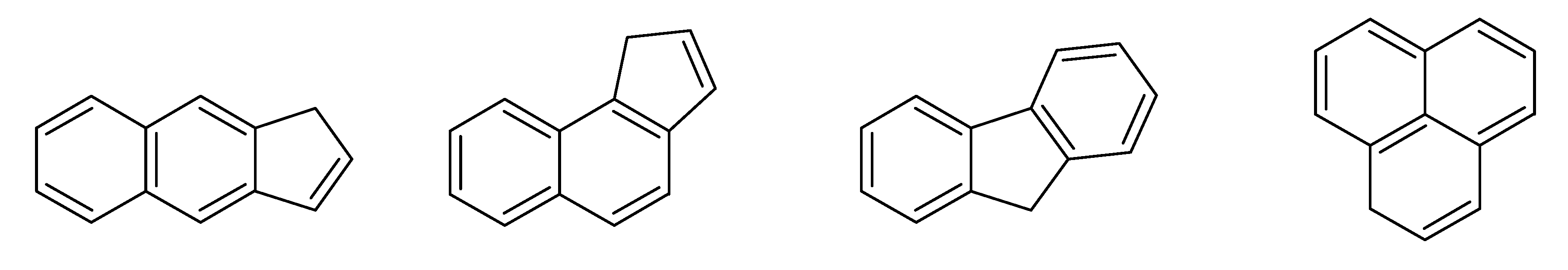
- bondsa 11–12: There are 11 or 12 aromatic bonds, corresponding to two condensed aromatic 6–rings (naphthalene skeleton) or two disjoint aromatic 6–rings.
- –
- ringsize 5–13: to avoid rings of size three and four.
- –
- –
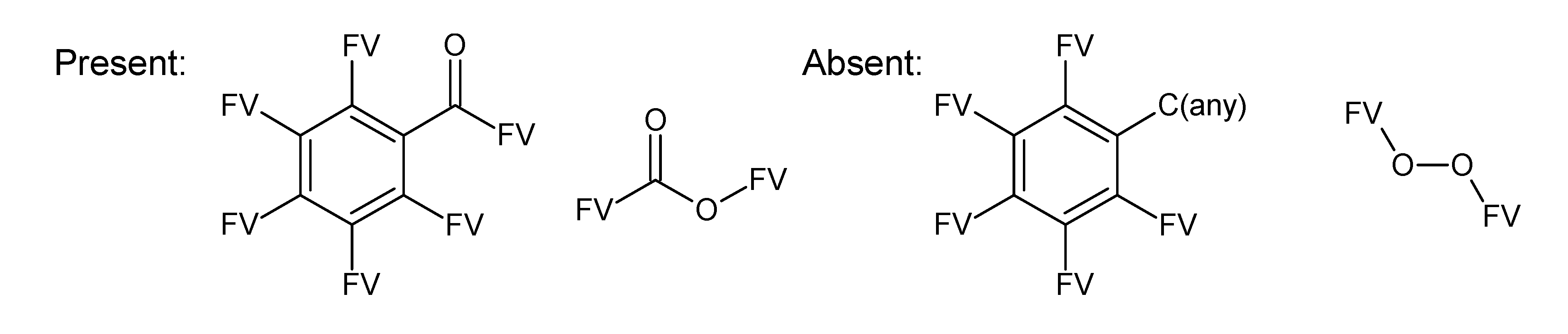
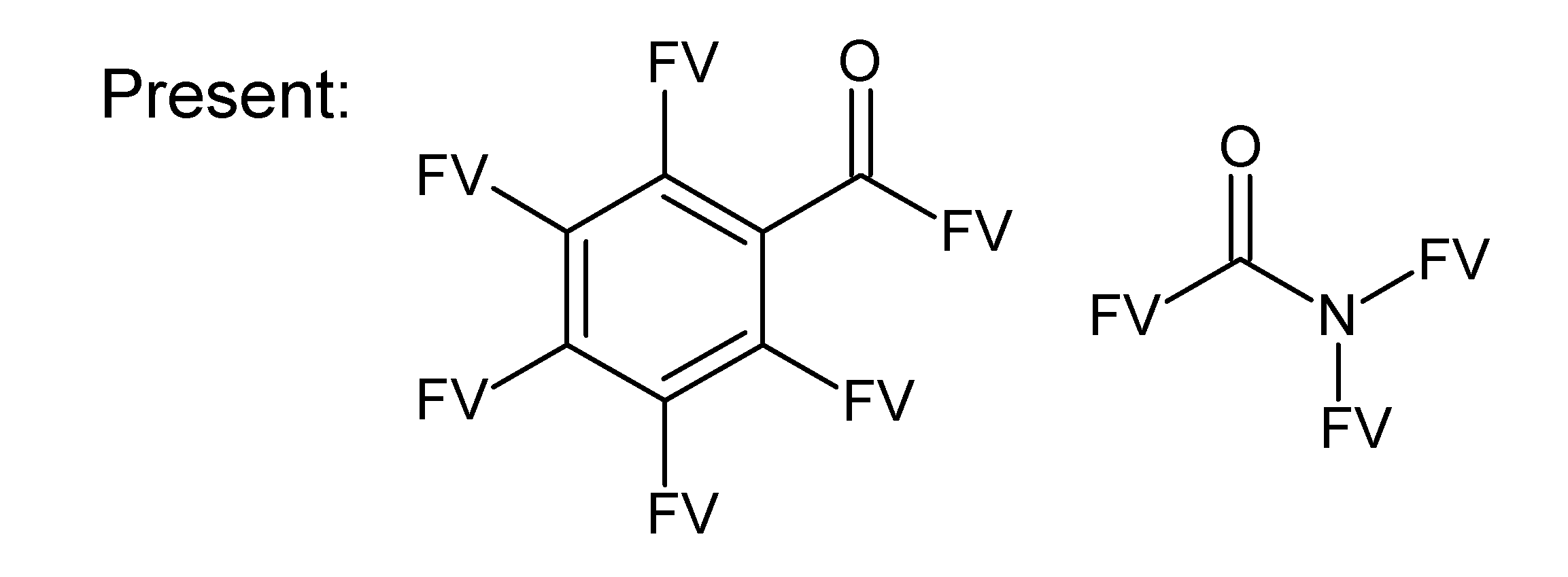
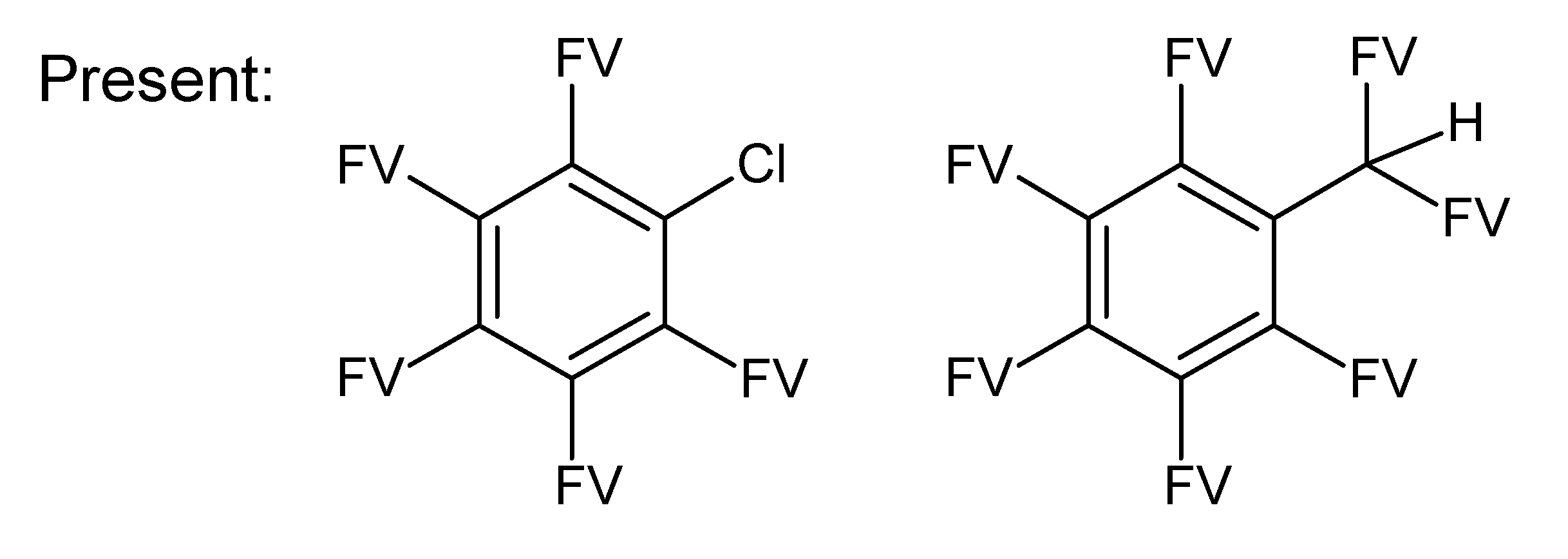
- badlist SideChainTerminals.sdf: a badlist of substructures that occur at the end of side chains, including –CH3, =CH2, –NH2 and =NH, to prevent the occurrence of side chains.
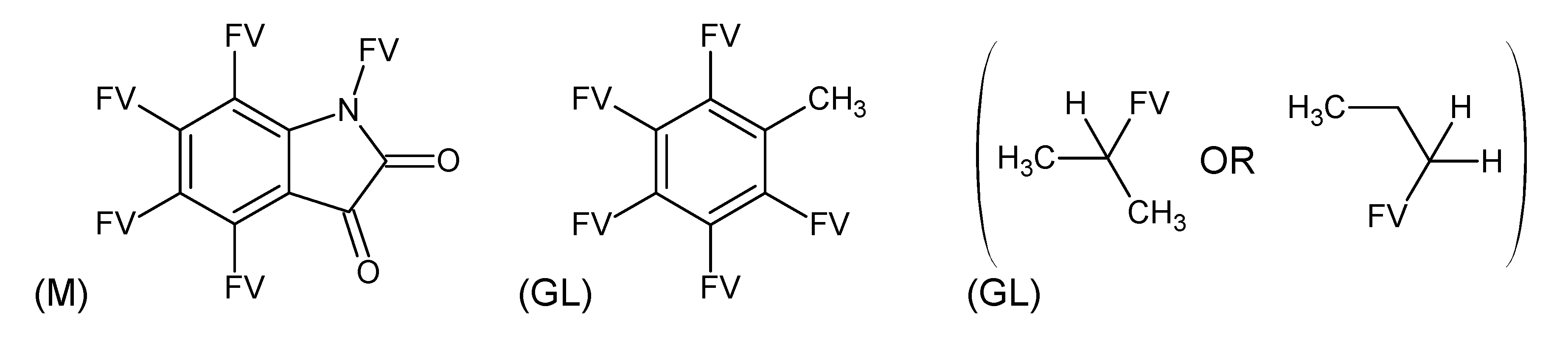
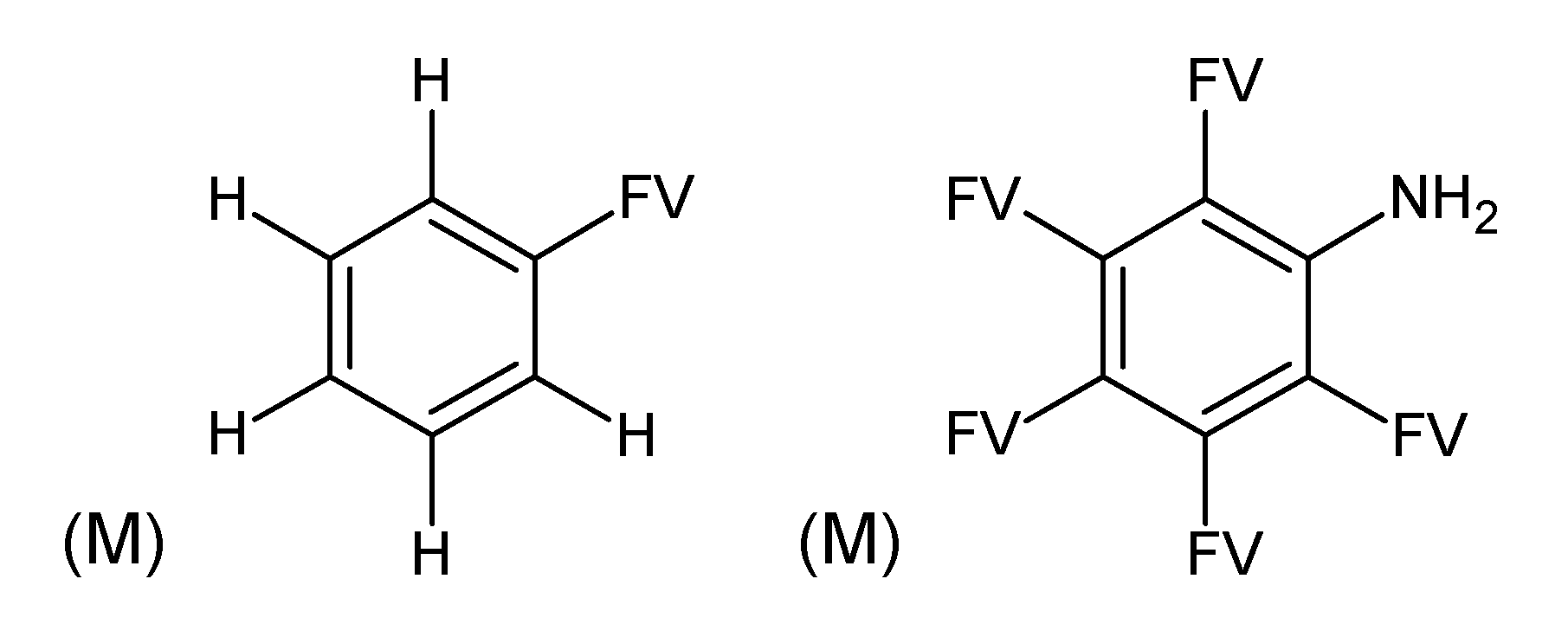
3.3. Category 3: Best Molecular Formula with GC–MS
| Challenge Number | Molecular Formula | PubChem CID | Formulas Entered | Rank of Corrrect | Possible Structures | Structures Entered | Rank of Correct | RRP |
|---|---|---|---|---|---|---|---|---|
| 1 | C8H4O3 | 6552 | 5 | 1 | 4,161,969 | 93 | 19(2) | 0.201 |
| 2 | C8H5NO2 | 6550 | 2 | 1 | 38,484,571 | 80 | 1 | 0 |
| 3 | C7H7ClO | 26799 | 1 | 1 | 62,643 | 3 | 3 | 1 |
| 4 | C7H7ClO | 12823 | 1 | 1 | 62,643 | 3 | 2 | 0.5 |
| 5 | C6H4Cl2 | 13866817 | 1 | 1 | 1323 | 3 | 1(2) | 0.25 |
| 6 | C12H10 | 6478 | 2 | 1 | 37,720,012 | 187 | 5 | 0.021 |
| 7 | C7H5ClO2 | 6079 | 1 | 1 | 507,196 | 3 | 1 | 0 |
| 8 | C13H10 | 6592 | 1 | 1 | >1E8 (40%) | 90 | 1 | 0 |
| 9 | C8H7ClO2 | 11402 | 1 | 1 | 5,160,746 | 3 | 3 | 1 |
| 10 | C6H3Cl3O | 21106172 | 1 | 1 | 19,969 | 6 | 2 | 0.2 |
| 11 | C6H12NO4PS2 | 16412 | 15 | 1 | >1E8 (4%) | 45 | 1 | 0 |
| 12 | C6H6Cl6 | 10468511 | 1 | 4 | 1421 | 1 | 1 | NA |
| 13 | C3H6S3 | 15959 | 5 | 1 | 102 | 13 | 13 | 1 |
| 14 | C3H9O3PS | 8686 | 1 | 1 | 19,054 | 1 | 1 | NA |
| 15 | C12H8O | 551 | 1 | 1 | >1E8 (30%) | 19 | 1 | 0 |
| 16 | C3H9PS2O2 | 29165 | 8 | 1 | 27,776 | 1 | 1 | NA |
3.4. Category 4: Best Structure with GC–MS
4. Conclusions and Perspectives
Conflict of Interest
Acknowledgements
A. Appendix
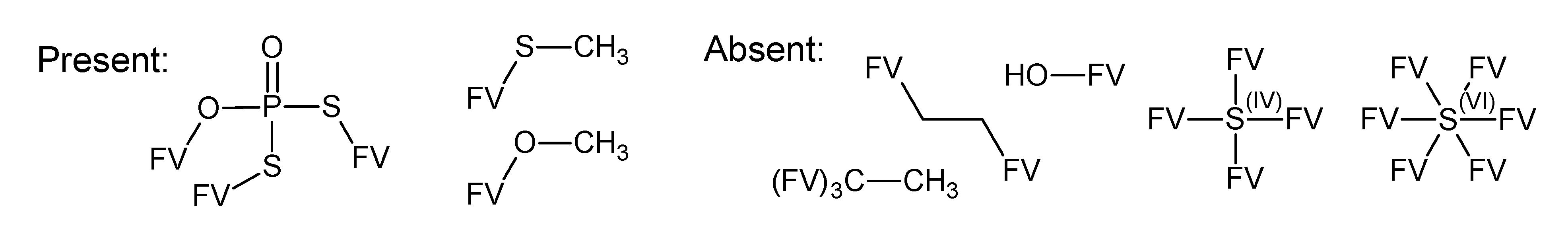
A.1. Substructures for CASMI Category 4: GC Challenges
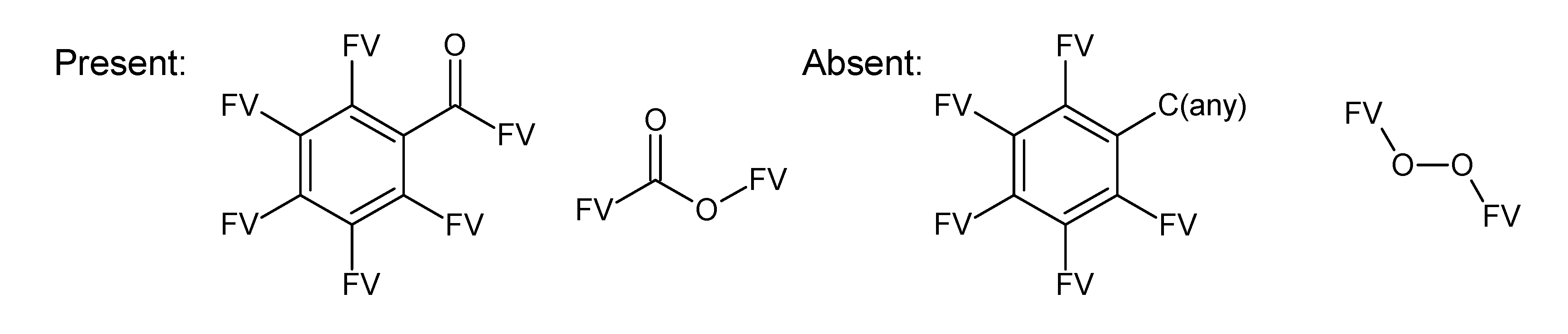
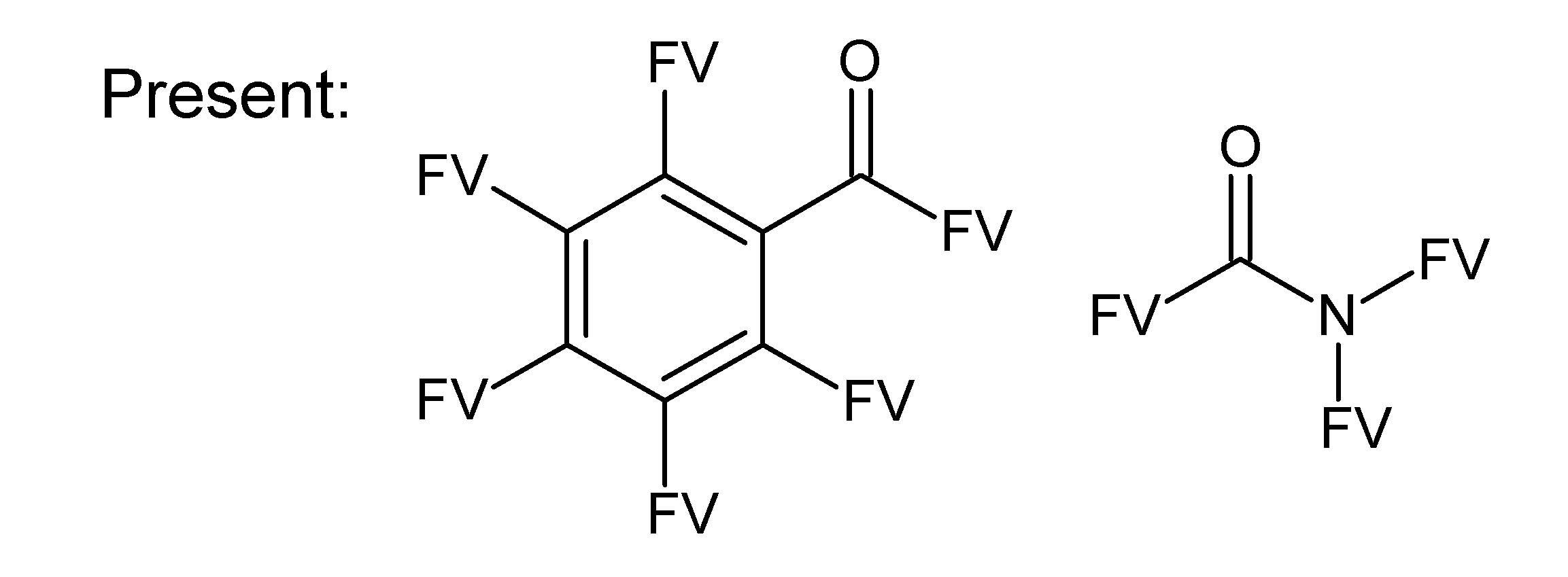
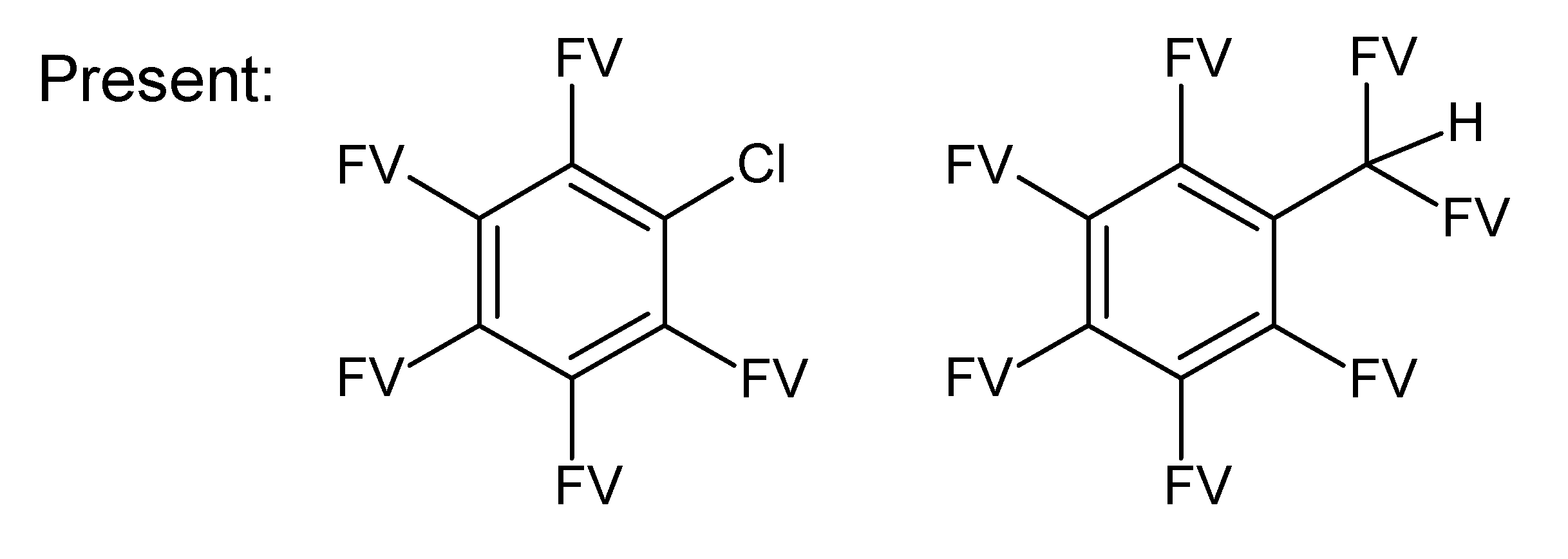
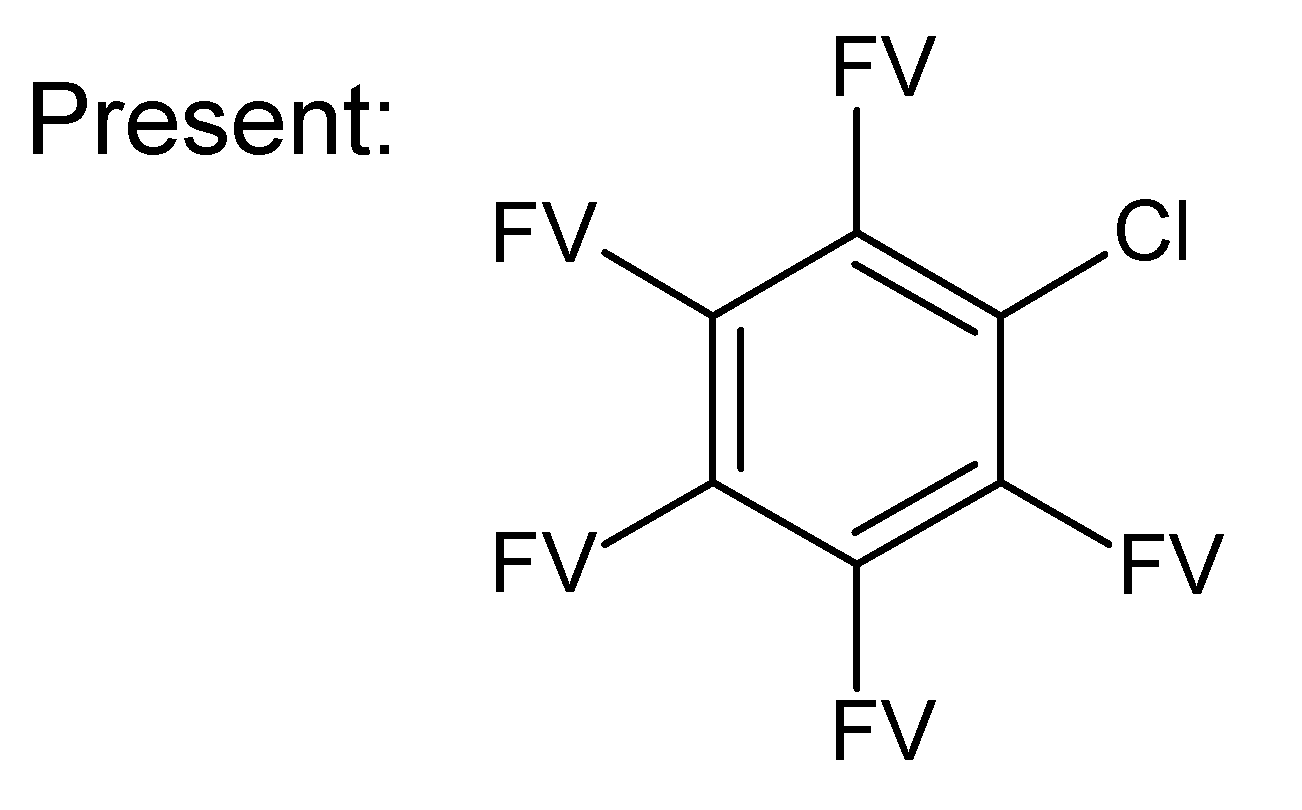
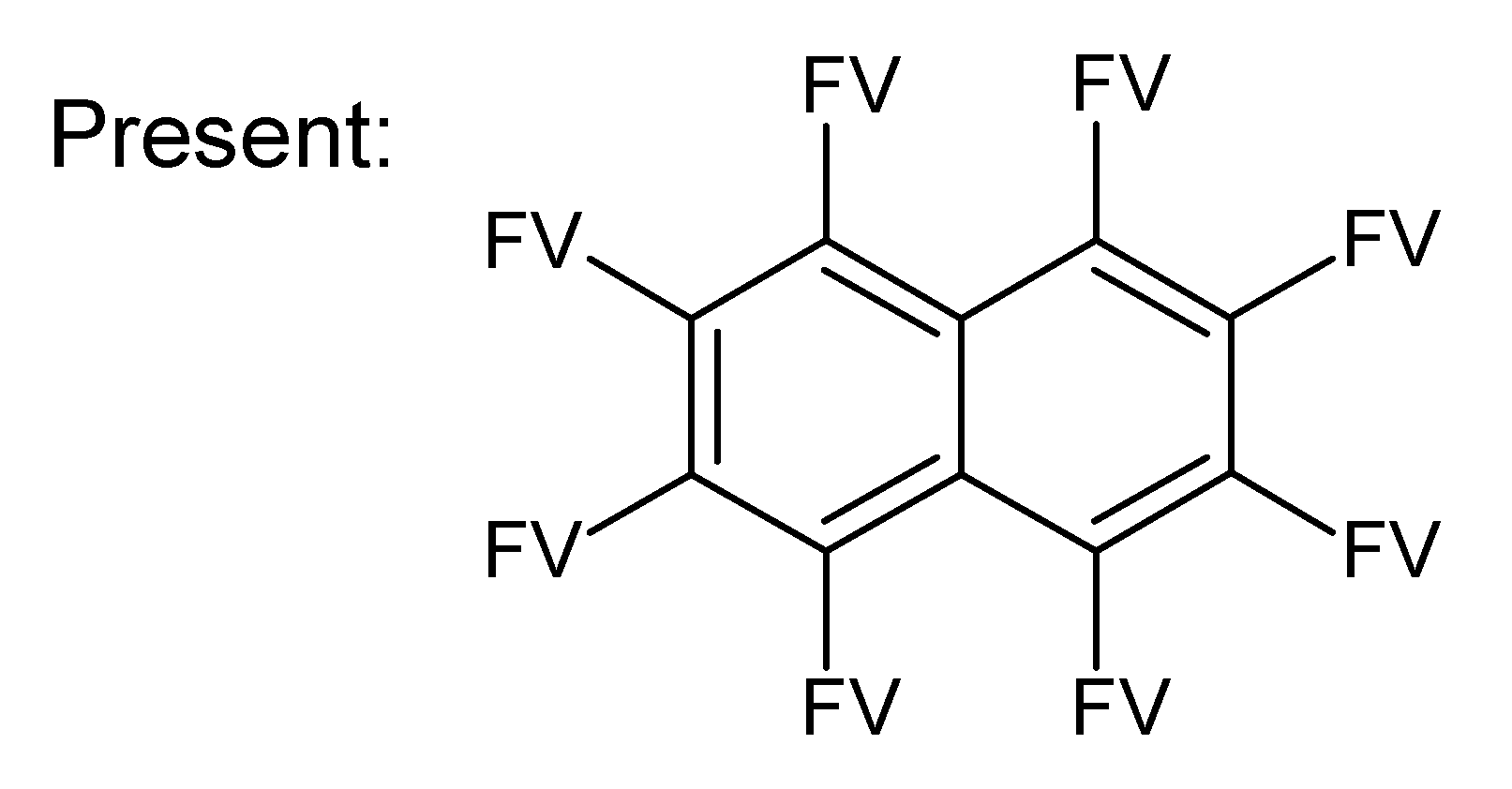
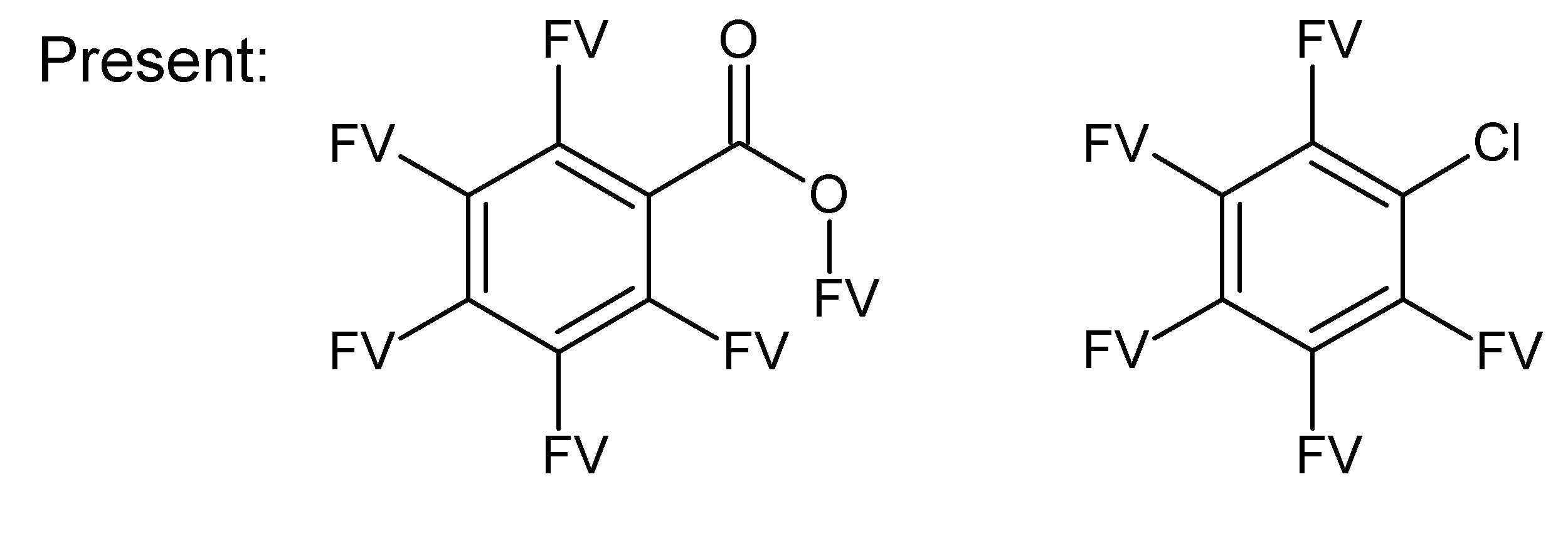
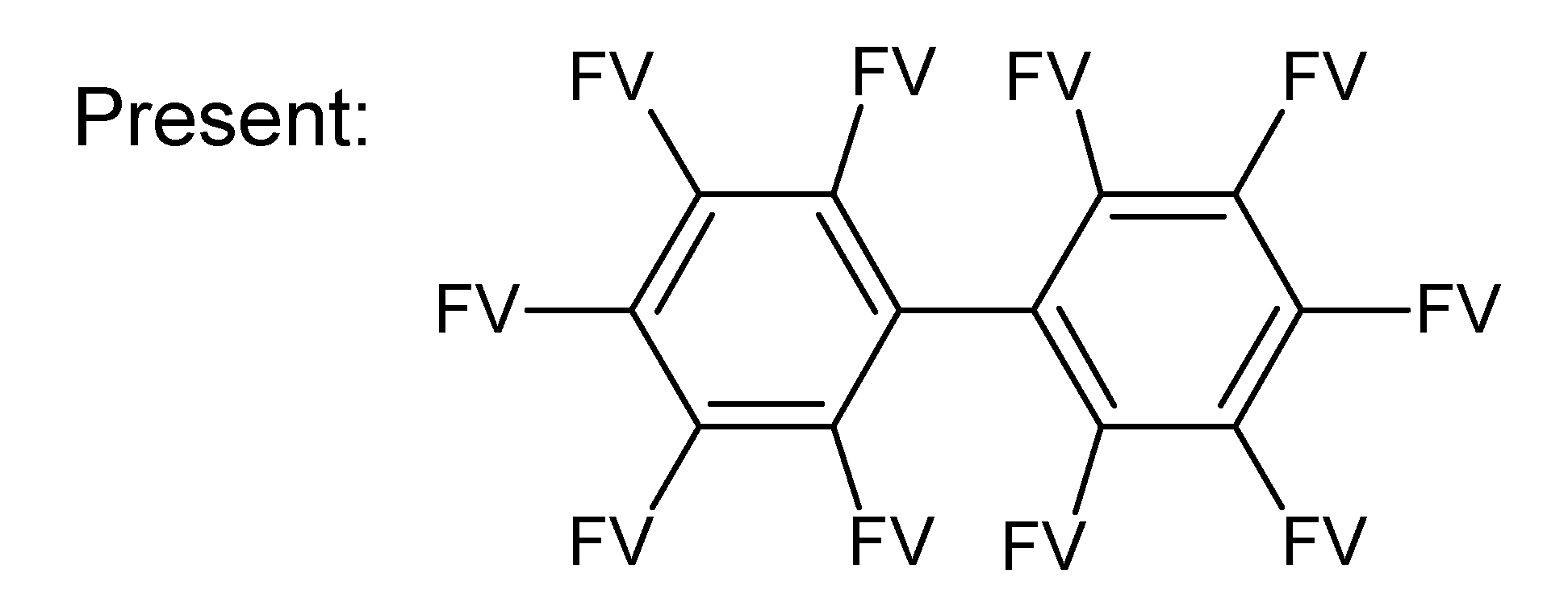
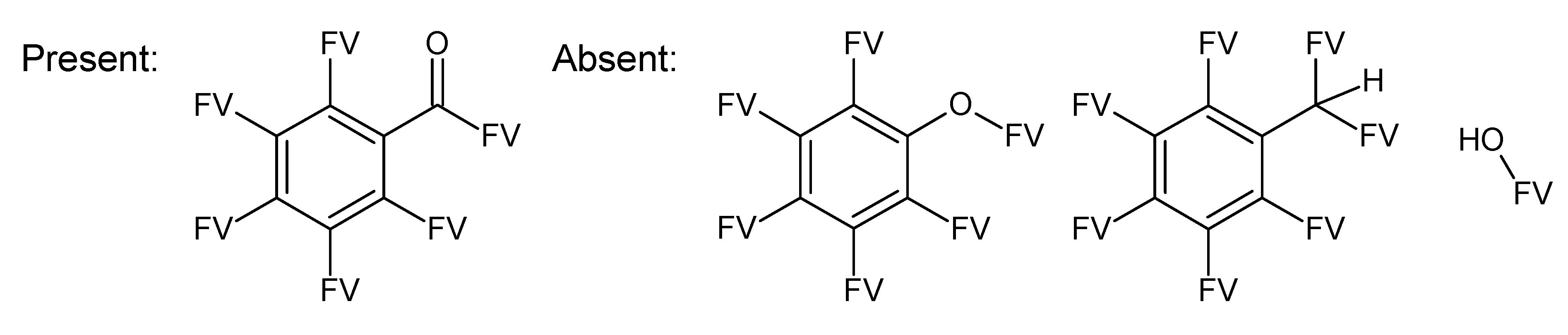
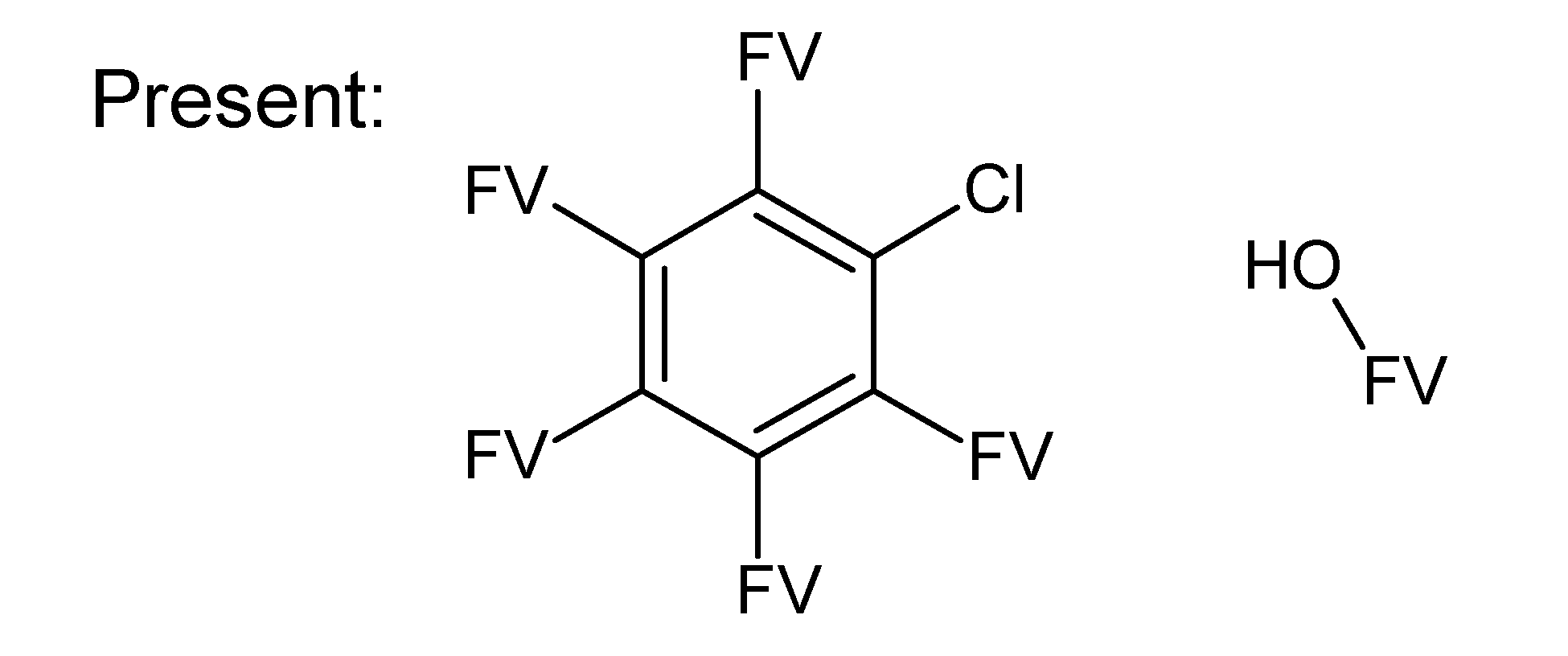
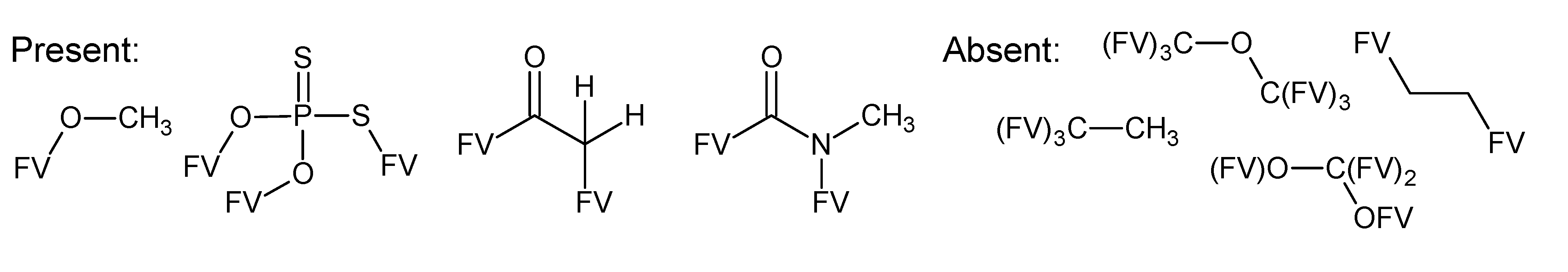

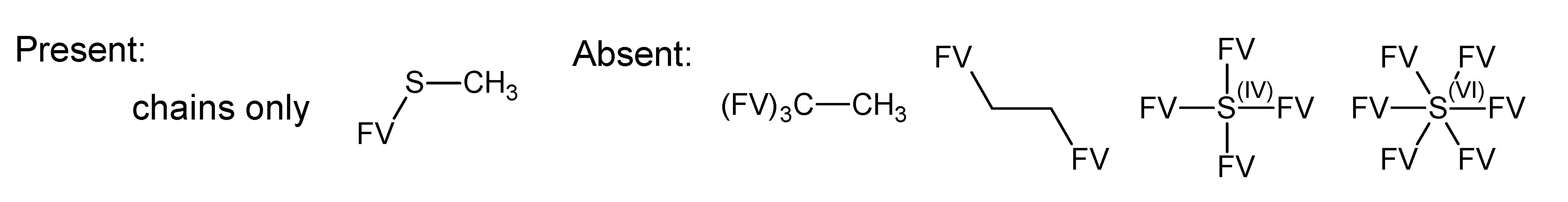

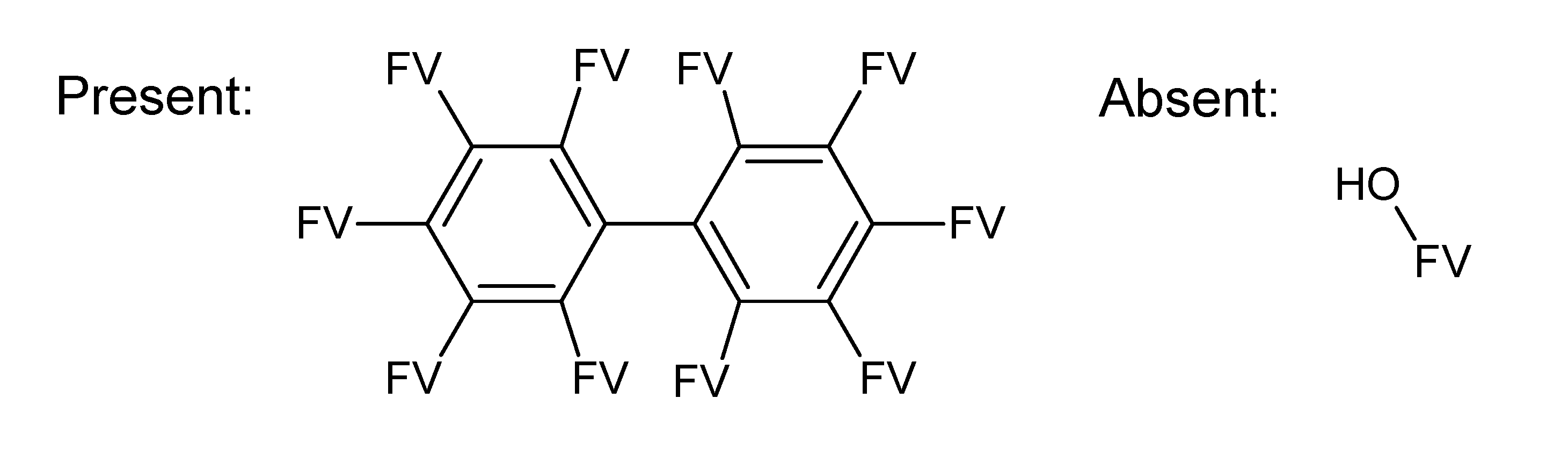
References
- Meringer, M.; Reinker, S.; Zhang, J.; Muller, A. MS/MS data improves automated determination of molecular formulas by mass spectrometry. MATCH Commun. Math. Comput. Chem. 2011, 65, 259–290. [Google Scholar]
- Gugisch, R.; Kerber, A.; Kohnert, A.; Laue, R.; Meringer, M.; Rücker, C.; Wassermann, A. MOLGEN 3.5 Reference Guide. 2009. Available online: http://molgen.de/documents/molgen35.pdf (accessed on 28 February 2013).
- Benecke, C.; Grüner, T.; Kerber, A.; Laue, R.; Wieland, T. MOLecular structure GENeration with MOLGEN, new features and future developments. Fresenius J. Anal. Chem. 1997, 359, 23–32. [Google Scholar] [CrossRef]
- Gugisch, R.; Kerber, A.; Kohnert, A.; Laue, R.; Meringer, M.; Rücker, C.; Wassermann, A. MOLGEN 5.0 Reference Guide. 2009. Available online: http://molgen.de/documents/manual50.pdf (accessed on 28 February 2013).
- Gugisch, R.; Kerber, A.; Kohnert, A.; Laue, R.; Meringer, M.; Rücker, C.; Wassermann, A. MOLGEN 5.0, a Molecular Structure Generator in Advances in Mathematical Chemistry. In Advances in Mathematical Chemistry; Basak, S.C., Restrepo, G., Villaveces, J.L., Eds.; Bentham Science Publishers: Sharjah, UAE, 2013; in press. [Google Scholar]
- Wolf, S.; Schmidt, S.; Müller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinform. 2010, 11, 148. [Google Scholar] [CrossRef]
- Heinonen, M.; Rantanen, A.; Mielikäinen, T.; Kokkonen, J.; Kiuru, J.; Ketola, R.A.; Rousu, J. FiD: A software for ab initio structural identification of product ions from tandem mass spectrometric data. Rapid Commun. Mass Spectrom. 2008, 22, 3043–3052. [Google Scholar] [CrossRef]
- HighChem. Mass Frontier Version 6.0; HighChem/Thermo Scientific: Bratislava, Slovakia, 2013. [Google Scholar]
- Kerber, A.; Laue, R.; Meringer, M.; Rücker, C. MOLGEN–QSPR, a software package for the search of quantitative structure property relationships. MATCH Commun. Math. Comput. Chem. 2004, 51, 187–204. [Google Scholar]
- Kerber, A.; Laue, R.; Meringer, M.; Varmuza, K. MOLGEN–MS: Evaluation of Low Resolution Electron Impact Mass Spectra with MS Classification and Exhaustive Structure Generation. In Advances in Mass Spectrometry; Gelpi, E., Ed.; Wiley: West Sussex, UK, 2001; Volume 15, pp. 939–940. [Google Scholar]
- Meringer, M. Mathematical Models for Conbinatorial Chemistry and Molecular Structure Elucidation; (in German). Logos–Verlag Berlin: Berlin, Germany, 2004; p. 390. [Google Scholar]
- Schymanski, E.L. Integrated Analytical and Computer Tools for Toxicant Identification in Effect–Directed Analysis. PhD thesis, Faculty for Chemistry and Physics, Technical University Bergakadamie Freiberg and Helmholtz Center for Environmental Research-UFZ, Leipzig, Germany, 2011; p. 122. [Google Scholar]
- Lindsay, R.K.; Buchanan, B.G.; Feigenbaum, E.A.; Lederberg, J. Applications of Artificial Intelligence for Organic Chemistry: The DENDRAL Project; McGraw–Hill: New York, NY, USA, 1980. [Google Scholar]
- Varmuza, K.; Werther, W. Mass spectral classifiers for supporting systematic structure elucidation. J. Chem. Inf. Comput. Sci. 1996, 36, 323–333. [Google Scholar] [CrossRef]
- Kerber, A.; Laue, R.; Grüner, T.; Meringer, M. MOLGEN 4.0. MATCH Commun. Math. Comput. Chem. 1998, 37, 205–208. [Google Scholar]
- Kerber, A.; Meringer, M.; Rücker, C. CASE via MS: Ranking structure candidates by mass spectra. Croatica Chem. Acta 2006, 79, 449–464. [Google Scholar]
- Grüner, T.; Kerber, A.; Laue, R.; Liepelt, M.; Meringer, M.; Varmuza, K.; Werther, W. Bestimmung von Summenformeln aus Massenspektren durch Erkennung überlagerter Isotopenmuster. MATCH Commun. Math. Comput. Chem. 1998, 37, 163–177. [Google Scholar]
- Kerber, A.; Laue, R.; Meringer, M.; Rücker, C.; Schymanski, E.L. Mathematical Chemistry and Chemoinformatics: Structure Generation, Elucidation and Quantitative Structure-Property Relationships; Walter de Gruyter: Berlin, Germany, to appear in 2013.
- Schymanski, E.L.; Meinert, C.; Meringer, M.; Brack, W. The use of MS classifiers and structure generation to assist in the identification of unknowns in effect–directed analysis. Anal. Chim. Acta. 2008, 615, 136–147. [Google Scholar] [CrossRef]
- NIST/EPA/NIH. NIST 2011 Mass Spectral Library; National Institute of Standards and Technology, US Secretary of Commerce: Gaithersburg, Maryland, USA, 2011. [Google Scholar]
- Schymanski, E.L.; Meringer, M.; Brack, W. Automated strategies to identify compounds on the basis of GC/EI–MS and calculated properties. Anal. Chem. 2011, 83, 903–912. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Gallampois, C.M.J.; Krauss, M.; Meringer, M.; Neumann, S.; Schulze, T.; Wolf, S.; Brack, W. Consensus structure elucidation combining GC/EI-MS, structure generation, and calculated properties. Anal. Chem. 2012, 84, 3287–3295. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Neumann, S. CASMI: Challenges and solutions. Metabolites Year, in press. [Google Scholar]
- Schymanski, E.L.; Neumann, S. CASMI: And the winner is .. Metabolites Year, in press. [Google Scholar]
- Meringer, M. MOLGEN–MS/MS Software User Manual. München, Germany, 2011. Available online: http://molgen.de/documents/MolgenMsMs.pdf (accessed on 28 February 2013).
- Stravs, M.A.; Schymanski, E.L.; Singer, H.P.; Hollender, J. Automatic recalibration and processing of tandem mass spectra using formula annotation. J. Mass Spectrom. 2013, 48, 89–99. [Google Scholar] [CrossRef]
- Dalby, A.; Nourse, J.G.; Hounshell, W.D.; Gushurst, A.K.I.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef]
- Grüner, T.; Kerber, A.; Laue, R.; Meringer, M.; Varmuza, K.; Werther, W. MOLGEN–MS version 1.0.1.2. University of Bayreuth, Germany, 2009. Available online: http://www.molgen.de (accessed on 28 February 2013). Trial version available online.
- Schymanski, E.L.; Neumann, S. Critical Assessment of Small Molecule Identification Contest. 2012. Available online: http://www.casmi-contest.org/challenges-cat3-4.shtml/ (accessed on 24 May 2013).
- Schymanski, E.L.; Neumann, S. Critical Assessment of Small Molecule Identification Contest Rules. 2012. Available online: http://casmi-contest.org/rules.shtml (accessed on 20 May 2013).
- Schymanski, E.L.; Meringer, M.; Brack, W. Matching structures to mass spectra using fragmentation patterns: Are the results as good as they look? Anal. Chem. 2009, 81, 3608–3617. [Google Scholar] [CrossRef]
- Oberacher, H. Applying tandem mass spectral libraries for solving the CASMI LC/MS challenge 2012. Metabolites 2013, 3, 312–324. [Google Scholar] [CrossRef]
- Ruttkies, C.; Gerlich, M.; Neumann, S. Tackling challenging challenges with MetFrag and MetFusion. Metabolites Year, in press. [Google Scholar]
- Rasche, F.; Svatos, A.; Maddula, R.K.; Böttcher, C.; Böcker, S. Computing fragmentation trees from tandem mass spectrometry data. Anal. Chem. 2011, 83, 1243–1251. [Google Scholar] [CrossRef]
- Sheldon, M.T.; Mistrik, R.; Croley, T.R. Determination of ion structures in structurally related compounds using precursor ion fingerprinting. J. Am. Soc. Mass Spectrom. 2009, 20, 370–376. [Google Scholar] [CrossRef]
- Hildebrandt, C.; Wolf, S.; Neumann, S. Database supported candidate search for metabolite identification. J. Integr. Bioinform. 2011. [Google Scholar] [CrossRef]
- Peironcely, J.E.; Rojas-Chertó, M.; Tas, A.; Vreeken, R.J.; Reijmers, T.; Coulier, L.; Hankemeier, T. An automated pipeline for de novo metabolite identification using mass spectrometry-based metabolomics. Anal. Chem. 2013, 85, 3576–3583. [Google Scholar] [CrossRef]
- Peironcely, J.E.; Rojas-Chertó, M.; Fichera, D.; Reijmers, T.; Coulier, L.; Faulon, J.L.; Hankemeier, T. OMG: Open molecule generator. J. Cheminform. 2012. [Google Scholar] [CrossRef]
- Dixon, R.A.; Strack, D. Phytochemistry meets genome analysis, and beyond. Phytochemistry 2003, 62, 815–816. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Meringer, M.; Schymanski, E.L. Small Molecule Identification with MOLGEN and Mass Spectrometry. Metabolites 2013, 3, 440-462. https://doi.org/10.3390/metabo3020440
Meringer M, Schymanski EL. Small Molecule Identification with MOLGEN and Mass Spectrometry. Metabolites. 2013; 3(2):440-462. https://doi.org/10.3390/metabo3020440
Chicago/Turabian StyleMeringer, Markus, and Emma L. Schymanski. 2013. "Small Molecule Identification with MOLGEN and Mass Spectrometry" Metabolites 3, no. 2: 440-462. https://doi.org/10.3390/metabo3020440
APA StyleMeringer, M., & Schymanski, E. L. (2013). Small Molecule Identification with MOLGEN and Mass Spectrometry. Metabolites, 3(2), 440-462. https://doi.org/10.3390/metabo3020440