Abstract
The goals of metabolic engineering are well-served by the biological information provided by metabolomics: information on how the cell is currently using its biochemical resources is perhaps one of the best ways to inform strategies to engineer a cell to produce a target compound. Using the analysis of extracellular or intracellular levels of the target compound (or a few closely related molecules) to drive metabolic engineering is quite common. However, there is surprisingly little systematic use of metabolomics datasets, which simultaneously measure hundreds of metabolites rather than just a few, for that same purpose. Here, we review the most common systematic approaches to integrating metabolite data with metabolic engineering, with emphasis on existing efforts to use whole-metabolome datasets. We then review some of the most common approaches for computational modeling of cell-wide metabolism, including constraint-based models, and discuss current computational approaches that explicitly use metabolomics data. We conclude with discussion of the broader potential of computational approaches that systematically use metabolomics data to drive metabolic engineering.
Abbreviations

| Abbreviation | Meaning | Abbreviation | Meaning |
|---|---|---|---|
| CE-MS | Capillary Electrophoresis-Mass Spectrometry | MOMA | Minimization Of Metabolic Adjustment |
| CHO | Chinese Hamster Ovary | NET | Network-Embedded Thermodynamic |
| COBRA | Constraints Based Reconstruction and Analysis | NMR | Nuclear Magnetic Resonance |
| dFBA | Dynamic Flux Balance Analysis | OMNI | Optimal Metabolic Network Identification |
| EMUs | Elementary Metabolite Units | ODE | Ordinary Differential Equation |
| FBA | Flux Balance Analysis | PLS | Partial Least Squares |
| GC-MS | Gas Chromatography-Mass Spectrometry | PLS-DA | Partial Least Squares Discriminant Analysis |
| HCA | Hierarchical Clustering Analysis | PCA | Principal Components Analysis |
| HPLC | High-Performance Liquid Chromatography | QP | Quadratic Programming |
| idFBA | Integrated-Dynamic Flux Balance Analysis | rFBA | Regulatory Flux Balance Analysis |
| iFBA | Integrated Flux Balance Analysis | SBRT | Systems Biology Research Tool |
| IOMA | Integrative “Omics”-Metabolic Analysis | TMFA | Thermodynamic Metabolic Flux Analysis |
| LP | Linear Programming | TAL | Transaldolase |
| LC-MS | Liquid Chromatography-Mass Spectrometry | TKL | Transketolase |
| MASS | Mass Action Stoichiometric Simulation | TCA | Tricarboxylic Acid |
| MCA | Metabolic Control Analysis | VIP | Variable Importance in the Projection |
| MFA | Metabolic Flux Analysis | VHG | Very High Gravity |
1. Introduction
Organisms such as Saccharomyces cerevisiae and Aspergillus niger have a long history of commercial use in natural fermentation processes to produce chemicals such as ethanol and citric acid. Traditional bioprocess engineering entails the design and optimization of the equipment and procedures necessary to efficiently manufacture these and other biologically derived products. The development of recombinant DNA technologies enabled the direct manipulation and expansion of the metabolic capabilities of S. cerevisiae and A. niger (as well as other organisms such as Escherichia coli and Bacillus subtilis), which resulted in the emergence of metabolic engineering as a field distinct from bioprocess engineering [1]. Metabolic engineering is the (usually genetic) control of the metabolic activities of a living organism to establish and optimize the production of desirable metabolites – the class of small molecules that comprise the primary resources and intermediates of all cellular activity. With widespread and growing interest in environmentally sustainable industrial technologies, metabolic engineering is poised to provide an effective and efficient means for producing various small molecule chemicals from clean and renewable sources, such as biofuels derived from lignocellulosic feedstock [2,3,4,5,6,7,8,9,10,11,12].
Frequently, metabolic engineering studies use targeted analysis of a few carefully selected intracellular or secreted extracellular compounds to drive or assess the progress of their efforts [2,9,13,14,15,16,17,18,19,20,21,22,23,24]. High-Performance Liquid Chromatography (HPLC) and enzymatic assays have typically been the methods of choice to generate this data, used in engineering S. cerevisiae [2,13,21,23], E. coli [16,19], Clostridium acetobutylicum [14,18], and other organisms. These measurements may be direct readouts of the performance of an engineered strain [2], or they may be interpreted as performance and response characteristics (for example, trehalose as a marker for stress response in yeast [21,23]). These analyses are typically focused on effects at the level of individual pathways [2,19,21,25].
Another technique used to characterize metabolic pathways during metabolic engineering is Metabolic Flux Analysis (MFA). MFA provides more information than measurement of just a few metabolites, and is a staple technique of many who work in metabolic engineering [14,18,20,22,24,26,27,28,29,30,31,32]. In MFA, isotopically labeled metabolites (typically using 13C labels) are leveraged to calculate fluxes – the rate at which material is processed through a metabolic pathway – from knowledge of carbon-carbon transitions in each reaction and the measured isotopomer distribution in each metabolite [1]. Ongoing research in MFA includes continued improvement of 13C protocols and analytical platforms [33,34,35,36], improvements to software for MFA calculations [32,37], use of network stoichiometry to determine the minimal set of required metabolite measurements [38], and study of Elementary Metabolite Units (EMUs) for more efficient analysis of flux patterns [31,39,40].
Metabolic engineering seeks to maximize the production of selected metabolites in a cell, whether produced by the organisms’ natural metabolic activities or by entire exogenous pathways introduced through genetic engineering. Strategic, small-scale measurements and flux calculations have to date been indispensable tools for metabolic engineering. However, the development of systems-level analyses – precipitated by whole-genome sequencing and the rapid accumulation of data on RNA, protein and metabolite levels – has provided new opportunities to more completely understand the effects of strain manipulations. Genetic modifications often have additional effects outside the immediately targeted pathway, and a better understanding of the nature and extent of these perturbations would lead to more effective strategies for redesigning strains, as well as improved ability to understand why a proposed design may fail to achieve its predicted performance.
Aided by recent advancements in analytical platforms that allow for the simultaneous measurement of a wide spectrum of metabolites, metabolomics (the analysis of the total metabolic content of living systems) is approaching the level of maturity of preceding “global analysis” fields like proteomics and transcriptomics [41,42]. Metabolomics approaches have already found some success in clinical applications, where studies have demonstrated their efficacy in identifying clinically relevant biomarkers in diseases such as cancer [43,44,45]. Surprisingly, though, the application of metabolomics approaches to problems in metabolic engineering has been somewhat scarce.
Here, we review examples of recent strategies to integrate metabolomics datasets into metabolic engineering. First, we briefly cover the fundamentals of metabolomics. We then discuss strategies for assessing metabolic engineering strain designs, and how metabolomics methods can extend these strategies. We follow with discussion of computational tools for metabolic engineering, with an emphasis on how these methods are used to design strains and predict their performance as well as how metabolomics datasets are currently applied to computational modeling. We conclude with a brief summary of the state of the field and the potential that integrating metabolomics presents.
2. Metabolomics Background
The development of metabolomics, the newest of the global analysis methods, has much in common with its predecessor fields of genomics, transcriptomics, and proteomics [41,42]. The analytical platforms used for metabolomics have now developed to the point that metabolomics datasets can serve as an excellent complement to standard metabolic engineering approaches. The goals of metabolic engineering ultimately focus on producing desired metabolites, and metabolomics offers a means of broadly and directly assessing how well a strain meets those goals. What follows is a cursory overview of metabolomics technologies and the most common ways that metabolomics data are interpreted and analyzed, provided as context for how metabolomics data can be used towards metabolic engineering efforts.
2.1. Analytical Platforms
One of the primary difficulties facing the development of metabolomics has been the staggering diversity of metabolites. Metabolites are substantially more chemically diverse than the subunit-based chemistries of DNA, RNA, and proteins, impeding the progress of metabolomics as a truly “omics” field that measures all metabolites. The entire genome and transcriptome can be (at least theoretically) surveyed using single platforms, from simple PCR to more exhaustive sequencing and microarrays, whereas metabolomics requires multiple analytical platforms to achieve complete coverage of all metabolites.
Common approaches involve coupling of a chromatographic separation to mass spectrometry, including gas chromatography-mass spectrometry (GC-MS) [6,25,28,29,34,46,47,48,49,50,51,52,53,54,55,56,57,58], liquid chromatography-mass spectrometry (LC-MS) [26,33,34,49,52,54,56,59,60,61,62,63], and capillary electrophoresis-mass spectrometry (CE-MS) [34,63,64,65]. Other common platforms include nuclear magnetic resonance (NMR) [28,43,66,67] and an assortment of direct injection-mass spectrometry methods [43,48,51,53,67]. Protocols for using these platforms are under constant development, and span sample processing and work-up [51,56,68], efforts to improve the quantitative reliability of measurements [33,56,61], and data processing software [69,70,71,72,73,74,75,76,77,78]. These software tools, along with those presented in subsequent sections of this manuscript, are summarized in Table 1 (though we emphasize that this list is far from exhaustive). A more extensive review of these platforms is available from Dunn et al. [79].
2.2. Data Analysis
As the youngest of the global analysis methods, metabolomics has drawn heavily from the data analysis techniques developed for transcriptomics and proteomics. Like these two fields, the datasets generated by metabolomics suffer from a “curse of dimensionality,” where there are far more variables than there are samples. Methods taken from transcriptomics and proteomics, as well as some derived from the field of chemometrics, have been used extensively to analyze metabolomics data as a result [41,42] (some examples given in Figure 1). Though metabolomics datasets are of high dimension, the connected nature of biochemical pathways and networks can often lead to strong underlying patterns in the data; multivariate techniques have proven effective at identifying these underlying factors even if individual effect sizes are too small to be detected by univariate analyses (e.g., Figure 1a), and so here we highlight several of the methods most widely adopted for metabolomics studies.
One of the most prominent methods for analysis of metabolomics data is Principal Components Analysis (PCA) (Figure 1b). This technique identifies the natural “axes” of variation in the dataset by constructing a series of orthogonal component axes from the original metabolite features. Each component is a weighted combination of the original metabolite measurements that provides the maximum possible variance in a single composite variable; the components are all mutually orthogonal. The weights of the original features for each component (“loadings”) and the projections of the samples onto the components (“scores”) can reveal putative biomarkers or lead to simplified separation between biological sample classes, respectively [41,48,49,57,68,80,81,82,83,84,85,86]. Notably, this is an unsupervised technique; PCA uses no information about sample classes in its calculations, and the user can try to identify clusters of data points before projecting class information onto the score plot.
A few examples of using PCA to reveal underlying patterns in metabolomics datasets include the characterization of extracellular culture conditions in Chinese Hamster Ovary (CHO) cell batch cultures [85], a study of the response of S. cerevisiae to very high gravity (VHG) fermentations [80], comparisons of metabolomes across mutant strains of S. cerevisiae [42,48,68,82], and analyses of Pseudomonas putida growth on various carbon sources [49,81]. In this context, the loadings from the components that capture the separation between sample classes (e.g., culture condition or strain) on the score plot provide information about which metabolites are important to each class. The magnitude of each metabolite’s loading coefficient, and the groups of metabolites with high loadings in components that capture separation, can be used to infer biological significance.
Much of the value of PCA comes from its dimensional reduction capabilities: typically the first few components contain biologically relevant information, and higher components contain variance due to noise or biological variability. The number of components that are “significant” is an open question, and depends predominantly on the dataset or even the specific downstream processing and applications [87]. Since the principal component scores are “optimal” lower-dimension projections of the original data, they can be used in place of the original data in subsequent analysis, such as Hierarchical Clustering Analysis (HCA, Figure 1c) [85]. For example, Barrett et al. performed PCA on a flux balance analysis solution space to identify a lower-dimension set of key reactions that form the underlying basis of the solution space [84].
Partial Least Squares (PLS) regression and discriminant analysis (PLS-DA, Figure 1d-f) are also common tools in metabolomics analysis. They are multivariate analogs of linear regression and linear discriminant analysis, respectively. They are constructed in a manner similar to PCA, but require response variables (e.g. titer, viability, or conversion) or a class label, respectively, to determine the component axes [88]. Again, assessment of metabolite loading coefficients in PLS-DA axes allows biological interpretability. In one representative application, Cajka et al. used PLS-DA to identify a set of compounds that could discriminate between different beers by their origin [67]. Kamei et al. used OPLS-DA, a variant of PLS-DA that constructs distinct predictive and orthogonal components that describe between-class and within-class variance, respectively, [89,90], to assess the effects of knockouts related to replicative lifespan in S. cerevisiae [86]. They found that a component corresponding to separation between short-lived and long-lived strains identified differences in TCA cycle metabolites as predictors of longevity. These are just a few examples of the increasingly prevalent applications of PLS-based techniques in the field.
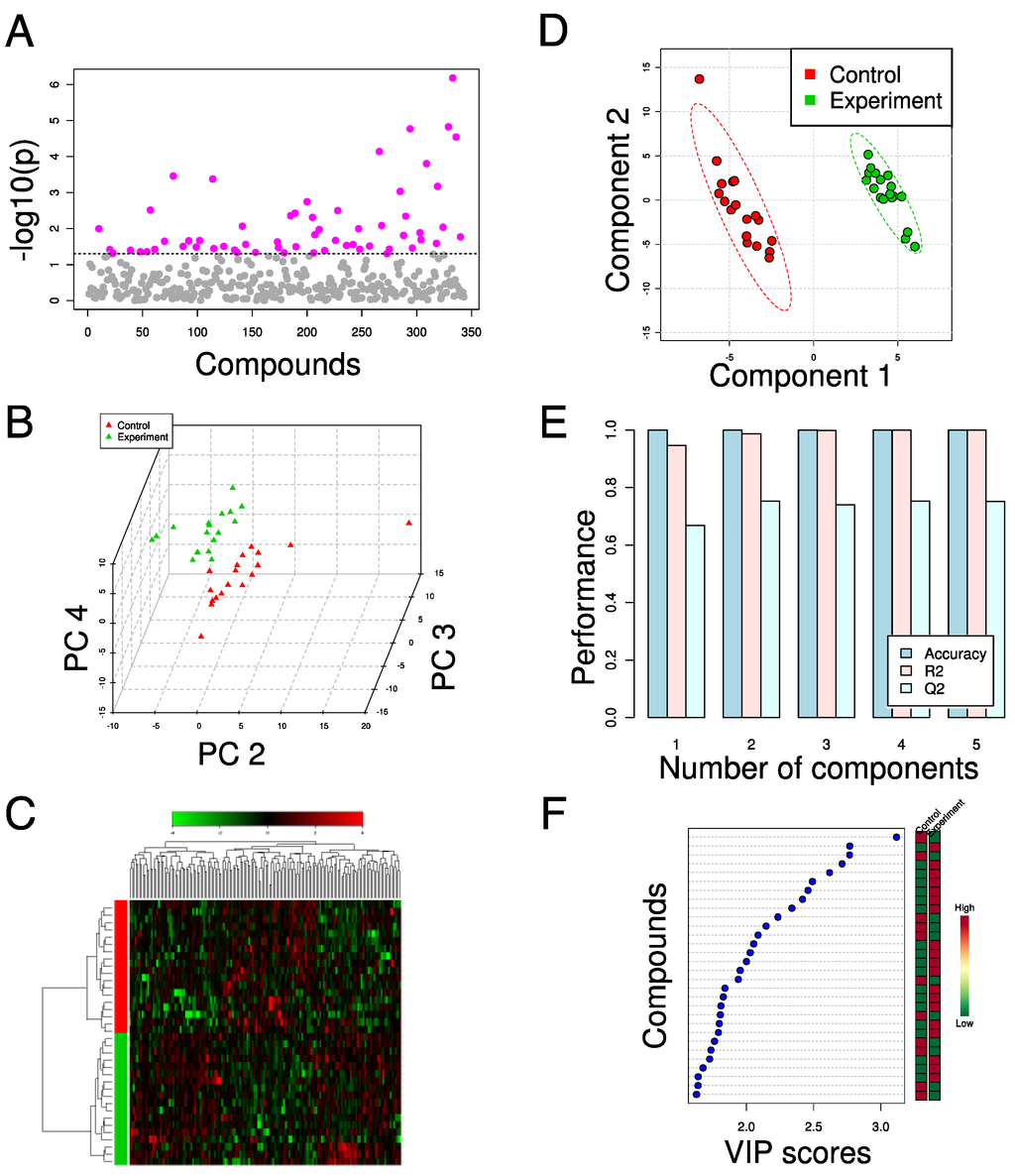
Figure 1.
Examples of data analysis techniques for metabolomics. The effects of glucose deprivation on a cancer cell line were measured with GC-MS and analyzed in MetaboAnalyst [78] (unpublished data). (a) Pairwise t-tests of metabolites identify statistical significance of differences in individual compounds between control and experiment. The dotted line indicates p < 0.05 (no multiple hypotheses testing corrections). (b) PCA score plot reveals separation between control and experiment samples in components 2, 3, and 4. Component 1 (not shown) corresponds to analytical batch separation. (c) HCA (Ward method, Pearson’s correlation) and heatmap using the 150 most significant compounds as determined by t-test. Compounds along top, samples along left side. (d) PLS-DA score plot shows separation achieved using components 1 and 2. Dashed circles indicate the 95% confidence interval for each class. (e) Leave-one-out cross-validation shows that the majority of the predictive capacity is derived from the first two PLS-DA components. R2 and Q2 denote, respectively, the goodness of fit and goodness of prediction statistics. (f) Contribution of individual compounds to PLS-DA component 1. The 30 most important compounds and their relative abundance in control and experiment are shown, sorted by the Variable Importance in the Projection (VIP) [91] for the first component.
Complete and effective use of a metabolomics dataset necessitates not only careful design of experiment and data processing methods, but also a thorough validation of conclusions from data analysis (e.g. apparent clusters in principal component space). For example, discussion of p-value distributions by Hojer-Pedersen et al. touches upon the importance of multiple hypothesis testing corrections in metabolomics studies, such as Bonferroni or false discovery rate corrections [82,92]. As a supervised method, PLS-DA is particularly susceptible to over-fitting, and so cross-validation is critical [93]. Statistical issues aside, non-biological factors can also lead to separation in principal component space, with sources of variance potentially including derivatization protocols [68,80,81], analytical platform [48], chromatographic drift or batch effects [94] and data processing methods [81]. Broadhurst and Kell review other potential pitfalls in greater detail [95].
3. Applications of Metabolomics in Metabolic Engineering
Metabolomics continues to be exploited for numerous biomedical applications, ranging from the study of differences between clinically isolated and industrial yeast strains [83], to blood or urine-based biomarkers for many human diseases, including diabetes [64,96], gallstone diseases [97], and multiple types of cancer [43,44,45] (Blekherman et al. provide a more comprehensive review of the applications of metabolomics to cancer biomarker discovery [98]). Metabolomics also has the potential for a significant biotechnological impact in metabolic engineering: as the goal of metabolic engineering is to manipulate metabolite production, metabolomics naturally lends itself to that goal. Moreover, organisms such as S. cerevisiae and E. coli have been studied extensively, providing a rich biological context in which the metabolome of strains derived from both rational design and directed evolution strategies can be interpreted and understood. Nonetheless, the use of metabolomics in metabolic engineering is not as prevalent as one might expect.
3.1. Metabolomics Data as an Extension of Small-Scale, Targeted Analysis
The simplest and most direct use of metabolomics datasets is as an extension of existing small-scale metabolite analyses; metabolomics inherently enables a more comprehensive assessment of a strain than a handful of narrowly selected measurements. Studies employing this approach typically either compare strains and culture conditions, or seek to monitor the time-course evolution of many metabolite concentrations in parallel. These studies use a combination of measured growth and production parameters in conjunction with direct examination of the metabolomics data (e.g. significant increases or decreases in metabolite levels) in the context of known biochemical pathways to determine the effects of mutations and culture conditions. For example, if one overexpresses the enzyme that is the first step in a linear biosynthetic pathway and finds that the first few metabolites accumulate significantly but subsequent metabolites do not, this may suggest a rate-limiting step further down the pathway that needs to be upregulated. Broader knowledge of metabolite levels beyond the target pathway can serve to determine the wider-ranging effects of a given metabolic engineering perturbation and can suggest candidate supplementary perturbations (to address, for example, cofactor imbalances).
One example of a strain- or condition-comparison approach is a study of an arcA mutant in E. coli by Toya et al., which compared parent and mutant responses to aerobic, anaerobic and nitrate-rich media conditions [65]. Through analysis of fold-changes in the metabolome, transcriptome, and 13C MFA-derived fluxes, they found significant differences in tricarboxylic acid (TCA) cycle metabolism and ATP production among conditions. Similarly, Christen et al. compared the metabolomic profiles of seven yeast species to assess differences in aerobic fermentation on glucose [62]. While 13C MFA suggested differences between TCA cycle fluxes and consistent flux through glycolysis, there was a much wider variation of metabolite levels across species – especially in amino acid pool compositions. They also found that across species, these values correlated poorly with fluxes.
In an example of time-course analysis, Hasunuma et al. studied the effects of acetic and formic acid, chemicals commonly found in lignocellulosic hydrolysates, on xylose-utilizing strains of S. cerevisiae [10]. They engineered strains to overexpress transaldolase (TAL) or transketolase (TKL), which are thought to control rate-limiting steps in the pentose phosphate pathway during xylose utilization. Differences in intracellular levels of pentose phosphate pathway metabolites between the controls and the mutants as determined from fold changes confirmed the pentose phosphate pathway as a key chokepoint. Compared to the parent strain, they found that the single mutants both exhibited improved growth characteristics and ethanol production, while the double mutant did not. A separate similar study also explored differences in xylose-utilizing strains [9].
Other work with S. cerevisiae has investigated the transient effects of redox perturbations [99] or relief from glucose limitation [26,100], as well as differences between S. cerevisiae and Pichia pastoris [56]. However, examples span a variety of organisms and culture conditions, from xylose utilization in A. niger [6] to the effects of extended culture periods [101] and low phenylacetic acid conditions after key pathway knockouts [29] on penicillin biosynthesis in Penicillium chrysogenum.
3.2. General Strategies for Integrating Metabolomics into Metabolic Engineering
The simple approaches used to exploit the results of targeted measurements can be scaled up to metabolomics datasets, but they often do not take full advantage of structures or patterns in the data at the systems level. Many in the field of metabolic engineering have used multivariate techniques to interrogate metabolomics datasets on more complicated questions about strain performance and metabolite allocation. Due to the complexity of biological systems, the answers to these questions are often non-intuitive and increasingly difficult to identify without taking such a systems-scale approach.
3.2.1. Adaptive Evolution and High Throughput Libraries: Locating the Cause of Improved Phenotypes
While rational design approaches were the original driving force in metabolic engineering, directed evolution and high throughput screens of mutant libraries have since become increasingly commonplace [3,4,8,11,12,102,103,104,105,106,107,108,109]. One of the main difficulties involved in these two “inverse metabolic engineering” approaches is the identification of the mutations responsible for the improved phenotype [110]. These frequently non-intuitive changes can often best be pinpointed with a direct, systems-scale readout of the metabolic state [107].
Common techniques employed in such approaches include HCA, PCA, and PLS-DA. These methods generate clusters or loadings that identify key metabolite differences, which in turn suggest what genetic changes may have been selected for. For example, Hong et al. used PCA and clustering analysis of metabolomics data, supplemented with transcriptional data, to investigate strains of S. cerevisiae that had been selected via directed evolution for improved galactose uptake [107]. They identified up-regulation of PGM2 as a common method for indirectly improving flux through the Leloir pathway for galactose utilization via relieved feed-forward inhibition of galactose-1-phospate and activation of reserve carbohydrate metabolism. The enrichment analysis methods used for the transcriptional data are discussed in further detail in the next section.
A study by Yoshida et al. examined metabolic differences between S. cerevisiae and Saccharomyces pastorianus in regards to SO2 and H2S production [111]. They generated metabolomic and transcriptomic datasets under typical beer fermentation conditions, and identified lowered transcription levels and increased levels of upstream metabolites for two reactions in S. cerevisiae that they subsequently hypothesized to be rate-limiting fluxes for desired production of H2S and SO2. They verified their prediction by exposing S. pastorianus to known inhibitors for the corresponding enzymes, which recapitulated this effect. They used this knowledge to engineer and test S. cerevisiae strains that relieved these bottlenecks, and then developed a mutant with similar properties via a directed evolution process for commercial use.
Similarly, Wisselink et al. investigated a xylose-utilizing strain of S. cerevisiae developed by introduction of L-arabinose pathway genes to an existing xylose-utilizing strain, followed by directed evolution to improve L-arabinose utilization [112]. They used fold-change and enrichment analysis of transcripts, coupled with thermodynamic analysis of reactions using metabolite mass action ratios and reaction driving forces, to identify an acquired response of the GAL regulon to arabinose levels as contributing to these improvements. They confirmed this discovery with subsequent knockouts of GAL2.
Other examples of using metabolomics for after-the-fact assessment of engineered strains include studying the effects of repeated exposure to vacuum fermentation conditions on S. cerevisiae [113], comparing evolved strains with knockouts proposed by the OptKnock algorithm [47], and identifying the differences between several yeast species during aerobic fermentation on glucose [62].
3.2.2. Other Global Analysis Approaches: Harnessing Proteomics, Transcriptomics, and Genomics for Metabolic Engineering
While the goal of metabolic engineering is to introduce a change on the metabolic level, many of these changes are necessarily implemented by introducing genetic modifications to affect transcriptional levels. As such, analysis of biological layers beyond the metabolome, such as the transcriptome and proteome, can provide further, and sometimes crucial, insight into the wide-reaching effects of an alteration.
A number of techniques widely used in metabolomics (such as PCA and HCA) are also well-established for many of these other “omics” datasets, though there are a number of other techniques that until recently were more specific to transcriptional or proteomic analyses. One of the most prominent examples of this is enrichment analysis, originally developed for transcriptomics datasets. Enrichment analysis uses information about the frequency of occurrence or the ranking of sets of gene names or functions in a given list of genes to examine the biological relevance of observed changes [114]. For example, the number of genes from a given pathway occurring in a list of interest (say, high-importance variables in PCA or a cluster from HCA) is assessed to see if that number of genes would be expected to be found in an arbitrary list of genes purely at random; this comparison is made using a hypergeometric distribution. If a list is statistically significantly “enriched” for a set of genes, one then may hypothesize that the list of genes plays an important role in the underlying biological process. This technique has recently been extended to metabolomics datasets [78,115]. A combination of enrichment, multivariate, and univariate analyses comprise the bulk of the strategies currently used in metabolic engineering to analyze “omics” datasets in parallel.
In metabolic engineering, use of metabolomics is comparatively much less common than using other global analysis approaches, perhaps attributable to the maturity of fields like transcriptomics and proteomics compared to metabolomics. Proteomics, transcriptomics and genomics have frequently been combined with small-scale metabolite measurements for metabolic engineering purposes. Examples of this include functional genomics with targeted metabolite measurements for isoprenoid production in E. coli [116], as well as the combination of the proteome, transcriptome and targeted metabolite measurements for E. coli carbon storage regulation [63], penicillin production in P. chrysogenum [101], glucose repression in S. cerevisiae [52], and relief from glucose deprivation in S. cerevisiae [100].
Flux measurements have also been commonly combined with other “omics” datasets, such as with the transcriptome and proteome in an analysis of lysine-producing Corynebacterium glutamicum during different stages of batch culture [46]. Other examples include the use of transcript data and 13C MFA to understand metabolic behavior in A. niger [7], transcriptional changes [117] and 13C MFA-derived fluxes [18] of C. acetobutylicum in response to stresses in bioreactors, and analysis of both systems-level MFA calculations [30] and Elementary Flux Modes [118] in tandem with transcriptomics data to assess metabolic control in S. cerevisiae.
The above examples at most used small-scale metabolite measurements, but a handful of studies have combined analysis of full metabolomics datasets with other “omics” datasets. Previously described analysis of adaptations from directed evolution generally fit this category: transcriptional measurements using microarrays [47,107,111,112] and genomic analysis [83] have each been combined with metabolomics to pinpoint the source of the observed phenotype.
In other applications, Piddock et al. assessed high gravity beer brewing conditions to determine the effect of the protease enzyme Flavourzyme on the free amino nitrogen content of the wort, and the resulting metabolomic and transcriptomic differences in a strain of brewer’s yeast [55]. A collaborative study by Canelas et al. investigated the growth characteristics of two strains of S. cerevisiae under two standard growth conditions. [66]. Datasets included intra- and extra-cellular metabolomes, the transcriptome, and the proteome, with an emphasis on reliable and quantitative measurements. They identified the specific growth rate and biomass yield differences between the strains that they attributed to differences in protein metabolism using analysis of variance (which determines statistical significance from comparisons of within-class and between-class variances) and gene enrichment analysis. Work by Dikicioglu et al. examined the combined metabolomic and transcriptomic response of S. cerevisiae to transient perturbations in glucose and ammonium concentrations [58]. Their work used HCA, t-tests, and analysis of fold-changes to study the short-term reorganization of the metabolome and transcriptome in response to temporary relief of nutrient-deprived conditions.
The emergence of genome-scale investigations has led to a deluge of information about all molecular layers in the cell. This in turn has provided a broader context in which metabolic engineering strategies can be evaluated. However, we note that many of the techniques discussed so far have focused on systematic assessment of the results of metabolic engineering strategies, rather than on systematic methods of designing strains to begin with. While some of these studies have used the insight gained from their evaluations to in turn design new strains, more systematic approaches to strain design are being developed – some of which are now capable of exploiting metabolomics datasets.
4. Computational Methods for Combining Metabolomics and Metabolic Engineering
One of the difficulties in applying metabolomics datasets to strain design is the volume of data produced in a metabolomics experiment. Computational approaches are well suited to systematically integrate large volumes of biochemical knowledge and data. As shown in Figure 2, they serve dual purposes: they can combine existing biochemical knowledge with strain design objectives to execute putative metabolic engineering strategies in silico before taking the time and expense to execute them in vivo, and they can close the loop on experimental design by producing hypotheses that, when tested, can be used iteratively to refine broader biochemical knowledge and models. This in turn leads to improved predictive power for subsequent rounds of metabolic engineering design.
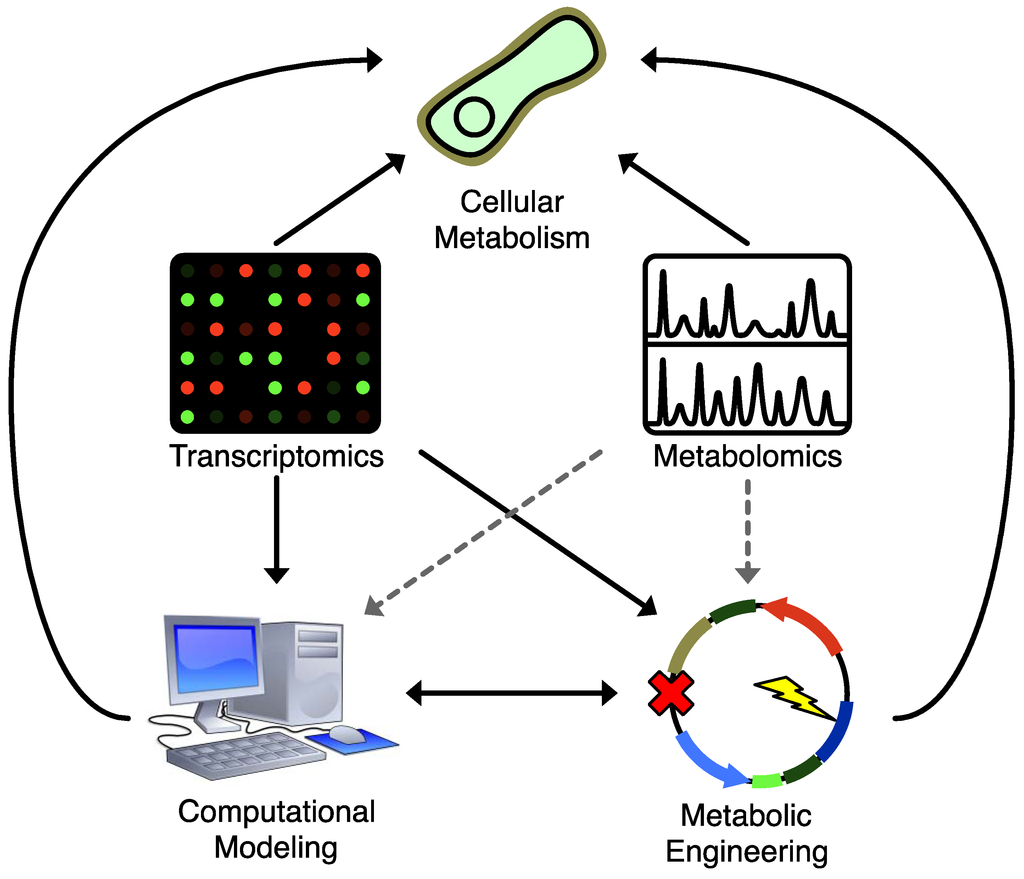
Figure 2.
Applications of various techniques to understanding and manipulating cellular metabolism. Solid lines represent widely used strategies, dashed lines represent underused strategies. Both metabolomics and transcriptional profiling provide a direct readout that helps enable a deeper understanding of cellular metabolism, but only transcriptional profiling has seen widespread application to enhance standard computational modeling and metabolic engineering strategies. Integrating metabolomics data into metabolic engineering and computational modeling strategies would help bridge gaps in biochemical knowledge and improve our ability to control cellular metabolism.
One of the most powerful ways to extend this concept would be to include metabolomic data in the design and fitting of computational models. While there are many well-developed metabolic modeling strategies, most of these approaches have not yet been adapted to effectively leverage the additional information that metabolomics can offer. Nonetheless, these strategies have made substantial contributions to metabolic engineering. We discuss these computational approaches to establish how they have been used to date in metabolic engineering, to suggest how metabolomics can contribute to their effectiveness, and to highlight current efforts to integrate the two.
4.1. Constraint-Based Models
The most basic models for metabolic engineering use simplified equations for bioreactor kinetics to empirically fit relationships between characteristics such as metabolite uptake or secretion and specific growth rate. While these models are useful as tools for investigating specific behaviors of existing strains, their small-scale and coarse-grained nature precludes broader application to directing engineering strategies, as well as the possibility of substantively integrating metabolomics data even when available [13].
Early biochemical modeling strategies initially sought to move beyond such simplistic approaches by compiling knowledge of metabolic pathways and enzyme kinetics into detailed mechanistic models to predict the dynamic behavior of metabolite concentrations [119]. However, numerous issues hampered these efforts. Incomplete knowledge of the regulatory structure or the form of reaction rate equations can limit the accuracy of these types of models [120]. More importantly, the necessary kinetic parameters are for the most part unknown, or have only been measured in vitro for specific organisms (although recent efforts have sought to develop methods to determine kinetic parameters relevant to in vivo conditions via selected intracellular metabolite measurements [121,122]). Additionally, many models in systems biology also exhibit “sloppy” behaviors in regard to parametric sensitivity, where model performance is sensitive only to certain parameter combinations and consistent parameter estimation is difficult even with sufficient data [123].
To attempt to overcome these issues, “constraint-based” approaches that calculate metabolic fluxes primarily from stoichiometry were developed. This change of focus from dynamic metabolite levels to fluxes made sense, as the idea of optimizing and controlling metabolic fluxes has long been a fundamental part of metabolic engineering. These approaches allow flux calculations without the difficulties arising from parametric uncertainty by predicting flux distributions from the structure of the biochemical network and constraints on the feasible range of fluxes [124,125].
4.1.1. Flux Balance Analysis: The Prototypical Constraint-based Model
Flux Balance Analysis (FBA) is, in short, a modeling technique that uses metabolic network stoichiometry, a set of feasible flux ranges, and a cellular objective function to calculate an optimal flux distribution for a metabolic network [124,125]. To achieve this, FBA invokes a pseudo-steady state assumption. (Based on the fact that metabolic reactions occur more quickly than upstream cellular processes like signal transduction, transcription, and translation, this may often be a reasonable simplification.) As a result, the non-linear set of differential equations describing the stoichiometric metabolite mass balance is reduced to a set of linear equations in terms of unknown reaction fluxes. Since the number of reactions exceeds the number of metabolites in biologically relevant metabolic networks, the system is underdetermined. Bounds on the solution space and an objective function are required to fully specify a unique solution. Selection of a linear objective function, such as maximization of a flux corresponding to biomass generation, specifies a linear programming (LP) problem (which can be efficiently solved numerically). By construction, FBA does not use information on metabolite concentrations themselves in its calculations.
A few examples of basic FBA for metabolic engineering include estimation of flux distributions from MFA measurements [31,126], prediction of knockout performances to assess proposed strain designs [5], use of artificial (virtual) metabolites to better capture flux ratios from 13C MFA [127], systematic evaluation of different objective functions in E. coli [128], and evaluation of Elementary Flux Modes from flux measurements [129] and transcription datasets [118]. A study of E. coli metabolism comparing growth in aerobic and anaerobic culture by Chen et al. highlights the complementary nature of FBA and MFA [130]. They note that FBA often performs well when predicting extracellular fluxes, but intracellular flux distributions are more difficult; it is important to compare fluxes derived from FBA calculations against actual fluxes as derived from measurements and MFA. Tools such as the COnstraints Based Reconstruction and Analysis (COBRA) toolbox for MATLAB [131], CellNetAnalyzer [132], the Systems Biology Research Tool (SBRT) [133], and OptFlux [134] are available for simplified implementation of constraint-based modeling methods (and are listed in Table 1).
4.1.2. Model Reconstructions
A prerequisite step in FBA is the reconstruction of genome-scale metabolic networks for the organism of interest. Reviews by Fiest et al. [135] and by Thiele and Palsson [136] describe this process in detail, and new strategies for efficiently or even automatically performing parts of this process are being continuously developed (listed in Table 1) [137,138,139,140]. The reconstruction process involves the synthesis of a genome-scale model from established biochemical and genomic knowledge, stored in publically accessible databases. These databases span various organisms and layers of biological information (e.g., biochemical pathways, transcription factors, and nutrient transport mechanisms). Examples of relevant databases for this process (as well as several for metabolomics in general) are also listed in Table 1.
A few recent examples particularly relevant to metabolic engineering and metabolomics include models of A. niger [141], C. acetobutylicum [138], and Clostridium beijerinckii [142], updated reconstructions of E. coli [143,144], and addition of lipid metabolism to a model for S. cerevisiae [145]. Notably, a reconstruction of Mycoplasma genitalium [126] has recently been incorporated into a whole-cell computational model by Karr et al. [146], who have suggested development of a similar model for E. coli as a possible next step. Network reconstructions for several dozen species are publicly available, and programs such as MetRxn have been developed to aid comparison across model reconstructions [147]. Some examples of these are shown in Table 1.
4.1.3. Applications of Constraint-based Models in General to Metabolic Engineering
The original FBA framework has been supplemented with dozens of refinements broadly referred to as constraint-based models. While these models retain the optimization problem framework based on stoichiometric constraints, the flux constraints or objective function are altered. We direct the reader to reviews on the topic of FBA by Lee, Gianchanani, and others for more complete discussion of these methods [148,149], though it is instructive to analyze a few representative classes relevant to metabolic engineering. Furthermore, we note that these approaches do not generally make use of metabolite measurements.
The most basic refinements are straightforward extensions of FBA, from adding a simplified representation of transcriptional regulatory constraints, to integrating uptake/effluxes and comparing against extracellular concentration profiles. Examples from this family include regulatory FBA (rFBA) [150,151,152], dynamic FBA (dFBA) [153,154], integrated FBA (iFBA) [154,155], and integrated-dynamic FBA (idFBA) [156]. While these refinements demonstrate improved accuracy over basic FBA, most dynamic examples only make use of targeted extracellular concentration profiles as a means of constraining their dynamic elements. Using metabolomics data to constrain FBA solutions could provide these strategies direct information about intracellular metabolite levels in place of relying purely on the calculated fluxes to infer intracellular behaviors. Similarly, regulatory versions of FBA have looked at transcriptional regulation, but not regulation at the metabolic level (e.g., allosteric regulation). Metabolomics can provide information necessary to capture these effects.
Another class of refinements to FBA comprises methods intended to better reduce the discrepancy between model predictions and experimental observations. Optimal metabolic network identification (OMNI) is used to identify discrepancies between measured and predicted fluxes, and then determine changes that need to be made to the model to better match the measurements [157]. As a proof of concept, it identified bottleneck reactions that, once removed from the model, accounted for reduced growth rates in evolved E. coli strains compared to the original model predictions. The GrowMatch algorithm uses growth and lethality data to correct a network reconstruction [158], and its application to a reconstruction of S. cerevisiae led to improved knockout lethality predictions. [159]. A number of these corrections were only identified by taking into account double gene deletion data, and improvements included growth-medium specific regulatory constraints. These (and other [160,161]) methods would also directly benefit from the additional information that metabolomics datasets can provide about the intracellular state of metabolism.
More directly relevant to metabolic engineering applications is a class of refinements focused on predicting the result of metabolic network alterations. An early and well-known example of this is Minimization of Metabolic Adjustment (MOMA), which formulates a quadratic programming (QP) problem to find the feasible flux distribution nearest to the original FBA solution in response to a gene knockout [162]. OptKnock uses a bi-level optimization framework to balance an FBA objective function with a desired overproduction target [163] (the work by Hua et al. compares the results of an evolved strain against an OptKnock prediction [47]). OptGene uses a genetic algorithm to generate metabolic engineering strategies [164], and was used by Asadollahi et al. to design a strain exhibiting improved sesquiterpene production in S. cerevisiae [165]. Another extension is OptForce, which uses flux measurement data to generate a minimal set of engineering interventions required to guarantee the desired overproduction target [166]. In addition to recapitulating already proven strategies for succinate production in E. coli, it also identified several other successful and nonintuitive strategies. Again, while none of these methods integrate metabolomics data into their calculations, they could potentially be improved significantly if they could harness such data.
4.1.4. Thermodynamic Constraints: Integrating Metabolomics Data into Constraint-based Models
As reviewed above, the majority of constraint-based modeling strategies make negligible use of systems-scale metabolite data in their calculations. The requirement that organisms adhere not only to stoichiometric mass conservation, but also to thermodynamic restrictions on energy and entropy, provides one means of introducing metabolite concentrations into the constraints. Several constraint-based model techniques make use of metabolite or metabolomics data in this fashion.
Network-embedded Thermodynamic (NET) Analysis combines pre-determined flux directions with quantitative metabolomics datasets and the metabolite Gibbs energy of formations to determine the feasible ranges of Gibbs free energy of reaction throughout the system [167]. This method can assess the internal consistency of a metabolomics dataset, predict thermodynamically feasible ranges for unmeasured metabolites, and identify putative sites of transcriptional regulation. anNET is a MATLAB implementation of the algorithm designed to facilitate straightforward application of NET analysis [168].
Table 1.
Summary of Software Tools Presented in This Manuscript.
| Tool Name | Reference | Description |
|---|---|---|
| Metabolomics Data Processing | ||
| ChromA | [72] | GC-MS Peak Alignment |
| Metab | [75] | GC-MS Data Statistical Analysis Package |
| MetaboAnalyst 2.0 | [78] | Web-based Metabolomics Data Processing Pipeline |
| MetAlign | [76] | GC-MS and LC-MS Data Processing Pipeline |
| Mzmine 2 | [74] | MS Data Processing Pipeline |
| SpectConnect | [71] | GC-MS Peak Alignment |
| Xalign | [69] | LC-MS Data Pre-processing |
| XCMS Online | [77] | Web-based Untargeted Metabolomics Pipeline |
| Constraint-Based Modeling | ||
| anNET | [168] | MATLAB-based NET analysis |
| CellNetAnalyzer | [132] | MATLAB-based Metabolic and Signal Network Analysis |
| COBRA Toolbox | [131] | MATLAB-based FBA Toolbox Suite |
| OptFlux | [134] | Open Source, Modular Constraint-based Model Strain Design Software Toolbox |
| Systems Biology Research Tool | [133] | Open Source, Modular Systems Biology Computational Tool |
| Network Reconstruction | ||
| GapFind, GapFill | [137] | Automated Network Gap Identification and Hypothesis Generation |
| GeneForce | [139] | Regulatory Rule Correction for Integrated Metabolic and Regulatory Models |
| MetRxn | [147] | Web-based Knowledgebase Comparison Tool |
| Model SEED | [140] | Web-based Generation, Optimization and Analysis of Genome-scale Metabolic Models |
| Databases | ||
| BioCyc | [169] | Genome and pathway database for >2000 organisms |
| BRENDA | [170] | Comprehensive enzyme database, ~5000 enzymes |
| ChEBI | [171] | Biologically relevant small molecules and their properties |
| KEGG | [172] | Genomes, enzymatic pathways, and biological chemicals |
| MetaCyc | [173] | >1,900 metabolic pathways from >2,200 different organisms |
| PubChem | [174] | Biological activity and structures of small molecules |
NET analysis of a metabolomics dataset for S. cerevisiae revealed a thermodynamic inconsistency in whole-cell NAD/NADH ratio, which led Canelas et al. to engineer a strain with a bacterial mannitol-1-phosphate 5-dehydrogenase to allow them to determine cytosolic levels from measurements of cytosolic pH, fructose-6-phosphate, and mannitol-1-phosphate [175]. They found that the ratio calculated from this method was consistent in NET analysis and displayed a sensitive response to redox perturbations. Other studies have also used NET analysis to verify the thermodynamic consistency of their measurements in a variety of metabolic engineering contexts [9,59,176].
Henry et al. developed Thermodynamic Metabolic Flux Analysis (TMFA), a constraint-based modeling approach similar to NET analysis [177]. While both methods calculate the feasible Gibbs energy of reaction ranges in the network, TMFA does not require user-supplied flux directions and by design excludes thermodynamically infeasible futile cycles from the resulting calculations. (It is worth noting, though, that the elimination of infeasible futile loops from the FBA solution space can be performed separately and without concentration data [178,179]). Henry et al. also discuss the use of group contribution methods to calculate Gibbs energy of formation and the effect of the associated estimation uncertainties on TMFA results [177]. Recent improvements to group contribution methods should improve this approach’s accuracy [180,181].
Garg et al. applied TMFA to 13C MFA calculations to produce thermodynamically consistent fluxes [182]. The E. coli network reconstruction published by Fiest et al. includes thermodynamic information, and the manuscript includes an assessment of thermodynamic consistency using TMFA [143]. Canelas et al. used TMFA as one criterion for identifying near-equilibrium reactions in an effort to use metabolomics datasets to determine in vivo thermodynamic (and kinetic) parameters [54].
While TMFA and NET analysis have been the prevailing approaches used to incorporate metabolite concentrations and thermodynamic constraints into constraint-based modeling approaches, several other methods have been developed. For example, Bordel et al. developed a constraint-based model based on Ziegler’s principle for the maximization of entropy production that uses non-equilibrium thermodynamics to identify flux bottlenecks; they identified reactions that impeded previous efforts to improve glycolytic flux via overexpression of glycolysis enzymes in S. cerevisiae [183]. Hoppe et al. designed a constraint-based model that combines thermodynamic constraints similar to TMFA and NET analysis with a penalty function for deviations from concentration measurements. The minimization of both the overall flux and the penalty function are used as the optimization criteria. In applying this new approach, they found that increased network size leads to increased sensitivity of the resulting flux distribution to metabolite concentrations and Gibbs free energy changes [184].
4.2. Kinetic Models
Constraint-based models have successfully directed numerous metabolic engineering projects. However, by construction they often ignore or have trouble dealing with dynamic metabolite behaviors that may have significant impact on final product titers, and in general they only indirectly make use of metabolite concentration measurements. Improved knowledge of network structures and strategies for dealing with parametric uncertainty have made ordinary differential equation (ODE) based models of metabolic kinetics increasingly viable tools for strain design. These methods explicitly model intracellular concentrations, making them attractive and convenient frameworks for integrating metabolomics datasets.
4.2.1. Recent Developments in Kinetic Modeling Strategies
Kinetic models are built around explicit mathematical descriptions of enzyme-metabolite interactions. Natural choices for kinetic rate laws are mass-action kinetics and Michaelis-Menten kinetics, but a review by Heijnen highlights several approximate rate laws that require fewer parameters and are relevant to metabolic engineering applications [185]. Included are discussions of the S-system and power-law kinetic rate laws, long established by the early efforts at kinetic modeling that developed into Biochemical Systems Theory, and reviewed specifically in the context of metabolic networks recently by Voit [186,187,188,189].
In light of recent genome-scale reconstruction efforts, several investigators have sought to assess the properties of several of these approximate forms in the context of metabolic networks. Examples of this approach include study of the glycolytic pathway in S. cerevisiae using a local linearization method [190] and lin-log kinetics [191,192], as well as study of central carbon metabolism in E. coli to compare lin-log kinetics, convenience kinetics, power law kinetics, and Michaelis-Menten kinetics [193]. This last study found that mixed models of Michaelis-Menten and lin-log kinetics are well suited for large-scale networks where the true rate laws are often still unknown. While these studies are not examples of using kinetic models for metabolic engineering or with metabolomics per se, they demonstrate the principles underlying the effective implementation of kinetic modeling for those purposes.
4.2.2. Examples of Kinetic Models for Metabolic Engineering
Independent of metabolomics, kinetic models have already been applied in several recent metabolic engineering contexts. Rasler et al. constructed a dynamical model of cellular redox state in S. cerevisiae to assess response to oxidative stress [194]. Literature kinetic values provided a qualitative fit to experimental data, and they achieved significantly improved quantitative matching by partially fitting parameter values (allowing them to vary by up to 4-fold from the literature values). Chassagnole et al. constructed a kinetic model of central carbon metabolism in E. coli in conjunction with MCA [120]. Their model captured most of the concentration trends, but even with parameter fitting, structural limitations of the model precluded complete agreement. A similar study by Oh et al. constructed a model of lactic acid fermentation in Lactococcus lactis, and the subsequent results were also used for MCA [20].
Ensemble approaches are also promising, and are in part a response to issues of parametric “sloppiness” which can preclude precise determination of kinetic parameters [123]. These entail constructing a set of models that are structurally identical, but each using a different parameter set. Each model fits the training data comparably well, and the behavior of the whole ensemble is used to make predictions. Tran et al. developed a model for central metabolism in E. coli as a proof of concept [195]. Starting with an ensemble spanning the entire thermodynamically feasible range of parameter combinations, they observed convergence to consistent fold-change behavior as training data were introduced. This effort led to an ensemble model constructed by Rizk et al. that predicted the effect of gene knockouts on the production of aromatic compounds in E. coli [196], and a model by Contador et al. to predict flux data in L-lysine-producing E. coli [197]. While these examples do not use metabolomics, we note that metabolomics datasets could in principle be easily adapted to this method and would be quite well-suited for further constraining the parameter ensembles.
4.2.3. Integrating Metabolomics Datasets into Kinetic Models
None of the aforementioned ODE models explicitly look to integrate metabolomics data into their analyses, due in part to the only recent development of more quantitative metabolomics techniques. While scarce, there have been some efforts to use metabolomics data to improve or exploit ODE-based models of metabolism. For example, Klimacek et al. used published kinetic parameters with time-course metabolomics measurements to assess metabolic control in xylose-fermenting S. cerevisiae [9]. Similarly, a model of nitrogen assimilation in E. coli developed by Yuan et al. used kinetic parameters from the literature together with a genetic algorithm to fit undefined parameters to metabolomics data [60]. Proteomic and transcriptomic data were also used to adjust reaction rate predictions. While these examples both use metabolomics data, the modeling strategies focused only on capturing the dynamics of individual pathways and modules – not the whole metabolic network.
Two recent examples have attempted to apply metabolomics measurements to models of an entire metabolic network. Yizhak et al. developed a constraint-based modeling approach they referred to as integrative “omics”-metabolic analysis (IOMA), which solves a QP problem in a constraint-based model to fit a flux distribution to proteomic and metabolomics data [198]. Their approach introduces a reaction rate model based on Michaelis-Menten kinetics, and uses proteomic data in conjunction with metabolomics data to fit the kinetic parameters and satisfy the steady-state flux requirement in the unperturbed system. This results in a defined system of ODEs. When compared against FBA and MOMA to predict the effects of gene knockouts based on an erythrocyte kinetic model and published data for E. coli, IOMA demonstrated significantly improved recall, precision, and accuracy.
A mass action stoichiometric simulation (MASS) modeling strategy described by Jamshidi et al. follows a different scheme, by fitting thermodynamic equilibrium constants to a measured metabolomics dataset and a calculated flux distribution [199]. The kinetic rate law equations are then solved to obtain the forward reaction rate constants. As a proof of concept, they used this method to construct a human erythrocyte model, and compared it against a previously published model. They also included mechanisms to account for regulation of five enzymes using mass-action kinetics. They assessed metabolite and protein level trajectories in response to redox load simulations, and performed sensitivity analysis to determine the dominant time scales within the model. The MASS model demonstrated many of the key behaviors of the original erythrocyte model.
Notably, these last two methods both take advantage of constraint-based modeling strategies, but result in ODE-based kinetic models that can subsequently be used for strain design. This reflects the complementary nature of metabolite fluxes and concentrations, especially when faced with system-wide parametric uncertainty. However, to fully capture the wide-ranging dynamics that directly and indirectly contribute to the often subtle and nonintuitive behaviors exhibited in engineered strains, additional model detail is necessary. More advanced modeling strategies will need to find ways to integrate additional information, including proteomics and transcriptomics, to meet this need.
5. Conclusions
Metabolomics is the global analysis of the metabolic content of a living system. While it has found increasing application in fundamental biological research and in fields of clinical interest (e.g. disease biomarker discovery), there is surprisingly little use of metabolomics approaches to drive metabolic engineering efforts. Existing experimental approaches to supplement rational metabolic engineering efforts typically focus instead on the determination of flux with MFA techniques, or the use of enzyme assays and analytical platforms such as HPLC for highly targeted metabolite measurements.
While global analysis methods have been used to better predict and assess the effects of metabolic engineering modifications, the techniques most typically used have been transcriptomic or proteomic analyses – not metabolomics. While this may have previously been due to the relative immaturity of metabolomics techniques, the current technology in the field should allow for easy integration of metabolomics into metabolic engineering workflows.
Direct applications of metabolomics datasets to metabolic engineering include expanding the existing narrowly targeted analysis methods to a broader scope, identifying non-intuitive mutations in strains produced by directed evolution, and adding direct metabolic context to other global analysis datasets. Computational approaches have also begun to integrate metabolomics datasets through thermodynamic constraints in constraint-based models, or even more directly in the case of some kinetic models.
However, long-term strategies will need to find novel ways of incorporating the system-wide perspective provided by metabolomics and other global analysis methods. Such approaches will facilitate strain design based on increasingly detailed mechanistic descriptions and enable us to engineer strains towards any arbitrary product, not just those well-suited to high-throughput screens and directed evolution. Computational methods have a great deal of potential here. In the case of kinetic models, combining the metabolome and proteome can help address issues of in vivo parameter estimation. Ensemble models are proving to be one effective method of addressing issues of parametric uncertainty and model “sloppiness”, and metabolomics provides substantial data to better constrain feasible parameter sets. With proper alterations and structural changes, constraint-based models may be able to more explicitly incorporate metabolite concentrations into constraints to capture effects such as allosteric regulation. We expect that these and other recently introduced approaches to integrate metabolomics data will yield substantial improvements in the efficiency and accuracy of prospective strain designs, both accelerating the pace and expanding the scope of developments in metabolic engineering. Nonetheless, it is clear that the depth and breadth of metabolic insight afforded by metabolomics is currently not being sufficiently exploited, and it is likely in such advances that we will make our most significant next steps as metabolic engineers.
Acknowledgments
The authors thank M. Smith and K. Vermeersch for helpful feedback on the manuscript. This work was supported by a DARPA Young Faculty Award and NSF award #1125684. RAD was also supported by the NSF Stem Cell Biomanufacturing IGERT program.
Conflict of Interest
The authors declare no conflict of interest.
References
- Stephanopoulos, G. Metabolic Fluxes and Metabolic Engineering. Metab. Eng. 1999, 1, 1–11. [Google Scholar] [CrossRef]
- Zaldivar, J.Z.; Borges, A.B.; Johansson, B.J.; Smits, H.S.; Villas-Bôas, S.V.-B.; Nielsen, J.N.; Olsson, L.O. Fermentation performance and intracellular metabolite patterns in laboratory and industrial xylose-fermenting Saccharomyces cerevisiae. Appl. Microbiol. Biot. 2002, 59, 436–442. [Google Scholar] [CrossRef]
- Sonderegger, M.; Sauer, U. Evolutionary Engineering of Saccharomyces cerevisiae for Anaerobic Growth on Xylose. Appl. Environ. Microb. 2003, 69, 1990–1998. [Google Scholar] [CrossRef]
- Sonderegger, M.; Jeppsson, M.; Hahn-Hägerdal, B.; Sauer, U. Molecular Basis for Anaerobic Growth of Saccharomyces cerevisiae on Xylose, Investigated by Global Gene Expression and Metabolic Flux Analysis. Appl. Environ. Microb. 2004, 70, 2307–2317. [Google Scholar] [CrossRef]
- Bro, C.; Regenberg, B.; Förster, J.; Nielsen, J. In silico aided metabolic engineering of Saccharomyces cerevisiae for improved bioethanol production. Metab. Eng. 2006, 8, 102–111. [Google Scholar] [CrossRef]
- Meijer, S.; Panagiotou, G.; Olsson, L.; Nielsen, J. Physiological characterization of xylose metabolism in Aspergillus niger under oxygen-limited conditions. Biotechnol. Bioeng. 2007, 98, 462–475. [Google Scholar] [CrossRef]
- Panagiotou, G.; Andersen, M.R.; Grotkjær, T.; Regueira, T.B.; Hofmann, G.; Nielsen, J.; Olsson, L. Systems Analysis Unfolds the Relationship between the Phosphoketolase Pathway and Growth in Aspergillus nidulans. PLoS ONE 2008, 3, e3847. [Google Scholar]
- Wisselink, H.W.; Toirkens, M.J.; Wu, Q.; Pronk, J.T.; van Maris, A.J.A. Novel Evolutionary Engineering Approach for Accelerated Utilization of Glucose, Xylose, and Arabinose Mixtures by Engineered Saccharomyces cerevisiae Strains. Appl. Environ. Microb. 2009, 75, 907–914. [Google Scholar] [CrossRef]
- Klimacek, M.; Krahulec, S.; Sauer, U.; Nidetzky, B. Limitations in Xylose-Fermenting Saccharomyces cerevisiae, Made Evident through Comprehensive Metabolite Profiling and Thermodynamic Analysis. Appl. Environ. Microb. 2010, 76, 7566–7574. [Google Scholar] [CrossRef]
- Hasunuma, T.; Sanda, T.; Yamada, R.; Yoshimura, K.; Ishii, J.; Kondo, A. Metabolic pathway engineering based on metabolomics confers acetic and formic acid tolerance to a recombinant xylose-fermenting strain of Saccharomyces cerevisiae. Microb. Cell Fact. 2011, 10, 2. [Google Scholar] [CrossRef]
- Koppram, R.; Albers, E.; Olsson, L. Evolutionary engineering strategies to enhance tolerance of xylose utilizing recombinant yeast to inhibitors derived from spruce biomass. Biotechnol. Biofuels 2012, 5, 32. [Google Scholar] [CrossRef]
- Zhang, W.; Geng, A. Improved ethanol production by a xylose-fermenting recombinant yeast strain constructed through a modified genome shuffling method. Biotechnol. Biofuels 2012, 5, 46. [Google Scholar] [CrossRef]
- Kresnowati, M.T.A.P.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Dynamic in vivo metabolome response of Saccharomyces cerevisiae to a stepwise perturbation of the ATP requirement for benzoate export. Biotechnol. Bioeng. 2008, 99, 421–441. [Google Scholar] [CrossRef]
- Sillers, R.; Chow, A.; Tracy, B.; Papoutsakis, E.T. Metabolic engineering of the non-sporulating, non-solventogenic Clostridium acetobutylicum strain M5 to produce butanol without acetone demonstrate the robustness of the acid-formation pathways and the importance of the electron balance. Metab. Eng. 2008, 10, 321–332. [Google Scholar] [CrossRef]
- Song, H.; Jang, S.H.; Park, J.M.; Lee, S.Y. Modeling of batch fermentation kinetics for succinic acid production by Mannheimia succiniciproducens. Biochem. Eng. J. 2008, 40, 107–115. [Google Scholar] [CrossRef]
- Zhang, K.; Sawaya, M.R.; Eisenberg, D.S.; Liao, J.C. Expanding metabolism for biosynthesis of nonnatural alcohols. Proc. Natl. Acad. Sci. USA 2008, 105, 20653–20658. [Google Scholar]
- Hou, J.; Lages, N.F.; Oldiges, M.; Vemuri, G.N. Metabolic impact of redox cofactor perturbations in Saccharomyces cerevisiae. Metab. Eng. 2009, 11, 253–261. [Google Scholar] [CrossRef]
- Sillers, R.; Al-Hinai, M.A.; Papoutsakis, E.T. Aldehyde-alcohol dehydrogenase and/or thiolase overexpression coupled with CoA transferase downregulation lead to higher alcohol titers and selectivity in Clostridium acetobutylicum fermentations. Biotechnol. Bioeng. 2009, 102, 38–49. [Google Scholar] [CrossRef]
- Trinh, C.T.; Huffer, S.; Clark, M.E.; Blanch, H.W.; Clark, D.S. Elucidating mechanisms of solvent toxicity in ethanologenic Escherichia coli. Biotechnol. Bioeng. 2010, 106, 721–730. [Google Scholar] [CrossRef]
- Oh, E.L.; Mingshou; Park, Changhun; Park, Changhun; Oh, Han Bin; Lee, Sang Yup; Lee, Jinwon. Dynamic Modeling of Lactic Acid Fermentation Metabolism with Lactococcus lactis. J. Microbiol. Biotechn. 2011, 21, 162–169. [Google Scholar] [CrossRef]
- Pereira, F.B.; Guimarães, P.M.R.; Teixeira, J.A.; Domingues, L. Robust industrial Saccharomyces cerevisiae strains for very high gravity bio-ethanol fermentations. J. Biosci. Bioeng. 2011, 112, 130–136. [Google Scholar] [CrossRef]
- Trinh, C.T.; Li, J.; Blanch, H.W.; Clark, D.S. Redesigning Escherichia coli Metabolism for Anaerobic Production of Isobutanol. Appl. Environ. Microb. 2011, 77, 4894–4904. [Google Scholar] [CrossRef]
- Aboka, F.O.; van Winden, W.A.; Reginald, M.M.; van Gulik, W.M.; van de Berg, M.; Oudshoorn, A.; Heijnen, J.J. Identification of informative metabolic responses using a minibioreactor: a small step change in the glucose supply rate creates a large metabolic response in Saccharomyces cerevisiae. Yeast 2012, 29, 95–110. [Google Scholar] [CrossRef]
- Lu, M.; Lee, S.; Kim, B.; Park, C.; Oh, M.; Park, K.; Lee, S.Y.; Lee, J. Identification of Factors Regulating Escherichia coli 2,3-Butanediol Production by Continuous Culture and Metabolic Flux Analysis. J. Microbiol. Biotechn. 2012, 22, 659–667. [Google Scholar] [CrossRef]
- Villas-Bôas, S.G.; Åkesson, M.; Nielsen, J. Biosynthesis of glyoxylate from glycine in Saccharomyces cerevisiae. FEMS Yeast Res. 2005, 5, 703–709. [Google Scholar] [CrossRef]
- Wu, L.; van Dam, J.; Schipper, D.; Kresnowati, M.T.A.P.; Proell, A.M.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Short-Term Metabolome Dynamics and Carbon, Electron, and ATP Balances in Chemostat-Grown Saccharomyces cerevisiae CEN.PK 113-7D following a Glucose Pulse. Appl. Environ. Microb. 2006, 72, 3566–3577. [Google Scholar]
- Costenoble, R.; Müller, D.; Barl, T.; Van Gulik, W.M.; Van Winden, W.A.; Reuss, M.; Heijnen, J.J. 13C-Labeled metabolic flux analysis of a fed-batch culture of elutriated Saccharomyces cerevisiae. FEMS Yeast Res. 2007, 7, 511–526. [Google Scholar] [CrossRef]
- Kleijn, R.J.; Geertman, J.-M.A.; Nfor, B.K.; Ras, C.; Schipper, D.; Pronk, J.T.; Heijnen, J.J.; Van Maris, A.J.A.; Van Winden, W.A. Metabolic flux analysis of a glycerol-overproducing Saccharomyces cerevisiae strain based on GC-MS, LC-MS and NMR-derived 13C-labelling data. FEMS Yeast Res. 2007, 7, 216–231. [Google Scholar] [CrossRef]
- Nasution, U.; van Gulik, W.M.; Ras, C.; Proell, A.; Heijnen, J.J. A metabolome study of the steady-state relation between central metabolism, amino acid biosynthesis and penicillin production in Penicillium chrysogenum. Metab. Eng. 2008, 10, 10–23. [Google Scholar] [CrossRef]
- Moxley, J.F.; Jewett, M.C.; Antoniewicz, M.R.; Villas-Boas, S.G.; Alper, H.; Wheeler, R.T.; Tong, L.; Hinnebusch, A.G.; Ideker, T.; Nielsen, J.; Stephanopoulos, G. Linking high-resolution metabolic flux phenotypes and transcriptional regulation in yeast modulated by the global regulator Gcn4p. Proc. Natl. Acad. Sci. USA 2009, 106, 6477–6482. [Google Scholar]
- Suthers, P.F.; Chang, Y.J.; Maranas, C.D. Improved computational performance of MFA using elementary metabolite units and flux coupling. Metab. Eng. 2010, 12, 123–128. [Google Scholar] [CrossRef]
- Ravikirthi, P.; Suthers, P.F.; Maranas, C.D. Construction of an E. Coli genome-scale atom mapping model for MFA calculations. Biotechnol. Bioeng. 2011, 108, 1372–1382. [Google Scholar] [CrossRef]
- Wu, L.; Mashego, M.R.; van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar]
- Büscher, J.r.M.; Czernik, D.; Ewald, J.C.; Sauer, U.; Zamboni, N. Cross-Platform Comparison of Methods for Quantitative Metabolomics of Primary Metabolism. Anal. Chem. 2009, 81, 2135–2143. [Google Scholar] [CrossRef]
- Choi, J.; Antoniewicz, M.R. Tandem mass spectrometry: A novel approach for metabolic flux analysis. Metab. Eng. 2011, 13, 225–233. [Google Scholar] [CrossRef]
- Choi, J.; Grossbach, M.T.; Antoniewicz, M.R. Measuring Complete Isotopomer Distribution of Aspartate Using Gas Chromatography/Tandem Mass Spectrometry. Anal. Chem. 2012, 84, 4628–4632. [Google Scholar]
- Srour, O.; Young, J.; Eldar, Y. Fluxomers: a new approach for 13C metabolic flux analysis. BMC Syst. Biol. 2011, 5, 129. [Google Scholar] [CrossRef]
- Chang, Y.; Suthers, P.F.; Maranas, C.D. Identification of optimal measurement sets for complete flux elucidation in metabolic flux analysis experiments. Biotechnol. Bioeng. 2008, 100, 1039–1049. [Google Scholar] [CrossRef]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Elementary metabolite units (EMU): A novel framework for modeling isotopic distributions. Metab. Eng. 2007, 9, 68–86. [Google Scholar] [CrossRef]
- Young, J.D.; Walther, J.L.; Antoniewicz, M.R.; Yoo, H.; Stephanopoulos, G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng. 2008, 99, 686–699. [Google Scholar] [CrossRef]
- Raamsdonk, L.M.; Teusink, B.; Broadhurst, D.; Zhang, N.; Hayes, A.; Walsh, M.C.; Berden, J.A.; Brindle, K.M.; Kell, D.B.; Rowland, J.J.; Westerhoff, H.V.; van Dam, K.; Oliver, S.G. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotech. 2001, 19, 45–50. [Google Scholar] [CrossRef]
- Allen, J.; Davey, H.M.; Broadhurst, D.; Heald, J.K.; Rowland, J.J.; Oliver, S.G.; Kell, D.B. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotech. 2003, 21, 692–696. [Google Scholar] [CrossRef]
- Chen, H.; Pan, Z.; Talaty, N.; Raftery, D.; Cooks, R.G. Combining desorption electrospray ionization mass spectrometry and nuclear magnetic resonance for differential metabolomics without sample preparation. Rapid Commun. Mass Sp. 2006, 20, 1577–1584. [Google Scholar] [CrossRef]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; Nyati, M.K.; Ahsan, A.; Kalyana-Sundaram, S.; Han, B.; Cao, X.; Byun, J.; Omenn, G.S.; Ghosh, D.; Pennathur, S.; Alexander, D.C.; Berger, A.; Shuster, J.R.; Wei, J.T.; Varambally, S.; Beecher, C.; Chinnaiyan, A.M. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar]
- Huang, Z.; Chen, Y.; Hang, W.; Gao, Y.; Lin, L.; Li, D.; Xing, J.; Yan, X. Holistic metabonomic profiling of urine affords potential early diagnosis for bladder and kidney cancers. Metabolomics 2012, 1–11. [Google Scholar]
- Krömer, J.O.; Sorgenfrei, O.; Klopprogge, K.; Heinzle, E.; Wittmann, C. In-Depth Profiling of Lysine-Producing Corynebacterium glutamicum by Combined Analysis of the Transcriptome, Metabolome, and Fluxome. J. Bacteriol. 2004, 186, 1769–1784. [Google Scholar] [CrossRef]
- Hua, Q.; Joyce, A.R.; Fong, S.S.; Palsson, B.Ø. Metabolic analysis of adaptive evolution for in silico-designed lactate-producing strains. Biotechnol. Bioeng. 2006, 95, 992–1002. [Google Scholar] [CrossRef]
- Mas, S.; Villas-Bôas, S.G.; Edberg Hansen, M.; Åkesson, M.; Nielsen, J. A comparison of direct infusion MS and GC-MS for metabolic footprinting of yeast mutants. Biotechnol. Bioeng. 2007, 96, 1014–1022. [Google Scholar] [CrossRef]
- van der Werf, M.J.; Overkamp, K.M.; Muilwijk, B.; Koek, M.M.; van der Werff-van der Vat, B.J.C.; Jellema, R.H.; Coulier, L.; Hankemeier, T. Comprehensive analysis of the metabolome of Pseudomonas putida S12 grown on different carbon sources. Mol. Biosyst. 2008, 4, 315–327. [Google Scholar] [CrossRef]
- Kleijn, R.J.; Buescher, J.M.; Le Chat, L.; Jules, M.; Aymerich, S.; Sauer, U. Metabolic Fluxes during Strong Carbon Catabolite Repression by Malate in Bacillus subtilis. J. Biol. Chem. 2010, 285, 1587–1596. [Google Scholar]
- Taymaz-Nikerel, H.; de Mey, M.; Ras, C.; ten Pierick, A.; Seifar, R.M.; van Dam, J.C.; Heijnen, J.J.; van Gulik, W.M. Development and application of a differential method for reliable metabolome analysis in Escherichia coli. Anal. Biochem. 2009, 386, 9–19. [Google Scholar]
- Kümmel, A.; Ewald, J.C.; Fendt, S.-M.; Jol, S.J.; Picotti, P.; Aebersold, R.; Sauer, U.; Zamboni, N.; Heinemann, M. Differential glucose repression in common yeast strains in response to HXK2 deletion. FEMS Yeast Res. 2010, 10, 322–332. [Google Scholar] [CrossRef]
- Taymaz-Nikerel, H.; van Gulik, W.M.; Heijnen, J.J. Escherichia coli responds with a rapid and large change in growth rate upon a shift from glucose-limited to glucose-excess conditions. Metab. Eng. 2011, 13, 307–318. [Google Scholar] [CrossRef]
- Canelas, A.B.; Ras, C.; ten Pierick, A.; van Gulik, W.M.; Heijnen, J.J. An in vivo data-driven framework for classification and quantification of enzyme kinetics and determination of apparent thermodynamic data. Metab. Eng. 2011, 13, 294–306. [Google Scholar] [CrossRef]
- Piddocke, M.; Fazio, A.; Vongsangnak, W.; Wong, M.; Heldt-Hansen, H.; Workman, C.; Nielsen, J.; Olsson, L. Revealing the beneficial effect of protease supplementation to high gravity beer fermentations using "-omics" techniques. Microb. Cell Fact. 2011, 10, 27. [Google Scholar] [CrossRef]
- Carnicer, M.; Canelas, A.; ten Pierick, A.; Zeng, Z.; van Dam, J.; Albiol, J.; Ferrer, P.; Heijnen, J.; van Gulik, W. Development of quantitative metabolomics for Pichia pastoris. Metabolomics 2012, 8, 284–298. [Google Scholar] [CrossRef]
- Carnicer, M.; Pierich, A.; Dam, J.; Heijnen, J.; Albiol, J.; Gulik, W.; Ferrer, P. Quantitative metabolomics analysis of amino acid metabolism in recombinant pichia pastoris under different oxygen availability conditions. Microb. Cell Fact. 2012, 11, 83. [Google Scholar]
- Dikicioglu, D.; Dunn, W.B.; Kell, D.B.; Kirdar, B.; Oliver, S.G. Short- and long-term dynamic responses of the metabolic network and gene expression in yeast to a transient change in the nutrient environment. Mol. Biosyst. 2012, 8, 1760–1774. [Google Scholar] [CrossRef]
- Fendt, S.-M.; Buescher, J.M.; Rudroff, F.; Picotti, P.; Zamboni, N.; Sauer, U. Tradeoff between enzyme and metabolite efficiency maintains metabolic homeostasis upon perturbations in enzyme capacity. Mol. Syst. Biol. 2010, 6, 356. [Google Scholar]
- Yuan, J.; Doucette, C.D.; Fowler, W.U.; Feng, X.-J.; Piazza, M.; Rabitz, H.A.; Wingreen, N.S.; Rabinowitz, J.D. Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol. Syst. Biol. 2009, 5, 302. [Google Scholar]
- Buescher, J.M.; Moco, S.; Sauer, U.; Zamboni, N. Ultrahigh Performance Liquid Chromatography−Tandem Mass Spectrometry Method for Fast and Robust Quantification of Anionic and Aromatic Metabolites. Anal. Chem. 2010, 82, 4403–4412. [Google Scholar] [CrossRef]
- Christen, S.; Sauer, U. Intracellular characterization of aerobic glucose metabolism in seven yeast species by 13C flux analysis and metabolomics. FEMS Yeast Res. 2011, 11, 263–272. [Google Scholar] [CrossRef]
- McKee, A.; Rutherford, B.; Chivian, D.; Baidoo, E.; Juminaga, D.; Kuo, D.; Benke, P.; Dietrich, J.; Ma, S.; Arkin, A.; Petzold, C.; Adams, P.; Keasling, J.; Chhabra, S. Manipulation of the carbon storage regulator system for metabolite remodeling and biofuel production in Escherichia coli. Microb. Cell Fact. 2012, 11, 79. [Google Scholar] [CrossRef]
- Hirayama, A.; Nakashima, E.; Sugimoto, M.; Akiyama, S.-i.; Sato, W.; Maruyama, S.; Matsuo, S.; Tomita, M.; Yuzawa, Y.; Soga, T. Metabolic profiling reveals new serum biomarkers for differentiating diabetic nephropathy. Anal. Bioanal. Chem. 2012, 404, 1–9. [Google Scholar] [CrossRef]
- Toya, Y.; Nakahigashi, K.; Tomita, M.; Shimizu, K. Metabolic regulation analysis of wild-type and arcA mutant Escherichia coli under nitrate conditions using different levels of omics data. Mol. Biosyst. 2012, 8, 2593–2604. [Google Scholar]
- Canelas, A.B.; Harrison, N.; Fazio, A.; Zhang, J.; Pitkanen, J.-P.; van den Brink, J.; Bakker, B.M.; Bogner, L.; Bouwman, J.; Castrillo, J.I.; Cankorur, A.; Chumnanpuen, P.; Daran-Lapujade, P.; Dikicioglu, D.; van Eunen, K.; Ewald, J.C.; Heijnen, J.J.; Kirdar, B.; Mattila, I.; Mensonides, F.I.C.; Niebel, A.; Penttila, M.; Pronk, J.T.; Reuss, M.; Salusjarvi, L.; Sauer, U.; Sherman, D.; Siemann-Herzberg, M.; Westerhoff, H.; de Winde, J.; Petranovic, D.; Oliver, S.G.; Workman, C.T.; Zamboni, N.; Nielsen, J. Integrated multilaboratory systems biology reveals differences in protein metabolism between two reference yeast strains. Nat. Commun. 2010, 1, 145. [Google Scholar]
- Cajka, T.; Riddellova, K.; Tomaniova, M.; Hajslova, J. Ambient mass spectrometry employing a DART ion source for metabolomic fingerprinting/profiling: a powerful tool for beer origin recognition. Metabolomics 2011, 7, 500–508. [Google Scholar] [CrossRef]
- Villas-Bôas, S.G.; Moxley, J.F.; Akesson, M.; Stephanopoulos, G.; Nielsen, J. High-throughput metabolic state analysis: the missing link in integrated functional genomics of yeasts. Biochem. J. 2005, 388, 669–677. [Google Scholar] [CrossRef]
- Zhang, X.; Asara, J.M.; Adamec, J.; Ouzzani, M.; Elmagarmid, A.K. Data pre-processing in liquid chromatography-mass spectrometry-based proteomics. Bioinformatics 2005, 21, 4054–4059. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O'Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Styczynski, M.P.; Moxley, J.F.; Tong, L.V.; Walther, J.L.; Jensen, K.L.; Stephanopoulos, G.N. Systematic Identification of Conserved Metabolites in GC/MS Data for Metabolomics and Biomarker Discovery. Anal. Chem. 2006, 79, 966–973. [Google Scholar]
- Hoffmann, N.; Stoye, J. ChromA: signal-based retention time alignment for chromatography-mass spectrometry data. Bioinformatics 2009, 25, 2080–2081. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 2010, 11, 395. [Google Scholar] [CrossRef]
- Aggio, R.; Villas−Bôas, S.G.; Ruggiero, K. Metab: an R package for high-throughput analysis of metabolomics data generated by GC-MS. Bioinformatics 2011, 27, 2316–2318. [Google Scholar] [CrossRef]
- Lommen, A.; Kools, H.J. MetAlign 3.0: performance enhancement by efficient use of advances in computer hardware. Metabolomics 2012, 8, 719–726. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef]
- Xia, J.; Mandal, R.; Sinelnikov, I.V.; Broadhurst, D.; Wishart, D.S. MetaboAnalyst 2.0—a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012, 40, W127–W133. [Google Scholar]
- Dunn, W.B.; Broadhurst, D.I.; Atherton, H.J.; Goodacre, R.; Griffin, J.L. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [Google Scholar]
- Devantier, R.; Scheithauer, B.; Villas-Bôas, S.G.; Pedersen, S.; Olsson, L. Metabolite profiling for analysis of yeast stress response during very high gravity ethanol fermentations. Biotechnol. Bioeng. 2005, 90, 703–714. [Google Scholar] [CrossRef]
- van den Berg, R.; Hoefsloot, H.; Westerhuis, J.; Smilde, A.; van der Werf, M. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics 2006, 7, 142. [Google Scholar] [CrossRef]
- Højer-Pedersen, J.; Smedsgaard, J.; Nielsen, J. The yeast metabolome addressed by electrospray ionization mass spectrometry: Initiation of a mass spectral library and its applications for metabolic footprinting by direct infusion mass spectrometry. Metabolomics 2008, 4, 393–405. [Google Scholar] [CrossRef]
- MacKenzie, D.A.; Defernez, M.; Dunn, W.B.; Brown, M.; Fuller, L.J.; de Herrera, S.R.M.S.; Günther, A.; James, S.A.; Eagles, J.; Philo, M.; Goodacre, R.; Roberts, I.N. Relatedness of medically important strains of Saccharomyces cerevisiae as revealed by phylogenetics and metabolomics. Yeast 2008, 25, 501–512. [Google Scholar] [CrossRef]
- Barrett, C.; Herrgard, M.; Palsson, B. Decomposing complex reaction networks using random sampling, principal component analysis and basis rotation. BMC Syst. Biol. 2009, 3, 30. [Google Scholar] [CrossRef]
- Chong, W.P.K.; Goh, L.T.; Reddy, S.G.; Yusufi, F.N.K.; Lee, D.Y.; Wong, N.S.C.; Heng, C.K.; Yap, M.G.S.; Ho, Y.S. Metabolomics profiling of extracellular metabolites in recombinant Chinese Hamster Ovary fed-batch culture. Rapid Commun. Mass Sp. 2009, 23, 3763–3771. [Google Scholar] [CrossRef]
- Kamei, Y.; Tamura, T.; Yoshida, R.; Ohta, S.; Fukusaki, E.; Mukai, Y. GABA metabolism pathway genes, UGA1 and GAD1, regulate replicative lifespan in Saccharomyces cerevisia. Biochem. Bioph. Res. Co. 2011, 407, 185–190. [Google Scholar] [CrossRef]
- Peres-Neto, P.R.; Jackson, D.A.; Somers, K.M. How many principal components? Stopping rules for determining the number of non-trivial axes revisited. Comput. Stat. Data. An. 2005, 49, 974–997. [Google Scholar] [CrossRef]
- Wold, S.; Ruhe, A.; Wold, H.; Dunn, I., W. The Collinearity Problem in Linear Regression. The Partial Least Squares (PLS) Approach to Generalized Inverses. SISC 1984, 5, 735–743. [Google Scholar]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemometr. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Wiklund, S.; Johansson, E.; Sjostrom, L.; Mellerowicz, E.J.; Edlund, U.; Shockcor, J.P.; Gottfries, J.; Moritz, T.; Trygg, J. Visualization of GC/TOF-MS-Based Metabolomics Data for Identification of Biochemically Interesting Compounds Using OPLS Class Models. Anal. Chem. 2007, 80, 115–122. [Google Scholar]
- Musumarra, G.; Barresi, V.; Condorelli, D.F.; Fortuna, C.G.; Scirè, S. Potentialities of multivariate approaches in genome-based cancer research: identification of candidate genes for new diagnostics by PLS discriminant analysis. J. Chemometr. 2004, 18, 125–132. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Westerhuis, J.; Hoefsloot, H.; Smit, S.; Vis, D.; Smilde, A.; van Velzen, E.; van Duijnhoven, J.; van Dorsten, F. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; Nicholls, A.W.; Wilson, I.D.; Kell, D.B.; Goodacre, R. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protocols 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Broadhurst, D.; Kell, D. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Culeddu, N.; Chessa, M.; Porcu, M.; Fresu, P.; Tonolo, G.; Virgilio, G.; Migaleddu, V. NMR-based metabolomic study of type 1 diabetes. Metabolomics 2012, 8, 1–8. [Google Scholar]
- Sonkar, K.; Behari, A.; Kapoor, V.; Sinha, N. 1H NMR metabolic profiling of human serum associated with benign and malignant gallstone diseases. Metabolomics 2012, 1–14. [Google Scholar]
- Blekherman, G.; Laubenbacher, R.; Cortes, D.; Mendes, P.; Torti, F.; Akman, S.; Torti, S.; Shulaev, V. Bioinformatics tools for cancer metabolomics. Metabolomics 2011, 7, 329–343. [Google Scholar] [CrossRef]
- Mashego, M.R.; Van Gulik, W.M.; Heijnen, J.J. Metabolome dynamic responses of Saccharomyces cerevisiae to simultaneous rapid perturbations in external electron acceptor and electron donor. FEMS Yeast Res. 2007, 7, 48–66. [Google Scholar] [CrossRef]
- Kresnowati, M.T.A.P.; van Winden, W.A.; Almering, M.J.H.; ten Pierick, A.; Ras, C.; Knijnenburg, T.A.; Daran-Lapujade, P.; Pronk, J.T.; Heijnen, J.J.; Daran, J.M. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Mol. Syst. Biol. 2006, 2, 49. [Google Scholar]
- Douma, R.; Batista, J.; Touw, K.; Kiel, J.; Krikken, A.; Zhao, Z.; Veiga, T.; Klaassen, P.; Bovenberg, R.; Daran, J.-M.; Heijnen, J.; van Gulik, W. Degeneration of penicillin production in ethanol-limited chemostat cultivations of Penicillium chrysogenum: A systems biology approach. BMC Syst. Biol. 2011, 5, 132. [Google Scholar] [CrossRef]
- Santos, C.N.S.; Stephanopoulos, G. Melanin-Based High-Throughput Screen for l-Tyrosine Production in Escherichia coli. Appl. Environ. Microb. 2008, 74, 1190–1197. [Google Scholar] [CrossRef]
- Tyo, K.E.; Zhou, H.; Stephanopoulos, G.N. High-Throughput Screen for Poly-3-Hydroxybutyrate in Escherichia coli and Synechocystis sp. Strain PCC6803. Appl. Environ. Microb. 2006, 72, 3412–3417. [Google Scholar] [CrossRef]
- Lee, S.K.; Chou, H.H.; Pfleger, B.F.; Newman, J.D.; Yoshikuni, Y.; Keasling, J.D. Directed Evolution of AraC for Improved Compatibility of Arabinose- and Lactose-Inducible Promoters. Appl. Environ. Microb. 2007, 73, 5711–5715. [Google Scholar]
- Atsumi, S.; Liao, J.C. Directed Evolution of Methanococcus jannaschii Citramalate Synthase for Biosynthesis of 1-Propanol and 1-Butanol by Escherichia coli. Appl. Environ. Microb. 2008, 74, 7802–7808. [Google Scholar] [CrossRef]
- Leonard, E.; Ajikumar, P.K.; Thayer, K.; Xiao, W.-H.; Mo, J.D.; Tidor, B.; Stephanopoulos, G.; Prather, K.L.J. Combining metabolic and protein engineering of a terpenoid biosynthetic pathway for overproduction and selectivity control. Proc. Natl. Acad. Sci. USA 2010, 107, 13654–13659. [Google Scholar]
- Hong, K.-K.; Vongsangnak, W.; Vemuri, G.N.; Nielsen, J. Unravelling evolutionary strategies of yeast for improving galactose utilization through integrated systems level analysis. Proc. Natl. Acad. Sci. USA 2011, 108, 12179–12184. [Google Scholar]
- Shen, C.R.; Lan, E.I.; Dekishima, Y.; Baez, A.; Cho, K.M.; Liao, J.C. Driving Forces Enable High-Titer Anaerobic 1-Butanol Synthesis in Escherichia coli. Appl. Environ. Microb. 2011, 77, 2905–2915. [Google Scholar] [CrossRef]
- Sun, Z.; Ning, Y.; Liu, L.; Liu, Y.; Sun, B.; Jiang, W.; Yang, C.; Yang, S. Metabolic engineering of the L-phenylalanine pathway in Escherichia coli for the production of S- or R-mandelic acid. Microb. Cell Fact. 2011, 10, 71. [Google Scholar] [CrossRef]
- Bailey, J.E.; Sburlati, A.; Hatzimanikatis, V.; Lee, K.; Renner, W.A.; Tsai, P.S. Inverse metabolic engineering: A strategy for directed genetic engineering of useful phenotypes. Biotechnol. Bioeng. 1996, 52, 109–121. [Google Scholar] [CrossRef]
- Yoshida, S.; Imoto, J.; Minato, T.; Oouchi, R.; Sugihara, M.; Imai, T.; Ishiguro, T.; Mizutani, S.; Tomita, M.; Soga, T.; Yoshimoto, H. Development of Bottom-Fermenting Saccharomyces Strains That Produce High SO2 Levels, Using Integrated Metabolome and Transcriptome Analysis. Appl. Environ. Microb. 2008, 74, 2787–2796. [Google Scholar]
- Wisselink, H.W.; Cipollina, C.; Oud, B.; Crimi, B.; Heijnen, J.J.; Pronk, J.T.; van Maris, A.J.A. Metabolome, transcriptome and metabolic flux analysis of arabinose fermentation by engineered Saccharomyces cerevisiae. Metab. Eng. 2010, 12, 537–551. [Google Scholar] [CrossRef]
- Ding, M.-Z.; Zhou, X.; Yuan, Y.-J. Metabolome profiling reveals adaptive evolution of Saccharomyces cerevisiae during repeated vacuum fermentations. Metabolomics 2010, 6, 42–55. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; Mesirov, J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar]
- Xia, J.; Wishart, D.S. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef]
- Kizer, L.; Pitera, D.J.; Pfleger, B.F.; Keasling, J.D. Application of Functional Genomics to Pathway Optimization for Increased Isoprenoid Production. Appl. Environ. Microb. 2008, 74, 3229–3241. [Google Scholar]
- Alsaker, K.V.; Paredes, C.; Papoutsakis, E.T. Metabolite stress and tolerance in the production of biofuels and chemicals: Gene-expression-based systems analysis of butanol, butyrate, and acetate stresses in the anaerobe Clostridium acetobutylicu. Biotechnol. Bioeng. 2010, 105, 1131–1147. [Google Scholar]
- Cakir, T.; Kirdar, B.; Onsan, Z.I.; Ulgen, K.; Nielsen, J. Effect of carbon source perturbations on transcriptional regulation of metabolic fluxes in Saccharomyces cerevisiae. BMC Syst. Biol. 2007, 1, 18. [Google Scholar] [CrossRef]
- Joshi, A.; Palsson, B.O. Metabolic dynamics in the human red cell: Part I—A comprehensive kinetic model. J. Theor. Biol. 1989, 141, 515–528. [Google Scholar] [CrossRef]
- Chassagnole, C.; Noisommit-Rizzi, N.; Schmid, J.W.; Mauch, K.; Reuss, M. Dynamic modeling of the central carbon metabolism of Escherichia coli. Biotechnol. Bioeng. 2002, 79, 53–73. [Google Scholar] [CrossRef]
- van Eunen, K.; Bouwman, J.; Daran-Lapujade, P.; Postmus, J.; Canelas, A.B.; Mensonides, F.I.C.; Orij, R.; Tuzun, I.; van den Brink, J.; Smits, G.J.; van Gulik, W.M.; Brul, S.; Heijnen, J.J.; de Winde, J.H.; Teixeira de Mattos, M.J.; Kettner, C.; Nielsen, J.; Westerhoff, H.V.; Bakker, B.M. Measuring enzyme activities under standardized in vivo-like conditions for systems biology. FEBS J 2010, 277, 749–760. [Google Scholar]
- van Eunen, K.; Kiewiet, J.A.L.; Westerhoff, H.V.; Bakker, B.M. Testing Biochemistry Revisited: How In Vivo Metabolism Can Be Understood from In Vitro Enzyme Kinetics. PLoS Comput. Biol. 2012, 8, e1002483. [Google Scholar] [CrossRef]
- Gutenkunst, R.N.; Waterfall, J.J.; Casey, F.P.; Brown, K.S.; Myers, C.R.; Sethna, J.P. Universally Sloppy Parameter Sensitivities in Systems Biology Models. PLoS Comput. Biol. 2007, 3, e189. [Google Scholar] [CrossRef]
- Savinell, J.M.; Palsson, B.O. Network analysis of intermediary metabolism using linear optimization. I. Development of mathematical formalism. J. Theor. Biol. 1992, 154, 421–454. [Google Scholar] [CrossRef]
- Varma, A.; Palsson, B.O. Metabolic Capabilities of Escherichia coli: I. Synthesis of Biosynthetic Precursors and Cofactors. J. Theor. Biol. 1993, 165, 477–502. [Google Scholar] [CrossRef]
- Suthers, P.F.; Burgard, A.P.; Dasika, M.S.; Nowroozi, F.; Van Dien, S.; Keasling, J.D.; Maranas, C.D. Metabolic flux elucidation for large-scale models using 13C labeled isotopes. Metab. Eng. 2007, 9, 387–405. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, T.Y.; Lee, D.-Y.; Lee, S.Y. Incorporating metabolic flux ratios into constraint-based flux analysis by using artificial metabolites and converging ratio determinants. J. Biotechnol. 2007, 129, 696–705. [Google Scholar]
- Schuetz, R.; Kuepfer, L.; Sauer, U. Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 2007, 3, 119. [Google Scholar]
- Nookaew, I.; Meechai, A.; Thammarongtham, C.; Laoteng, K.; Ruanglek, V.; Cheevadhanarak, S.; Nielsen, J.; Bhumiratana, S. Identification of flux regulation coefficients from elementary flux modes: A systems biology tool for analysis of metabolic networks. Biotechnol. Bioeng. 2007, 97, 1535–1549. [Google Scholar] [CrossRef]
- Chen, X.; Alonso, A.P.; Allen, D.K.; Reed, J.L.; Shachar-Hill, Y. Synergy between 13C-metabolic flux analysis and flux balance analysis for understanding metabolic adaption to anaerobiosis in E. coli. Metab. Eng. 2011, 13, 38–48. [Google Scholar]
- Hyduke, D.R.; Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; Kang, J.; Palsson, B.O. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protocols 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
- Klamt, S.; Saez-Rodriguez, J.; Gilles, E. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst. Biol. 2007, 1, 2. [Google Scholar]
- Wright, J.; Wagner, A. The Systems Biology Research Tool: evolvable open-source software. BMC Syst. Biol. 2008, 2, 55. [Google Scholar] [CrossRef]
- Rocha, I.; Maia, P.; Evangelista, P.; Vilaca, P.; Soares, S.; Pinto, J.; Nielsen, J.; Patil, K.; Ferreira, E.; Rocha, M. OptFlux: an open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 2010, 4, 45. [Google Scholar] [CrossRef]
- Feist, A.M.; Herrgard, M.J.; Thiele, I.; Reed, J.L.; Palsson, B.O. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Micro. 2009, 7, 129–143. [Google Scholar]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protocols 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Satish Kumar, V.; Dasika, M.; Maranas, C. Optimization based automated curation of metabolic reconstructions. BMC Bioinformatics 2007, 8, 212. [Google Scholar] [CrossRef]
- Senger, R.S.; Papoutsakis, E.T. Genome-scale model for Clostridium acetobutylicum: Part I. Metabolic network resolution and analysis. Biotechnol. Bioeng. 2008, 101, 1036–1052. [Google Scholar] [CrossRef]
- Barua, D.; Kim, J.; Reed, J.L. An Automated Phenotype-Driven Approach (GeneForce) for Refining Metabolic and Regulatory Models. PLoS Comput. Biol. 2010, 6, e1000970. [Google Scholar] [CrossRef]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotech. 2010, 28, 977–982. [Google Scholar]
- Andersen, M.R.; Nielsen, M.L.; Nielsen, J. Metabolic model integration of the bibliome, genome, metabolome and reactome of Aspergillus niger. Mol. Syst. Biol. 2008, 4, 178. [Google Scholar]
- Milne, C.; Eddy, J.; Raju, R.; Ardekani, S.; Kim, P.-J.; Senger, R.; Jin, Y.-S.; Blaschek, H.; Price, N. Metabolic network reconstruction and genome-scale model of butanol-producing strain Clostridium beijerinckii NCIMB 8052. BMC Syst. Biol. 2011, 5, 130. [Google Scholar] [CrossRef]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.O. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121. [Google Scholar]
- Orth, J.D.; Conrad, T.M.; Na, J.; Lerman, J.A.; Nam, H.; Feist, A.M.; Palsson, B.O. A comprehensive genome-scale reconstruction of Escherichia coli metabolism-2011. Mol. Syst. Biol. 2011, 7, 535. [Google Scholar]
- Nookaew, I.; Jewett, M.; Meechai, A.; Thammarongtham, C.; Laoteng, K.; Cheevadhanarak, S.; Nielsen, J.; Bhumiratana, S. The genome-scale metabolic model iIN800 of Saccharomyces cerevisiae and its validation: a scaffold to query lipid metabolism. BMC Syst. Biol. 2008, 2, 71. [Google Scholar] [CrossRef]
- Karr, Jonathan R.; Sanghvi, Jayodita C.; Macklin, Derek N.; Gutschow, Miriam V.; Jacobs, Jared M.; Bolival Jr, B.; Assad-Garcia, N.; Glass, John I.; Covert, Markus W. A Whole-Cell Computational Model Predicts Phenotype from Genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef]
- Kumar, A.; Suthers, P.; Maranas, C. MetRxn: a knowledgebase of metabolites and reactions spanning metabolic models and databases. BMC Bioinformatics 2012, 13, 6. [Google Scholar] [CrossRef]
- Lee, J.M.; Gianchandani, E.P.; Papin, J.A. Flux balance analysis in the era of metabolomics. Brief. Bioinf. 2006, 7, 140–150. [Google Scholar] [CrossRef]
- Gianchandani, E.P.; Chavali, A.K.; Papin, J.A. The application of flux balance analysis in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 372–382. [Google Scholar] [CrossRef]
- Covert, M.W.; Schilling, C.H.; Palsson, B. Regulation of Gene Expression in Flux Balance Models of Metabolism. J. Theor. Biol. 2001, 213, 73–88. [Google Scholar]
- Covert, M.W.; Palsson, B.Ø. Transcriptional Regulation in Constraints-based Metabolic Models of Escherichia coli. J. Biol. Chem. 2002, 277, 28058–28064. [Google Scholar]
- Meadows, A.L.; Karnik, R.; Lam, H.; Forestell, S.; Snedecor, B. Application of dynamic flux balance analysis to an industrial Escherichia coli fermentation. Metab. Eng. 2010, 12, 150–160. [Google Scholar] [CrossRef]
- Mahadevan, R.; Edwards, J.S.; Doyle Iii, F.J. Dynamic Flux Balance Analysis of Diauxic Growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Feng, X.; Xu, Y.; Chen, Y.; Tang, Y.J. Integrating Flux Balance Analysis into Kinetic Models to Decipher the Dynamic Metabolism of Shewanella oneidensis MR-1. PLoS Comput. Biol. 2012, 8, e1002376. [Google Scholar] [CrossRef]
- Covert, M.W.; Xiao, N.; Chen, T.J.; Karr, J.R. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 2008, 24, 2044–2050. [Google Scholar] [CrossRef]
- Lee, J.M.; Gianchandani, E.P.; Eddy, J.A.; Papin, J.A. Dynamic Analysis of Integrated Signaling, Metabolic, and Regulatory Networks. PLoS Comput. Biol. 2008, 4, e1000086. [Google Scholar] [CrossRef]
- Herrgård, M.J.; Fong, S.S.; Palsson, B.Ø. Identification of Genome-Scale Metabolic Network Models Using Experimentally Measured Flux Profiles. PLoS Comput. Biol. 2006, 2, e72. [Google Scholar] [CrossRef]
- Kumar, V.S.; Maranas, C.D. GrowMatch: An Automated Method for Reconciling In Silico/In Vivo Growth Predictions. PLoS Comput. Biol. 2009, 5, e1000308. [Google Scholar] [CrossRef]
- Zomorrodi, A.; Maranas, C. Improving the iMM904 S. cerevisiae metabolic model using essentiality and synthetic lethality data. BMC Syst. Biol. 2010, 4, 178. [Google Scholar] [CrossRef]
- Oh, Y.-G.; Lee, D.-Y.; Lee, S.Y.; Park, S. Multiobjective flux balancing using the NISE method for metabolic network analysis. Biotechnol. Progr. 2009, 25, 999–1008. [Google Scholar] [CrossRef]
- Adadi, R.; Volkmer, B.; Milo, R.; Heinemann, M.; Shlomi, T. Prediction of Microbial Growth Rate versus Biomass Yield by a Metabolic Network with Kinetic Parameters. PLoS Comput. Biol. 2012, 8, e1002575. [Google Scholar] [CrossRef]
- Segrè, D.; Vitkup, D.; Church, G.M. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. USA 2002, 99, 15112–15117. [Google Scholar]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef]
- Patil, K.; Rocha, I.; Forster, J.; Nielsen, J. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinformatics 2005, 6, 308. [Google Scholar]
- Asadollahi, M.A.; Maury, J.; Patil, K.R.; Schalk, M.; Clark, A.; Nielsen, J. Enhancing sesquiterpene production in Saccharomyces cerevisiae through in silico driven metabolic engineering. Metab. Eng. 2009, 11, 328–334. [Google Scholar] [CrossRef]
- Ranganathan, S.; Suthers, P.F.; Maranas, C.D. OptForce: An Optimization Procedure for Identifying All Genetic Manipulations Leading to Targeted Overproductions. PLoS Comput. Biol. 2010, 6, e1000744. [Google Scholar] [CrossRef]
- Kummel, A.; Panke, S.; Heinemann, M. Putative regulatory sites unraveled by network-embedded thermodynamic analysis of metabolome data. Mol. Syst. Biol. 2006, 2, 2006.0034. [Google Scholar]
- Zamboni, N.; Kummel, A.; Heinemann, M. anNET: a tool for network-embedded thermodynamic analysis of quantitative metabolome data. BMC Bioinformatics 2008, 9, 199. [Google Scholar] [CrossRef]
- Karp, P.D.; Ouzounis, C.A.; Moore-Kochlacs, C.; Goldovsky, L.; Kaipa, P.; Ahrén, D.; Tsoka, S.; Darzentas, N.; Kunin, V.; López-Bigas, N. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 2005, 33, 6083–6089. [Google Scholar]
- Schomburg, I.; Chang, A.; Schomburg, D. BRENDA, enzyme data and metabolic information. Nucleic Acids Res. 2002, 30, 47–49. [Google Scholar] [CrossRef]
- de Matos, P.; Alcántara, R.; Dekker, A.; Ennis, M.; Hastings, J.; Haug, K.; Spiteri, I.; Turner, S.; Steinbeck, C. Chemical Entities of Biological Interest: an update. Nucleic Acids Res. 2010, 38, D249–D254. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; Walk, T.C.; Zhang, P.; Karp, P.D. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. Chapter 12 PubChem: Integrated Platform of Small Molecules and Biological Activities. In Annu. Rep. Comput. Chem.; Ralph, A.W., David, C.S., Eds.; Elsevier, 2008; Volume 4, pp. 217–241. [Google Scholar]
- Canelas, A.B.; van Gulik, W.M.; Heijnen, J.J. Determination of the cytosolic free NAD/NADH ratio in Saccharomyces cerevisiae under steady-state and highly dynamic conditions. Biotechnol. Bioeng. 2008, 100, 734–743. [Google Scholar] [CrossRef]
- Jol, S.J.; Kümmel, A.; Terzer, M.; Stelling, J.; Heinemann, M. System-Level Insights into Yeast Metabolism by Thermodynamic Analysis of Elementary Flux Modes. PLoS Comput. Biol. 2012, 8, e1002415. [Google Scholar] [CrossRef]
- Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamics-Based Metabolic Flux Analysis. Biophys. J. 2007, 92, 1792–1805. [Google Scholar] [CrossRef]
- Price, N.D.; Famili, I.; Beard, D.A.; Palsson, B.Ø. Extreme Pathways and Kirchhoff's Second Law. Biophys. J. 2002, 83, 2879–2882. [Google Scholar] [CrossRef]
- Schellenberger, J.; Lewis, N.E.; Palsson, B.Ø. Elimination of Thermodynamically Infeasible Loops in Steady-State Metabolic Models. Biophys. J. 2011, 100, 544–553. [Google Scholar]
- Jankowski, M.D.; Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Group Contribution Method for Thermodynamic Analysis of Complex Metabolic Networks. Biophys. J. 2008, 95, 1487–1499. [Google Scholar] [CrossRef]
- Finley, S.D.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamic analysis of biodegradation pathways. Biotechnol. Bioeng. 2009, 103, 532–541. [Google Scholar] [CrossRef]
- Garg, S.; Yang, L.; Mahadevan, R. Thermodynamic analysis of regulation in metabolic networks using constraint-based modeling. BMC Research Notes 2010, 3, 125. [Google Scholar] [CrossRef]
- Bordel, S.; Nielsen, J. Identification of flux control in metabolic networks using non-equilibrium thermodynamics. Metab. Eng. 2010, 12, 369–377. [Google Scholar] [CrossRef]
- Hoppe, A.; Hoffmann, S.; Holzhutter, H.-G. Including metabolite concentrations into flux balance analysis: thermodynamic realizability as a constraint on flux distributions in metabolic networks. BMC Syst. Biol. 2007, 1, 23. [Google Scholar] [CrossRef]
- Heijnen, J.J. Approximative kinetic formats used in metabolic network modeling. Biotechnol. Bioeng. 2005, 91, 534–545. [Google Scholar] [CrossRef]
- Savageau, M.A. Biochemical systems analysis. I. Some mathematical properties of the rate law for the component enzymatic reactions. J. Theor. Biol. 1969, 25, 365–369. [Google Scholar] [CrossRef]
- Savageau, M.A. Mathematics of organizationally complex systems. Biomed. Biochim. Acta 1985, 44, 839–844. [Google Scholar]
- Voit, E.O.; Savageau, M.A. Accuracy of alternative representations for integrated biochemical systems. Biochemistry 1987, 26, 6869–6880. [Google Scholar] [CrossRef]
- Voit, E.O. Modelling metabolic networks using power-laws and S-systems. Essays Biochem. 2008, 45, 29–40. [Google Scholar] [CrossRef]
- Steuer, R.; Gross, T.; Selbig, J.; Blasius, B. Structural kinetic modeling of metabolic networks. Proc. Natl. Acad. Sci. USA 2006, 103, 11868–11873. [Google Scholar] [CrossRef]
- Nikerel, I.E.; van Winden, W.; van Gulik, W.; Heijnen, J. A method for estimation of elasticities in metabolic networks using steady state and dynamic metabolomics data and linlog kinetics. BMC Bioinformatics 2006, 7, 540. [Google Scholar] [CrossRef]
- Nikerel, I.E.; van Winden, W.A.; Verheijen, P.J.T.; Heijnen, J.J. Model reduction and a priori kinetic parameter identifiability analysis using metabolome time series for metabolic reaction networks with linlog kinetics. Metab. Eng. 2009, 11, 20–30. [Google Scholar] [CrossRef]
- Costa, R.S.; Machado, D.; Rocha, I.; Ferreira, E.C. Hybrid dynamic modeling of Escherichia coli central metabolic network combining Michaelis-Menten and approximate kinetic equations. Biosystems 2010, 100, 150–157. [Google Scholar] [CrossRef]
- Ralser, M.; Wamelink, M.; Kowald, A.; Gerisch, B.; Heeren, G.; Struys, E.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; Krobitsch, S. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 2007, 6, 10. [Google Scholar] [CrossRef]
- Tran, L.M.; Rizk, M.L.; Liao, J.C. Ensemble Modeling of Metabolic Networks. Biophys. J. 2008, 95, 5606–5617. [Google Scholar] [CrossRef]
- Rizk, M.L.; Liao, J.C. Ensemble Modeling for Aromatic Production in Escherichia coli. PLoS ONE 2009, 4, e6903. [Google Scholar]
- Contador, C.A.; Rizk, M.L.; Asenjo, J.A.; Liao, J.C. Ensemble modeling for strain development of l-lysine-producing Escherichia coli. Metab. Eng. 2009, 11, 221–233. [Google Scholar] [CrossRef]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar] [CrossRef]
- Jamshidi, N.; Palsson, B.Ø. Mass Action Stoichiometric Simulation Models: Incorporating Kinetics and Regulation into Stoichiometric Models. Biophys. J. 2010, 98, 175–185. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).