
RAGE Knockout Mitigates Diet-Induced Obesity and Metabolic Disruption

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Animals and Tissue Preparation
2.2. Body Composition and Adipocyte Morphology
2.3. Metabolic and Energy Expenditure Assessments
2.4. Mitochondrial Respiration Analysis
2.5. Inflammatory Marker Analysis
2.6. Statistical Analysis
3. Results
3.1. RAGE KO Mice Are Protected from Fat Gain on Western Diet
3.2. Adipocyte Size Differs Across Conditions
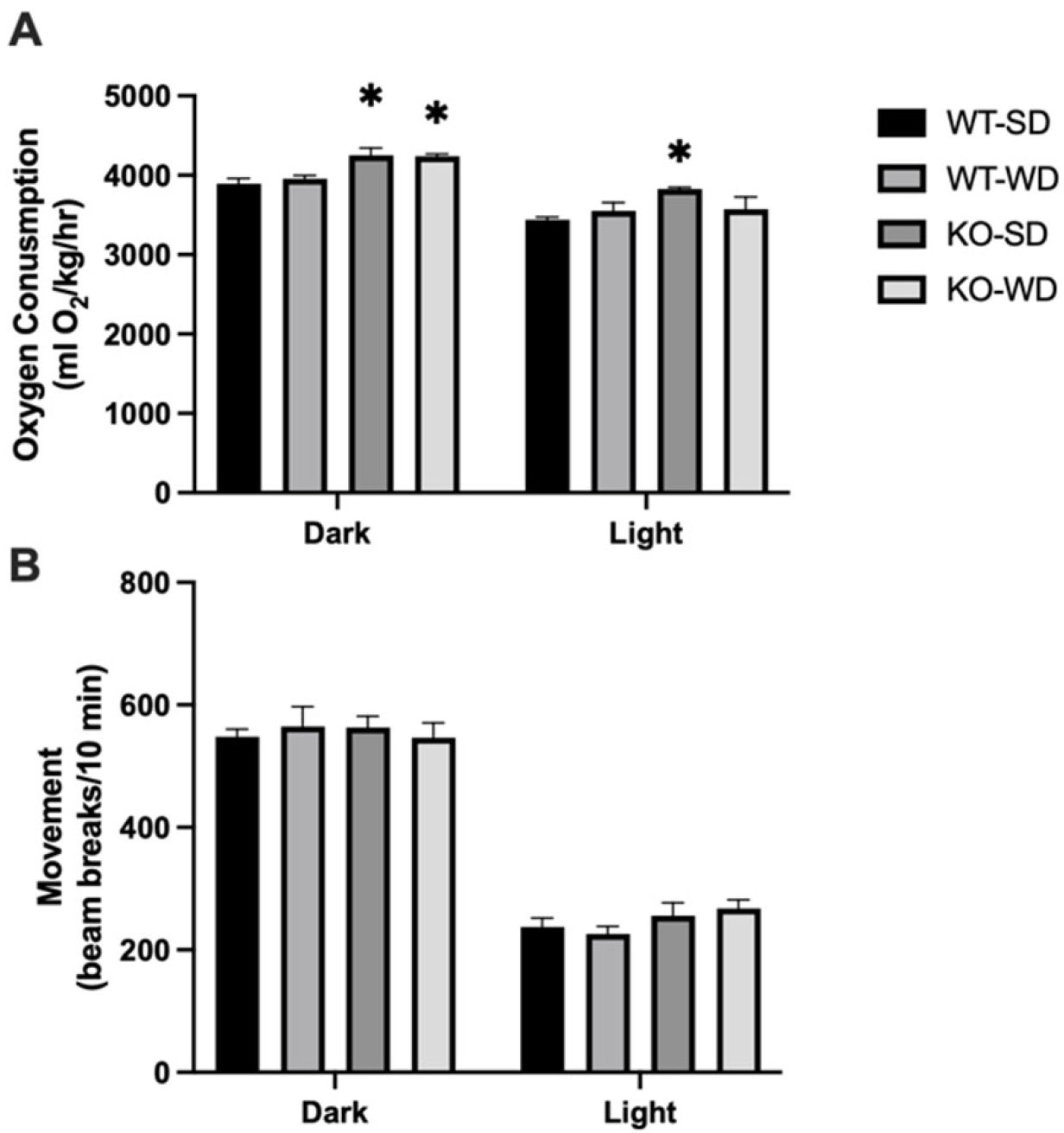
3.3. Increased Energy Expenditure in RAGE KO Mice
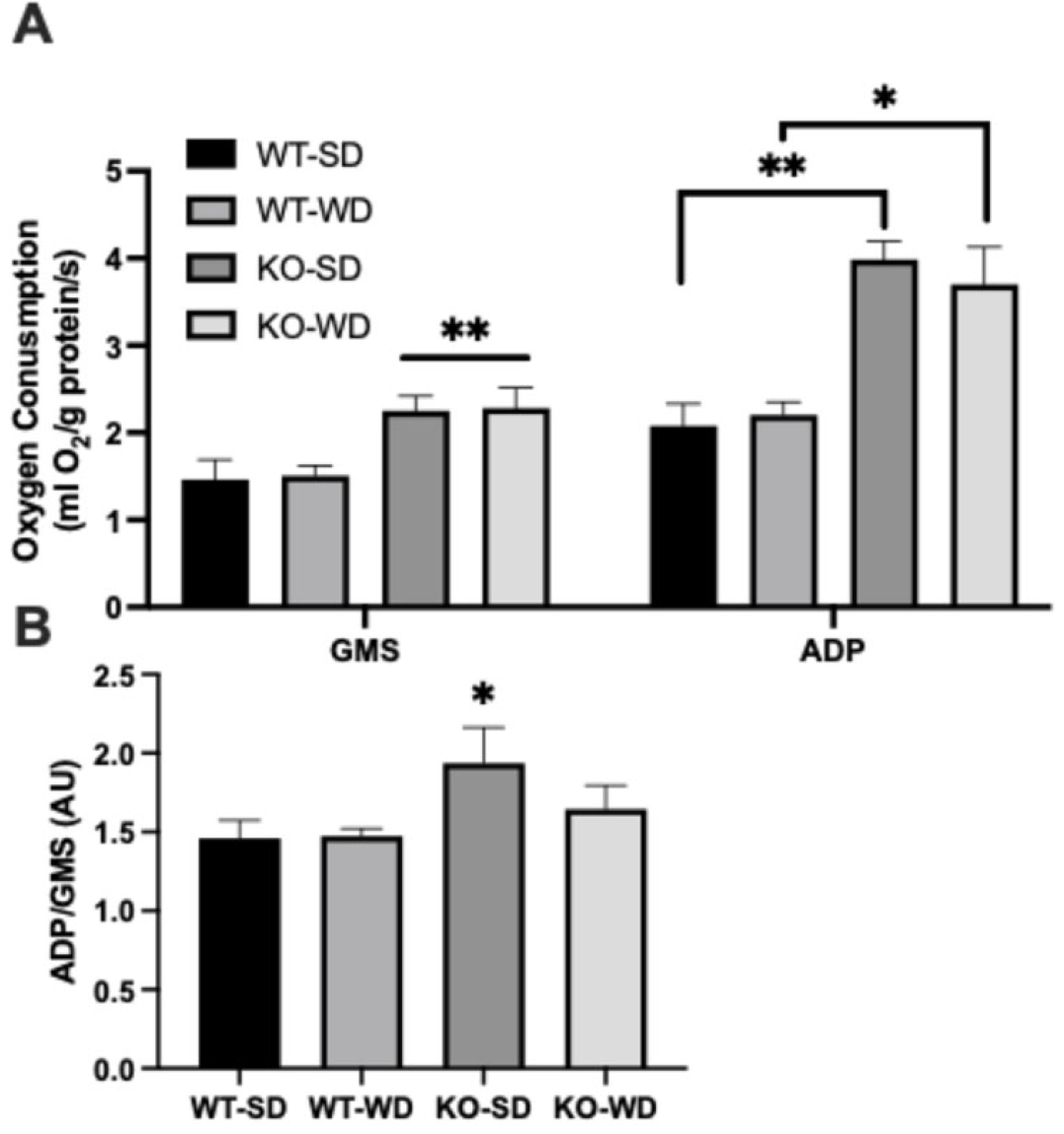
3.4. Altered Mitochondrial Bioenergetics in RAGE KO Mice
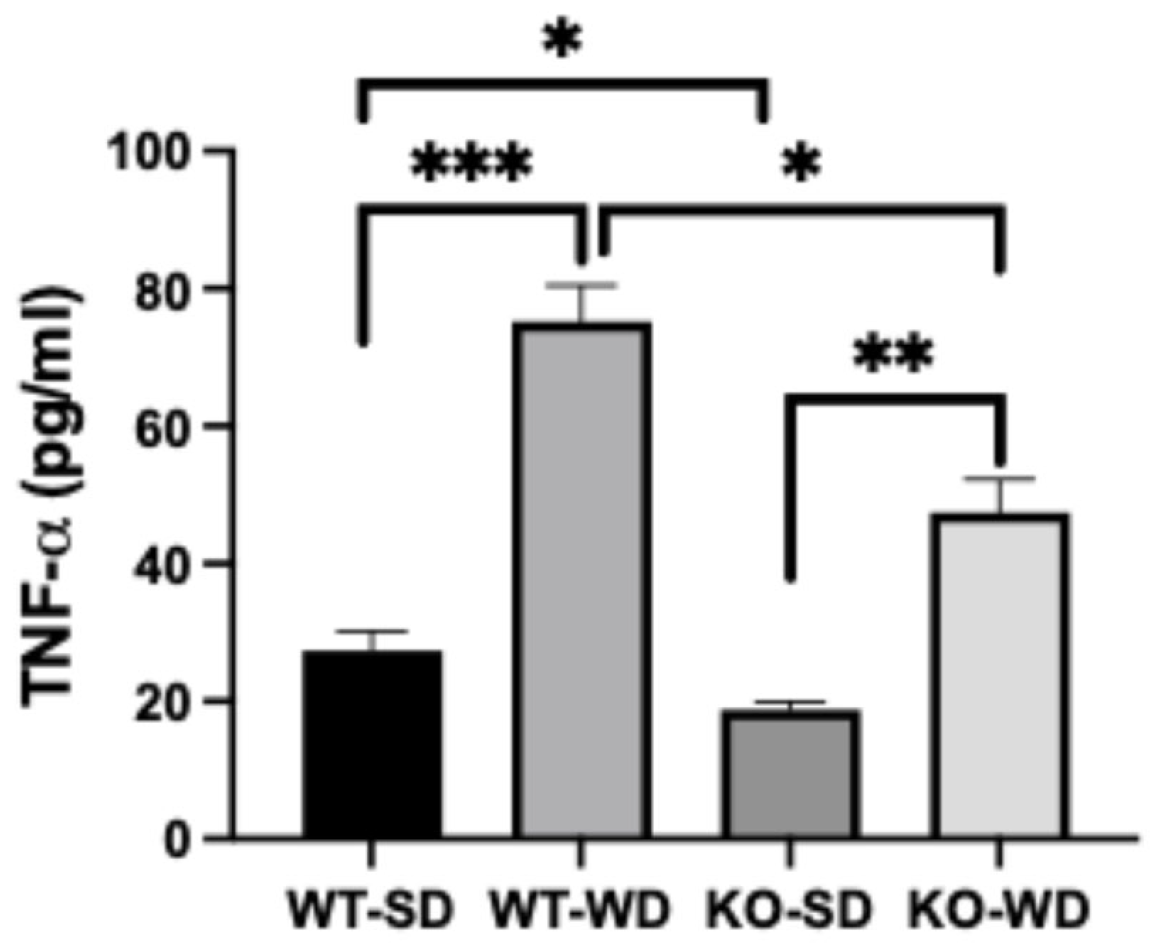
3.5. Reduced Systemic Inflammation in RAGE KO Mice
4. Discussion
4.1. RAGE Deletion Confers Protection Against Obesity and Inflammation
4.2. Enhanced Energy Expenditure and Mitochondrial Bioenergetics in RAGE KO Mice
4.3. Future Direction
4.4. Translational Potential and Therapeutic Implications
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bays, H.E.; Gonzalez-Campoy, J.M.; Bray, G.A.; Kitabchi, A.E.; Bergman, D.A.; Schorr, A.B.; Rodbard, H.W.; Henry, R.R. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert Rev. Cardiovasc. Ther. 2008, 6, 343–368. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Postina, R. Regulated proteolysis of RAGE and AbetaPP as possible link between type 2 diabetes mellitus and Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2009, 16, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Bro, S.; Flyvbjerg, A.; Binder, C.J.; Bang, C.A.; Denner, L.; Olgaard, K.; Nielsen, L.B. A neutralizing antibody against receptor for advanced glycation end products (RAGE) reduces atherosclerosis in uremic mice. Atherosclerosis 2008, 201, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Heier, M.; Margeirsdottir, H.D.; Gaarder, M.; Stensaeth, K.H.; Brunborg, C.; Torjesen, P.A.; Seljeflot, I.; Hanssen, K.F.; Dahl-Jorgensen, K. Soluble RAGE and atherosclerosis in youth with type 1 diabetes: A 5-year follow-up study. Cardiovasc. Diabetol. 2015, 14, 126. [Google Scholar] [CrossRef] [PubMed]
- Sanders, N.T.; Dutson, D.J.; Durrant, J.W.; Lewis, J.B.; Wilcox, S.H.; Winden, D.R.; Arroyo, J.A.; Bikman, B.T.; Reynolds, P.R. Cigarette smoke extract (CSE) induces RAGE-mediated inflammation in the Ca9-22 gingival carcinoma epithelial cell line. Arch. Oral Biol. 2017, 80, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, P.; Chatzopoulou, M.; Pegklidou, K.; Demopoulos, V.J. RAGE: A multi-ligand receptor unveiling novel insights in health and disease. Curr. Med. Chem. 2010, 17, 2232–2252. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Qian, J.; Zhang, Q.; Hu, Y.; Sun, D.; Jiang, L. Advanced glycation end products increased placental vascular permeability of human BeWo cells via RAGE/NF-kB signaling pathway. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 250, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Du, Z.; Shu, X.; Zhu, L.; Wu, J.; Gao, Q.; Wang, L.; Chen, N.; Li, Y.; Luo, M.; et al. Role of RAGE in obesity-induced adipose tissue inflammation and insulin resistance. Cell Death Discov. 2021, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Hurtado del Pozo, C.; Rosario, R.; Zou, Y.S.; Ananthakrishnan, R.; Xu, X.; Patel, P.R.; Benoit, V.M.; Yan, S.F.; Li, H.; et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes 2014, 63, 1948–1965. [Google Scholar] [CrossRef] [PubMed]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Hey-Mogensen, M.; Clausen, T.R. Targeting Mitochondrial Biogenesis and Mitochondrial Substrate Utilization to Treat Obesity and Insulin Resistance, Respectively—Two Data-Driven Hypotheses. Curr. Diabetes Rev. 2017, 13, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.S.; Decker, S.T.; Zhao, J.; Hoidal, J.R.; Heuckstadt, T.; Sanders, K.A.; Richardson, R.S.; Layec, G. The receptor for advanced glycation end products (RAGE) is involved in mitochondrial function and cigarette smoke-induced oxidative stress. Free Radic. Biol. Med. 2023, 195, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.E.; Campbell, K.M.; Kirkham, M.N.; Saito, E.R.; Remund, N.P.; Cayabyab, K.B.; Kim, I.J.; Heimuli, M.S.; Reynolds, P.R.; Arroyo, J.A.; et al. The Effect of Diesel Exhaust Particles on Adipose Tissue Mitochondrial Function and Inflammatory Status. Int. J. Mol. Sci. 2024, 25, 4322. [Google Scholar] [CrossRef] [PubMed]
- Curtis, K.L.; Homer, K.M.; Wendt, R.A.; Stapley, B.M.; Clark, E.T.; Harward, K.; Chang, A.; Clarke, D.M.; Arroyo, J.A.; Reynolds, P.R. Inflammatory Cytokine Elaboration Following Secondhand Smoke (SHS) Exposure Is Mediated in Part by RAGE Signaling. Int. J. Mol. Sci. 2023, 24, 5645. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Yin, J.Y.; Li, D.F.; Zhu, C.T.; Ye, J.P.; Pan, Y.Q. Effects of inflammatory and anti-inflammatory environments on the macrophage mitochondrial function. Sci. Rep. 2020, 10, 20324. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef] [PubMed]
- Todosenko, N.; Khaziakhmatova, O.; Malashchenko, V.; Yurova, K.; Bograya, M.; Beletskaya, M.; Vulf, M.; Gazatova, N.; Litvinova, L. Mitochondrial Dysfunction Associated with mtDNA in Metabolic Syndrome and Obesity. Int. J. Mol. Sci. 2023, 24, 12012. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmer, I.L.; Parker, G.; Chiu, A.T.; Beus, C.G.; Evans, E.P.; Radford, J.H.; Braithwaite, C.R.; van Slooten, R.D.; Cooper-Leavitt, E.T.; Moore, Z.E.; et al. RAGE Knockout Mitigates Diet-Induced Obesity and Metabolic Disruption. Metabolites 2025, 15, 524. https://doi.org/10.3390/metabo15080524
Palmer IL, Parker G, Chiu AT, Beus CG, Evans EP, Radford JH, Braithwaite CR, van Slooten RD, Cooper-Leavitt ET, Moore ZE, et al. RAGE Knockout Mitigates Diet-Induced Obesity and Metabolic Disruption. Metabolites. 2025; 15(8):524. https://doi.org/10.3390/metabo15080524
Chicago/Turabian StylePalmer, Isabelle L., Genevieve Parker, Alden T. Chiu, Colson G. Beus, Ethan P. Evans, Jack H. Radford, Cameron R. Braithwaite, Ryan D. van Slooten, Elijah T. Cooper-Leavitt, Zachary E. Moore, and et al. 2025. "RAGE Knockout Mitigates Diet-Induced Obesity and Metabolic Disruption" Metabolites 15, no. 8: 524. https://doi.org/10.3390/metabo15080524
APA StylePalmer, I. L., Parker, G., Chiu, A. T., Beus, C. G., Evans, E. P., Radford, J. H., Braithwaite, C. R., van Slooten, R. D., Cooper-Leavitt, E. T., Moore, Z. E., Clarke, D. M., Parrish, R. R., Arroyo, J. A., Reynolds, P. R., & Bikman, B. T. (2025). RAGE Knockout Mitigates Diet-Induced Obesity and Metabolic Disruption. Metabolites, 15(8), 524. https://doi.org/10.3390/metabo15080524