
Development and Validation of a New LC-MS/MS Method for the Assay of Plasmatic Peripheral Short- and Medium-Chain Fatty Acids for Metabolomics Applications

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Equipment
2.3. LC-MS Analysis Method
2.4. Calibration and Quality Control (QC) Standard Solutions for Plasma Sample Analysis
2.5. Preparation of Biological Plasma Samples
2.6. Validation of the Analytical Method
3. Results
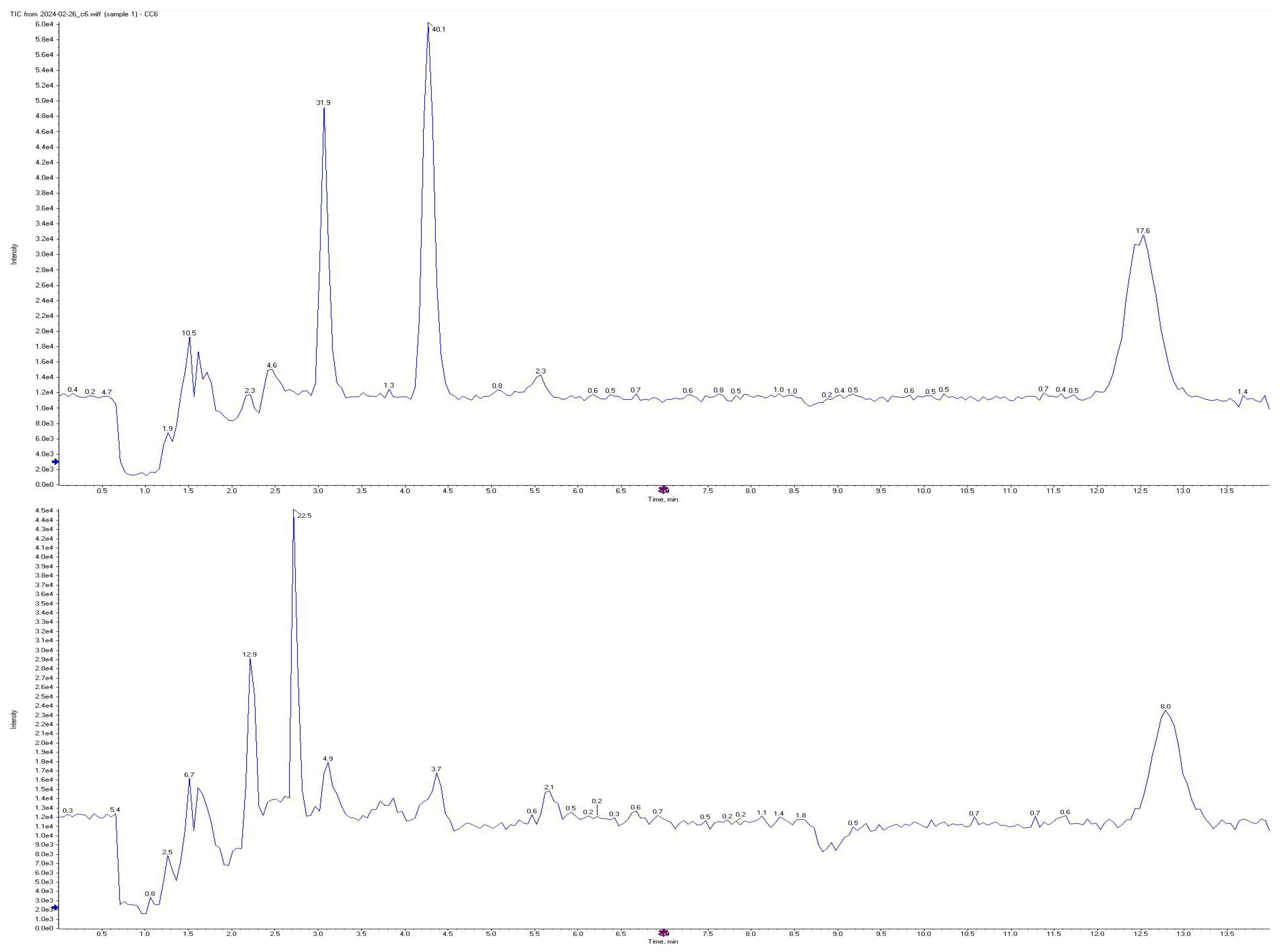
3.1. Sensitivity and Selectivity
3.2. Carryover
3.3. Linearity
3.4. Accuracy and Precision
3.5. Recovery for Plasma Sample Analysis
3.6. Reproducibility of Plasma Sample Preparation for Total PUFA Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SCFA | Short-chain fatty acids |
| MCFA | Medium-chain fatty acids |
| MS | Multiple sclerosis |
| LC-MS | Liquid chromatography coupled with mass spectrometry |
| LC-MS/MS | Liquid chromatography coupled with tandem mass spectrometry |
| HPLC | High performance liquid chromatography |
| 3-NPH | 3-Nitrophenylhydrazine |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| PYR | Pyridine |
| AA | Acetic acid |
| PA | Propionic (Propanoic) acid |
| BA | Butyric (Butanoic) acid |
| CA | Caproic (Hexanoic) acid |
| LLOQ | Lower limit of quantification |
| QC | Quality control sample(s) |
References
- Xiong, R.G.; Zhou, D.D.; Wu, S.X.; Huang, S.Y.; Saimaiti, A.; Yang, Z.J.; Shang, A.; Zhao, C.N.; Gan, R.Y.; Li, H.B. Health Benefits and Side Effects of Short-Chain Fatty Acids. Foods 2022, 11, 2863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, H.; Du, S.; Ren, J.; Qiao, G.; Ren, J. Rutin ameliorates calcium oxalate crystal-induced kidney injury through anti-oxidative stress and modulation of intestinal flora. Urolithiasis 2025, 53, 50. [Google Scholar] [CrossRef] [PubMed]
- Barcutean, L.; Farczadi, L.; Manescu, I.B.; Imre, S.; Maier, S.; Balasa, R. Short and Medium Chain Fatty Acids in a Cohort of Naïve Multiple Sclerosis Patients: Pre- and Post-Interferon Beta Treatment Assessment. Biologics 2024, 18, 349–361. [Google Scholar] [CrossRef]
- Pluznick, J.L. Gut microbiota in renal physiology: Focus on short-chain fatty acids and their receptors. Kidney Int. 2016, 90, 1191–1198. [Google Scholar] [CrossRef]
- Wu, S.; Bu, X.; Chen, D.; Wu, X.; Wu, H.; Caiyin, Q.; Qiao, J. Molecules-mediated bidirectional interactions between microbes and human cells. NPJ Biofilm. Microbiomes 2025, 11, 38. [Google Scholar] [CrossRef]
- Meesters, R.J.; van Eijk, H.M.; ten Have, G.A.; de Graaf, A.A.; Venema, K.; van Rossum, B.E.; Deutz, N.E. Application of liquid chromatography-mass spectrometry to measure the concentrations and study the synthesis of short chain fatty acids following stable isotope infusions. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 854, 57–62. [Google Scholar] [CrossRef]
- Zeng, M.; Cao, H. Fast quantification of short chain fatty acids and ketone bodies by liquid chromatography-tandem mass spectrometry after facile derivatization coupled with liquid-liquid extraction. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1083, 137–145. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, Z.; Wu, Y.; Shrestha, R.; Imai, H.; Uemura, N.; Hirano, K.I.; Chiba, H.; Hui, S.P. Development of a simultaneous quantitation for short-, medium-, long-, and very long-chain fatty acids in human plasma by 2-nitrophenylhydrazine-derivatization and liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1126–1127, 121771. [Google Scholar] [CrossRef]
- Song, H.E.; Lee, H.Y.; Kim, S.J.; Back, S.H.; Yoo, H.J. A Facile Profiling Method of Short Chain Fatty Acids Using Liquid Chromatography-Mass Spectrometry. Metabolites 2019, 9, 173. [Google Scholar] [CrossRef]
- Darlington, D.N. Conjugation of Short-Chain Fatty Acids to Bicyclic-Amines for Analysis by Liquid Chromatography Tandem Mass Spectroscopy. Molecules 2025, 30, 341. [Google Scholar] [CrossRef]
- Lee, D.; Kerry, M.S. A simple liquid chromatography method running in dual modes for quantification of short and medium chain fatty acids. J. Chromatogr. A 2024, 1714, 464566. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.Z.W.; Lee, S.X.Y.; Ooi, D.S.Q.; Ta, L.D.H.; Yap, G.C.; Tay, C.J.X.; Huang, C.H.; Tham, E.H.; Loo, E.X.L.; Shek, L.P.C.; et al. Sensitive LC-MS/MS method for the temporal profiling of bile acids, fatty acids and branched-chain alpha-keto acids in maternal plasma during pregnancy and cord blood plasma at delivery. Clin. Chim. Acta 2023, 547, 117449. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ruan, H.; Zong, Z.; Mao, F.; Wang, Y.; Jiao, Y.; Xu, L.; Yang, T.; Li, W.; Liu, X. A simulated strategy for analysis of Short- to Long- chain fatty acids in mouse serum beyond chemical standards. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2021, 1180, 122895. [Google Scholar] [CrossRef] [PubMed]
- Aydoğan, C. Recent advances and applications in LC-HRMS for food and plant natural products: A critical review. Anal. Bioanal. Chem. 2020, 412, 1973. [Google Scholar] [CrossRef]
- Bioanalytical Method Validation—Guidance for Industry; Unites Stated Food and Drug Administration: Silver Spring, MD, USA, 2018.
- Guideline on Bioanalytical Method Validation; European Medicines Agency: Amsterdam, The Netherlands, 2011.
- Nagatomo, R.; Ichikawa, A.; Kaneko, H.; Inoue, K. Comparison of 3-nitrophenylhydrazine, O-benzyl hydroxylamine, and 2-picolylamine derivatizations for analysis of short-chain fatty acids through liquid chromatography coupled with tandem mass spectrometry. Anal. Sci. 2024, 40, 843–851. [Google Scholar] [CrossRef]
- Chalova, P.; Tazky, A.; Skultety, L.; Minichova, L.; Chovanec, M.; Ciernikova, S.; Mikus, P.; Piestansky, J. Determination of short-chain fatty acids as putative biomarkers of cancer diseases by modern analytical strategies and tools: A review. Front. Oncol. 2023, 13, 1110235. [Google Scholar] [CrossRef]
- Li, C.; Liu, Z.; Bath, C.; Marett, L.; Pryce, J.; Rochfort, S. Optimised Method for Short-Chain Fatty Acid Profiling of Bovine Milk and Serum. Molecules 2022, 27, 436. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, Y.; Shrestha, R.; Gao, Z.; Zhao, Y.; Miura, Y.; Tamakoshi, A.; Chiba, H.; Hui, S.-P. Determination of total, free and esterified short-chain fatty acid in human serum by liquid chromatography-mass spectrometry. Ann. Clin. Biochem. 2019, 56, 190–197. [Google Scholar] [CrossRef]
- Cas, M.D.; Paroni, R.; Saccardo, A.; Casagni, E.; Arnoldi, S.; Gambaro, V.; Saresella, M.; Mario, C.; La Rosa, F.; Marventano, I.; et al. A straightforward LC-MS/MS analysis to study serum profile of short and medium chain fatty acids. J. Chromatogr. B-Anal. Technol. Biomed. LIFE Sci. 2020, 1154, 121982. [Google Scholar] [CrossRef]
- Jaochico, A.; Sangaraju, D.; Shahidi-Latham, S.K. A rapid derivatization based LC-MS/MS method for quantitation of short-chain fatty acids in human plasma and urine. Bioanalysis 2019, 11, 741–753. [Google Scholar] [CrossRef]
- Vagaggini, C.; Brai, A.; Bonente, D.; Lombardi, J.; Poggialini, F.; Pasqualini, C.; Barone, V.; Nicoletti, C.; Bertelli, E.; Dreassi, E. Development and validation of derivatization-based LC-MS/MS method for quantification of short-chain fatty acids in human, rat, and mouse plasma. J. Pharm. Biomed. Anal. 2023, 235, 115599. [Google Scholar] [CrossRef] [PubMed]
- Mayo-Martínez, L.; Lorenzo, M.P.; Martos-Moreno, G.Á.; Graell, M.; Barbas, C.; Rupérez, F.J.; Argente, J.; García, A. Short-chain fatty acids in plasma and feces: An optimized and validated LC-QqQ-MS method applied to study anorexia nervosa. Microchem. J. 2024, 200, 110255. [Google Scholar] [CrossRef]
- Saha, S.; Day-Walsh, P.; Shehata, E.; Kroon, P.A. Development and Validation of a LC-MS/MS Technique for the Analysis of Short Chain Fatty Acids in Tissues and Biological Fluids without Derivatisation Using Isotope Labelled Internal Standards. Molecules 2021, 26, 6444. [Google Scholar] [CrossRef] [PubMed]
- Shafaei, A.; Vamathevan, V.; Pandohee, J.; Lawler, N.G.; Broadhurst, D.; Boyce, M.C. Sensitive and quantitative determination of short-chain fatty acids in human serum using liquid chromatography mass spectrometry. Anal. Bioanal. Chem. 2021, 413, 6333–6342. [Google Scholar] [CrossRef]
- Song, W.-S.; Park, H.-G.; Kim, S.-M.; Jo, S.-H.; Kim, B.-G.; Theberge, A.B.; Kim, Y.-G. Chemical derivatization-based LC-MS/MS method for quantitation of gut microbial short-chain fatty acids. J. Ind. Eng. Chem. 2020, 83, 297–302. [Google Scholar] [CrossRef]
- Li, S.; Xiao, Q.; Sun, J.; Li, Z.; Zhang, M.; Tian, Y.; Zhang, Z.; Dong, H.; Jiao, Y.; Xu, F.; et al. A new chemical derivatization reagent sulfonyl piperazinyl for the quantification of fatty acids using LC-MS/MS. Talanta 2024, 277, 126378. [Google Scholar] [CrossRef]
- Barcutean, L.; Maier, S.; Burai-Patrascu, M.; Farczadi, L.; Balasa, R. The Immunomodulatory Potential of Short-Chain Fatty Acids in Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 3198. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SCFA | Molecular Weight (g/mol) | Parent Ion (m/z) | Fragment Ions (m/z) | Collision Energy (V) | Retention Time (min) |
|---|---|---|---|---|---|
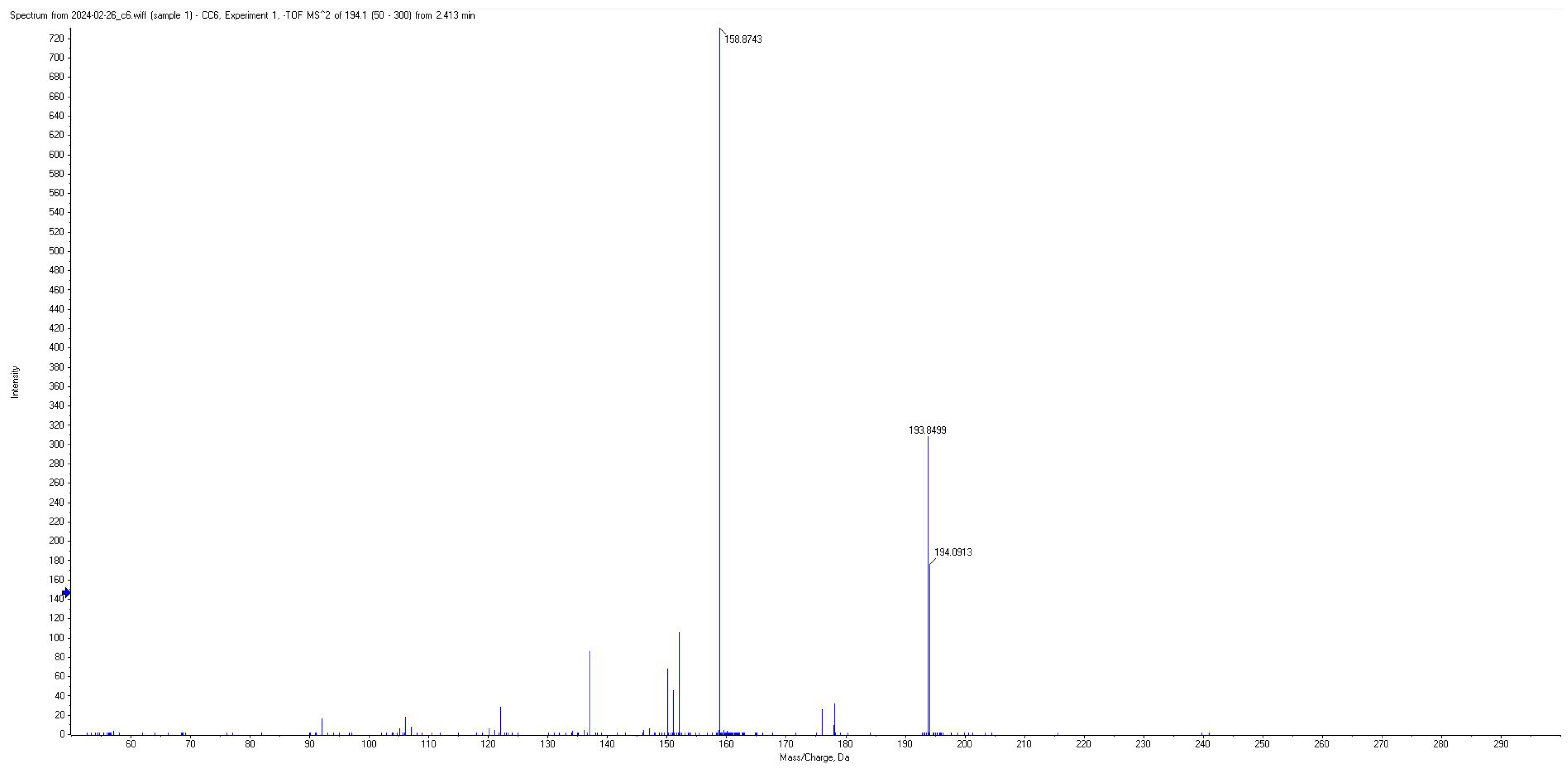
| AA | 60 | 194.07 | 152.05; 137.05; 122.02; 178.07; 150.05 | −20 | 2.2 |
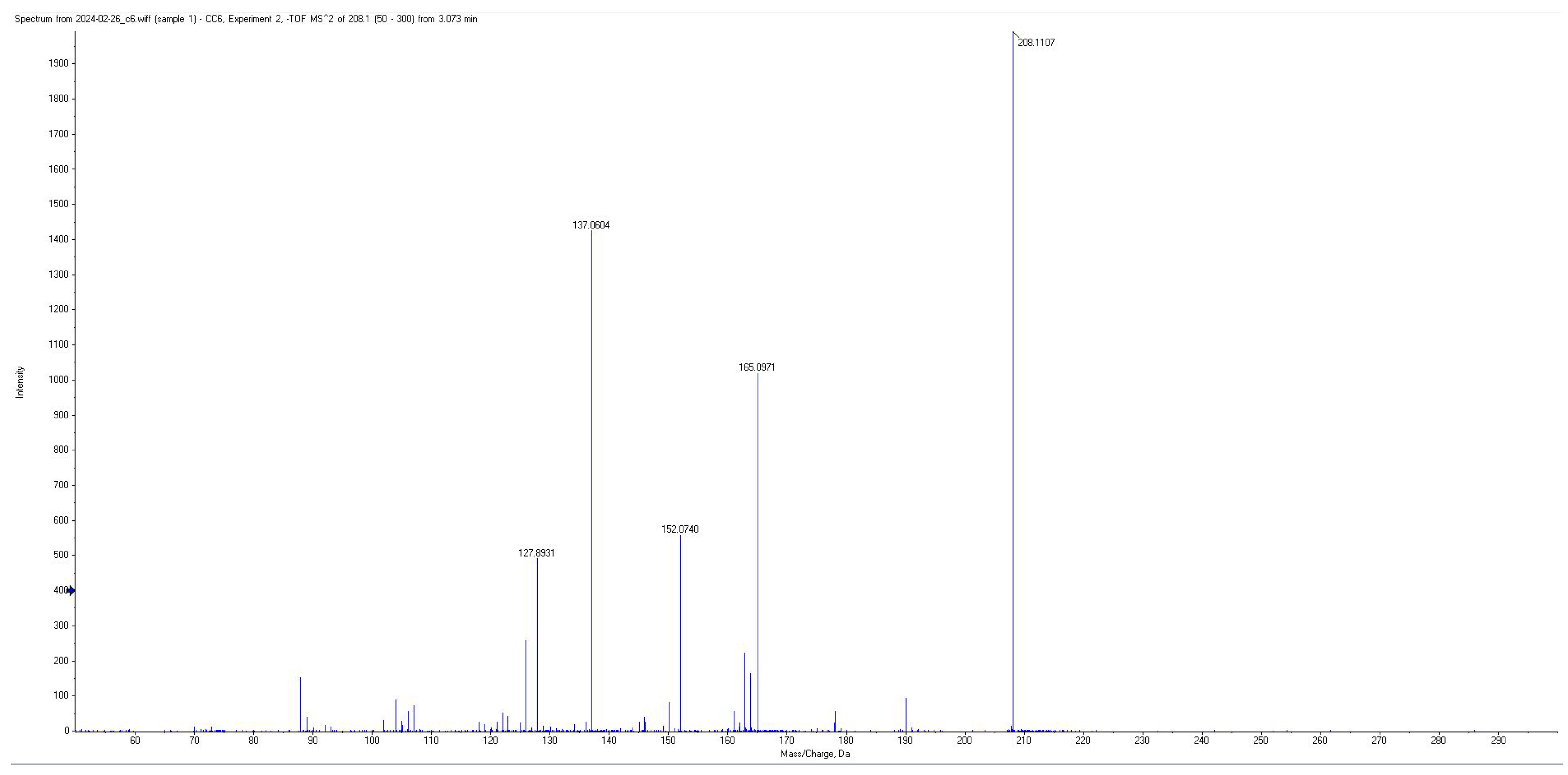
| PA | 74 | 208.09 | 152.05; 137.05; 150.05 | −20 | 2.7 |
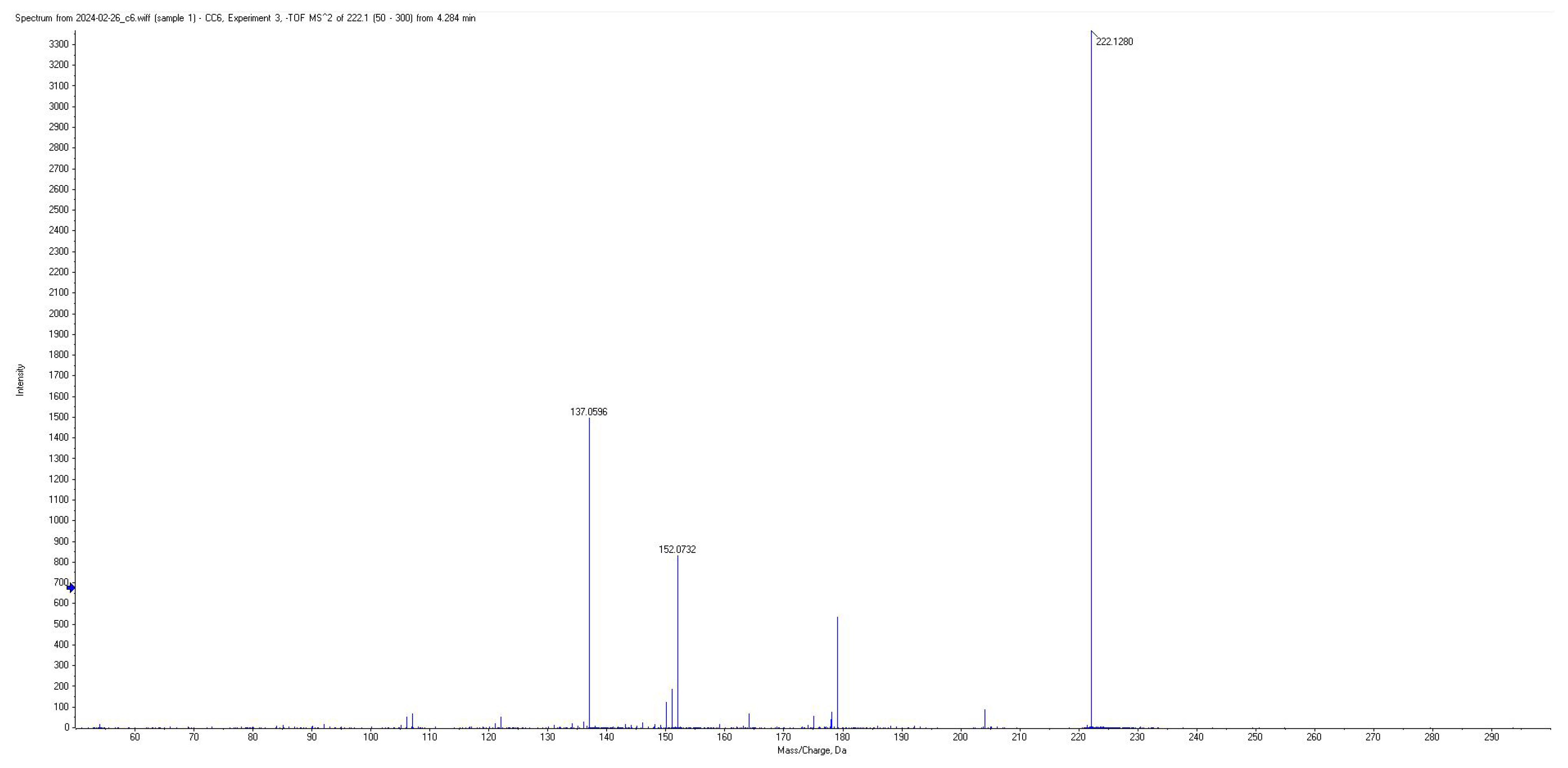
| BA | 88 | 222.07 | 152.05; 137.05; 122.02; 178.07; 150.05 | −20 | 4.0 |
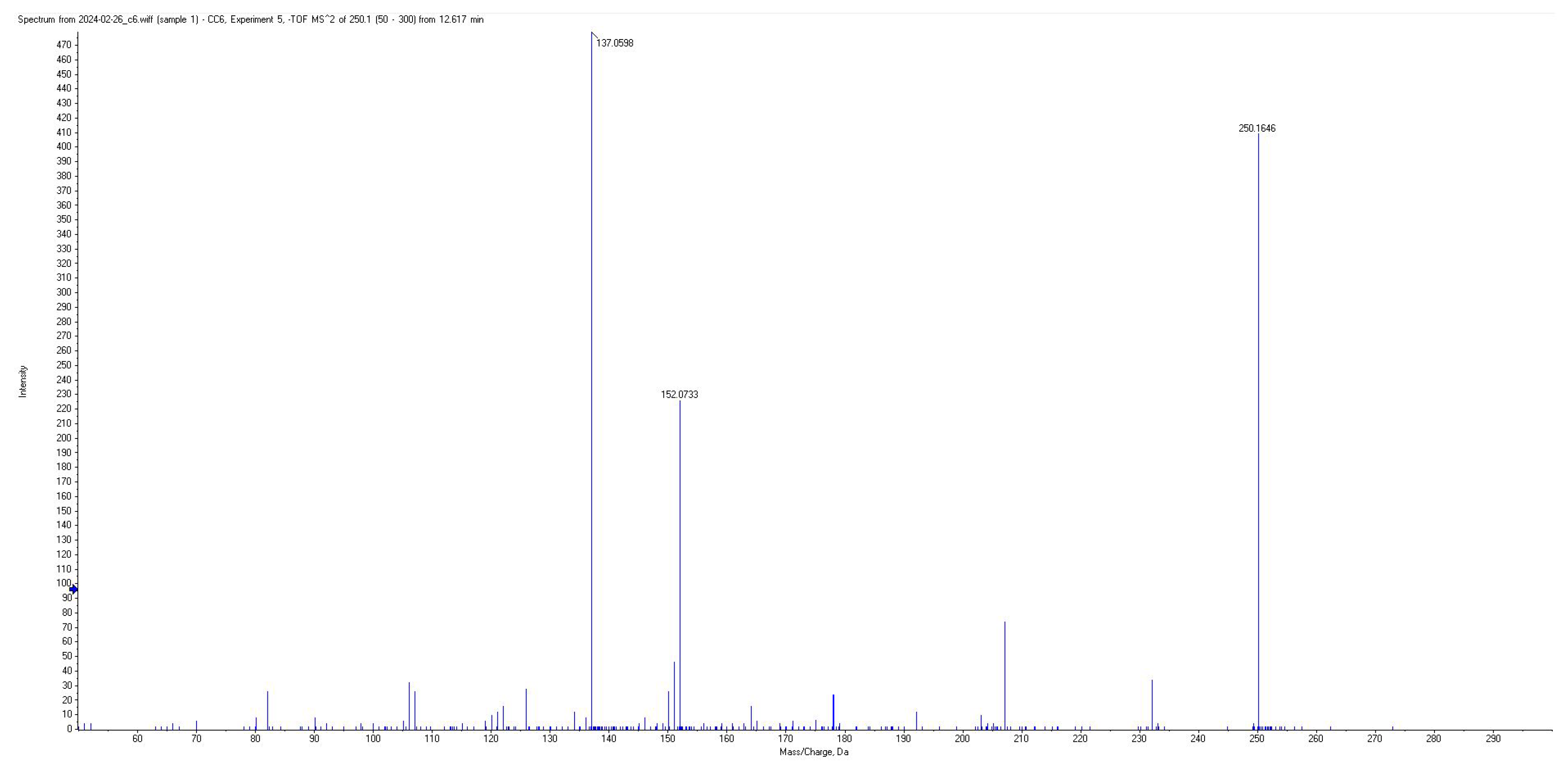
| CA | 116 | 250.13 | 152.05; 137.05; 178.07 | −20 | 13.0 |
| CA-d3 (internal standard) | 119 | 253.14 | 155.05; 140.05; 181.07 | −20 | 13.0 |
| Batch | AA | PA | BA | CA |
|---|---|---|---|---|
| Batch 1 | 0.99808 | 0.99430 | 0.99769 | 0.99831 |
| Batch 2 | 0.99476 | 0.99502 | 0.99490 | 0.99686 |
| Batch 3 | 0.99816 | 0.99672 | 0.99853 | 0.99966 |
| Batch 4 | 0.99916 | 0.99817 | 0.99860 | 0.99893 |
| Batch 5 | 0.99865 | 0.99934 | 0.99785 | 0.99897 |
| AA | PA | BA | CA | |
|---|---|---|---|---|
| Accuracy (%) | −12.6 ÷ 14.5 | −13.7 ÷ 14.0 | −14.7 ÷ 11.3 | −10.1 ÷ 9.3 |
| QC Solution | AA | PA | BA | CA | ||||
|---|---|---|---|---|---|---|---|---|
| Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | |
| QCA | 2.8 | 8.4 | −6.8 | 11.3 | 8.4 | 3.9 | 3.8 | 7.5 |
| QCB | 6.5 | 8.6 | −8.4 | 4.8 | −7.5 | 5.0 | 1.2 | 8.1 |
| QCC | −4.3 | 3.7 | −5.3 | 5.2 | −1.7 | 1.2 | −4.8 | 3.3 |
| QCD | −7.5 | 4.3 | 1.4 | 6.3 | 3.1 | 1.6 | −0.6 | 2.2 |
| QCE | 8.2 | 5.6 | 8.1 | 2.5 | 5.5 | 2.0 | 3.0 | 2.2 |
| QCF | 3.0 | 1.9 | 12.6 | 2.1 | 5.0 | 2.4 | 6.3 | 1.8 |
| QC Solution | AA | PA | BA | CA | ||||
|---|---|---|---|---|---|---|---|---|
| Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | |
| QCA | 1.6 | 7.2 | 3.3 | 11.9 | 3.1 | 8.3 | 3.5 | 11.6 |
| QCB | 4.1 | 11.4 | −6.2 | 6.3 | −7.9 | 5.4 | 2.8 | 7.9 |
| QCC | −2.3 | 1.1 | −6.2 | 4.9 | −1.1 | 3.3 | −4.1 | 3.3 |
| QCD | −5.7 | 5.8 | 0.4 | 5.3 | 2.2 | 1.8 | −2.1 | 2.6 |
| QCE | 6.9 | 6.4 | 7.4 | 6.6 | 4.0 | 2.0 | 3.2 | 3.8 |
| QCF | −0.9 | 3.1 | 6.4 | 1.4 | 4.4 | 2.4 | 4.0 | 2.9 |
| Within-Run | Between-Run | ||||||
|---|---|---|---|---|---|---|---|
| SCFA | Spike Std. Conc. (ng/mL) | Mean Conc. Plasma (ng/mL) | Mean Conc. Spiked Plasma (ng/mL) | Mean Recovery (%) | Mean Conc. Plasma (ng/mL) | Mean Conc. Spiked Plasma (ng/mL) | Mean Recovery (%) |
| AA | 50 | 905.97 | 431.18 | 90.22 | 849.08 | 425.51 | 94.65 |
| 500 | 905.97 | 763.76 | 108.64 | 849.08 | 742.11 | 110.02 | |
| PA | 50 | 40.42 | 41.70 | 92.24 | 42.20 | 42.27 | 92.78 |
| 500 | 40.42 | 308.38 | 114.13 | 42.20 | 301.44 | 111.19 | |
| BA | 50 | 36.46 | 40.10 | 92.76 | 37.51 | 39.08 | 89.32 |
| 500 | 36.46 | 302.87 | 112.91 | 37.51 | 298.31 | 110.99 | |
| CA | 50 | 7.68 | 30.34 | 104.87 | 7.22 | 32.60 | 113.96 |
| 500 | 7.68 | 286.72 | 112.95 | 7.22 | 282.70 | 111.47 | |
| CV (%) | |||
|---|---|---|---|
| SCFA | Within-Run (Day 1) | Within-Run (Day 2) | Between-Run |
| [Plasma 1/Plasma 2] | [Plasma 1/Plasma 2] | [Plasma 1/Plasma 2] | |
| AA | 9.6/2.4 | 9.4/2.3 | 6.7/2.6 |
| PA | 7.8/14.5 | 6.5/12.7 | 5.2/11.9 |
| BA | 9.8/2.5 | 10.0/2.6 | 7.2/8.6 |
| CA | 11.2/13.4 | 10.5/10.8 | 14.1/13.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farczadi, L.; Barcutean, L.; Maier, S.; Balasa, R.; Imre, S. Development and Validation of a New LC-MS/MS Method for the Assay of Plasmatic Peripheral Short- and Medium-Chain Fatty Acids for Metabolomics Applications. Metabolites 2025, 15, 403. https://doi.org/10.3390/metabo15060403
Farczadi L, Barcutean L, Maier S, Balasa R, Imre S. Development and Validation of a New LC-MS/MS Method for the Assay of Plasmatic Peripheral Short- and Medium-Chain Fatty Acids for Metabolomics Applications. Metabolites. 2025; 15(6):403. https://doi.org/10.3390/metabo15060403
Chicago/Turabian StyleFarczadi, Lenard, Laura Barcutean, Smaranda Maier, Rodica Balasa, and Silvia Imre. 2025. "Development and Validation of a New LC-MS/MS Method for the Assay of Plasmatic Peripheral Short- and Medium-Chain Fatty Acids for Metabolomics Applications" Metabolites 15, no. 6: 403. https://doi.org/10.3390/metabo15060403
APA StyleFarczadi, L., Barcutean, L., Maier, S., Balasa, R., & Imre, S. (2025). Development and Validation of a New LC-MS/MS Method for the Assay of Plasmatic Peripheral Short- and Medium-Chain Fatty Acids for Metabolomics Applications. Metabolites, 15(6), 403. https://doi.org/10.3390/metabo15060403