

Metabolic Effects of the Cancer Metastasis Modulator MEMO1

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
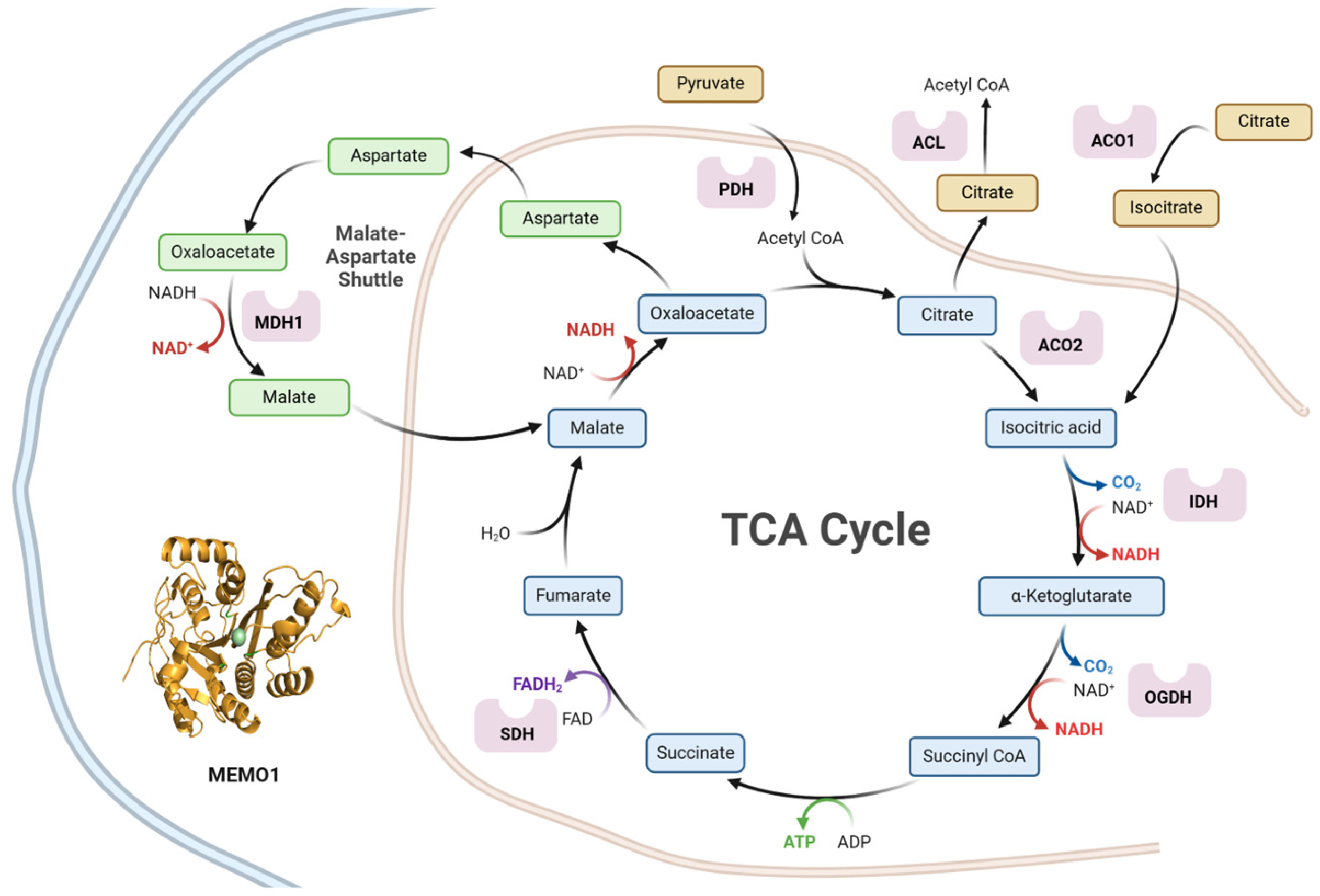
2.1. Identification of MEMO1 Genetic Interactions Related to the TCA Cycle
2.2. Cell Lines, shRNA Assay and Western Blot
2.3. Sample Preparation for LC-MS/MS
2.4. Metabolomic Analysis
3. Results
3.1. MEMO1 GOF-GIs Highlight a Link to Mitochondrial Energy Metabolism and Redox Homeostasis
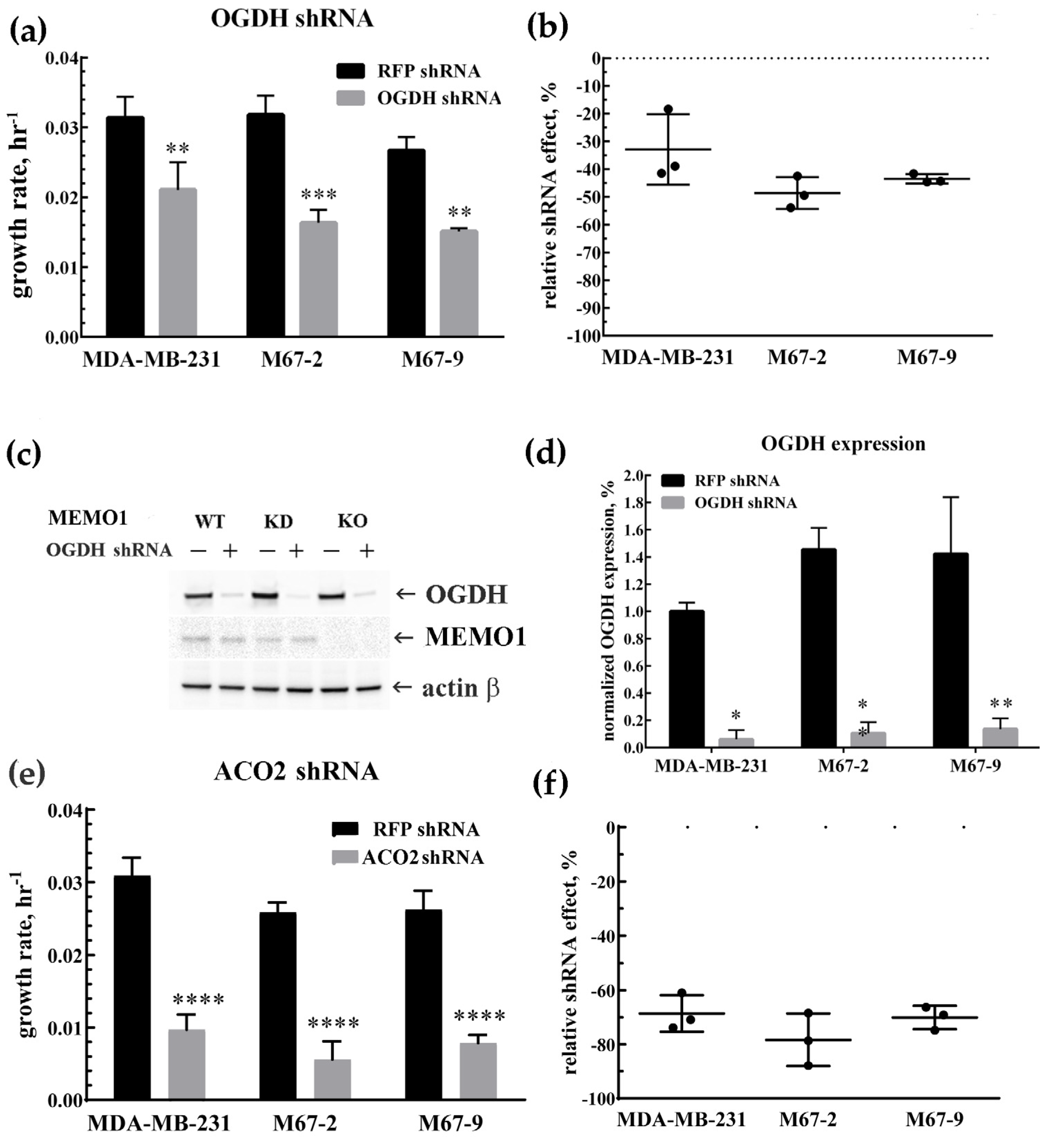
3.2. MDA-MB-231 Cells Metabolism Relies on the TCA Cycle: OGDH and ACO2 Knockdowns Inhbit Cell Proliferation
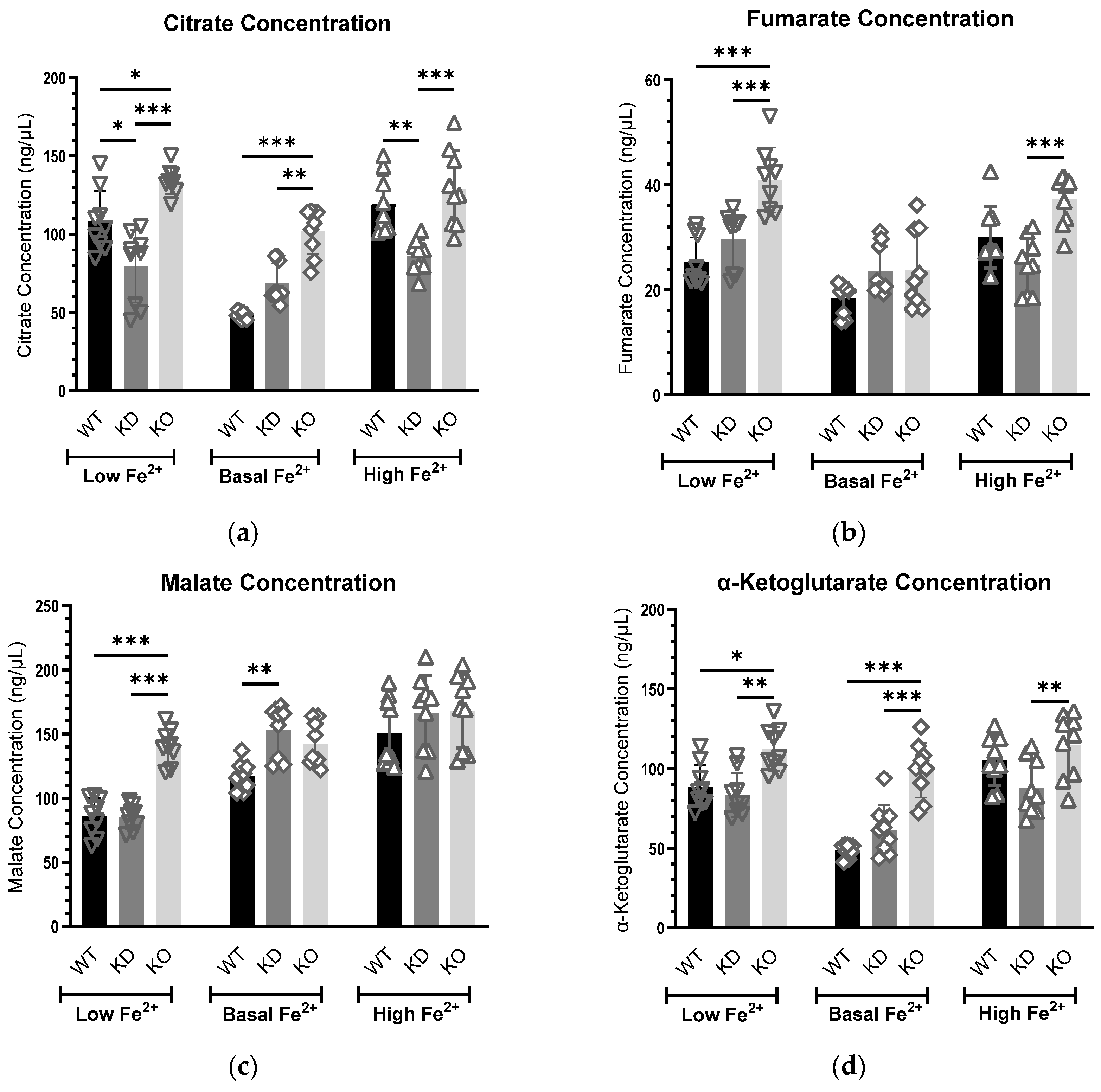
3.3. MEMO1-Mediated Modulation of Mitochondrial Metabolism via Iron Homeostasis
3.3.1. Citrate Concentrations Exhibit a Consistent Trend at the Basal Iron Level
3.3.2. Fumarate Levels Are Elevated in MEMO1-Deficient Cells Under Altered Iron Conditions
3.3.3. MEMO1 Regulates Malate Levels Primarily Under Iron Limitation
3.3.4. MEMO1 Loss Consistently Elevates α-Ketoglutarate Levels, Indicating Disrupted α-KG Metabolism
3.3.5. Iron Deficiency Affects Glutamate Metabolism in MEMO1 Knockout Cells
3.3.6. Pyruvate and Succinate Concentrations Remain Stable Despite Variations in MEMO1 Levels and Iron Availability
4. Discussion
4.1. Citrate
4.2. Fumarate
4.3. Malate
4.4. α-Ketoglutarate
4.5. Glutamate
4.6. Pyruvate and Succinate
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GOF | Gain-of-Function |
| GOF-GIs | Gain-of-Function Genetic Interactions |
| MEMO1-KD | MEMO1-knockdown |
| MEMO1-KO | MEMO1-knockout |
| DmPA | Dimethylaminophenacyl |
| TCA cycle | Tricarboxylic Acid cycle |
| OXPHOS | Oxidative Phosphorylation |
| α-KG | α-Ketoglutarate |
| LC-MS/MS | Liquid Chromatography–Tandem Mass Spectrometry |
| HPLC-MS/MS | High-Performance Liquid Chromatography–Tandem Mass Spectrometry |
| HILIC-MS/MS | Hydrophilic Interaction Liquid Chromatography–Tandem Mass Spectrometry |
| 12C2-DmPA-Br | 12C2-Dimethylaminophenacyl Bromide |
| 13C2-DmPA-Br | 13C2-Dimethylaminophenacyl Bromide |
| MRM | Multiple Reaction Monitoring |
| ESI | Electrospray Ionization |
| ISV | Ion Spray Voltage |
| Q-LTI | Quadrupole-Linear Ion Trap |
| GSEA | Gene Set Enrichment Analysis |
References
- Moor, M.B.; Haenzi, B.; Legrand, F.; Koesters, R.; Hynes, N.E.; Bonny, O. Renal Memo1 Differentially Regulates the Expression of Vitamin D-Dependent Distal Renal Tubular Calcium Transporters. Front. Physiol. 2018, 9, 874. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Plestant, C.; Yabuno-Nakagawa, K.; Li, J.; Lee, J.; Huang, C.W.; Lee, A.; Krupa, O.; Adhikari, A.; Thompson, S.; et al. Memo1-Mediated Tiling of Radial Glial Cells Facilitates Cerebral Cortical Development. Neuron 2019, 103, 836–852.e5. [Google Scholar] [CrossRef] [PubMed]
- Marone, R.; Hess, D.; Dankort, D.; Muller, W.J.; Hynes, N.E.; Badache, A. Memo mediates ErbB2-driven cell motility. Nat. Cell Biol. 2004, 6, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Hourihan, J.M.; Bland, M.S.; Obieglo, C.; Katic, I.; Moronetti Mazzeo, L.E.; Alcedo, J.; Blackwell, T.K.; Hynes, N.E. NADPH oxidase-mediated redox signaling promotes oxidative stress resistance and longevity through memo-1 in C. elegans. eLife 2017, 6, e19493. [Google Scholar] [CrossRef]
- Dolgova, N.; Uhlemann, E.E.; Boniecki, M.T.; Vizeacoumar, F.S.; Ara, A.; Nouri, P.; Ralle, M.; Tonelli, M.; Abbas, S.A.; Patry, J.; et al. MEMO1 binds iron and modulates iron homeostasis in cancer cells. eLife 2024, 13, e86354. [Google Scholar] [CrossRef]
- Sorokin, A.V.; Chen, J. MEMO1, a new IRS1-interacting protein, induces epithelial-mesenchymal transition in mammary epithelial cells. Oncogene 2013, 32, 3130–3138. [Google Scholar] [CrossRef]
- Frei, A.; MacDonald, G.; Lund, I.; Gustafsson, J.; Hynes, N.E.; Nalvarte, I. Memo interacts with c-Src to control Estrogen Receptor alpha sub-cellular localization. Oncotarget 2016, 7, 56170–56182. [Google Scholar] [CrossRef]
- MacDonald, G.; Nalvarte, I.; Smirnova, T.; Vecchi, M.; Aceto, N.; Dolemeyer, A.; Frei, A.; Lienhard, S.; Wyckoff, J.; Hess, D.; et al. Memo is a copper-dependent redox protein with an essential role in migration and metastasis. Sci. Signal 2014, 7, ra56. [Google Scholar] [CrossRef]
- Ru, Q.; Li, Y.; Chen, L.; Wu, Y.; Min, J.; Wang, F. Iron homeostasis and ferroptosis in human diseases: Mechanisms and therapeutic prospects. Signal Transduct. Target. Ther. 2024, 9, 271. [Google Scholar] [CrossRef]
- Inigo, M.; Deja, S.; Burgess, S.C. Ins and Outs of the TCA Cycle: The Central Role of Anaplerosis. Annu. Rev. Nutr. 2021, 41, 19–47. [Google Scholar] [CrossRef]
- Guertin, D.A.; Wellen, K.E. Acetyl-CoA metabolism in cancer. Nat. Rev. Cancer 2023, 23, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The Key Role of Anaplerosis and Cataplerosis for Citric Acid Cycle Function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed]
- Eniafe, J.; Jiang, S. The functional roles of TCA cycle metabolites in cancer. Oncogene 2021, 40, 3351–3363. [Google Scholar] [CrossRef]
- Arnold, P.K.; Finley, L.W.S. Regulation and function of the mammalian tricarboxylic acid cycle. J. Biol. Chem. 2023, 299, 102838. [Google Scholar] [CrossRef]
- Ohshima, K.; Morii, E. Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- McGuirk, S.; Audet-Delage, Y.; St-Pierre, J. Metabolic Fitness and Plasticity in Cancer Progression. Trends Cancer 2020, 6, 49–61. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Shimizu, K.; Matsuoka, Y. Regulation of glycolytic flux and overflow metabolism depending on the source of energy generation for energy demand. Biotechnol. Adv. 2019, 37, 284–305. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, N.; Chen, J.; Tao, Q.; Li, Q.; Li, J.; Chen, X.; Peng, C. The Tricarboxylic Acid Cycle Metabolites for Cancer: Friend or Enemy. Research 2024, 7, 0351. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.B.; Dias, S.M.G.; Dang, C.V. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. [Google Scholar] [CrossRef]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine Oxidation Maintains the TCA Cycle and Cell Survival during Impaired Mitochondrial Pyruvate Transport. Mol. Cell 2014, 56, 414–424. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes. Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Cai, Z.; Li, C.F.; Han, F.; Liu, C.; Zhang, A.; Hsu, C.C.; Peng, D.; Zhang, X.; Jin, G.; Rezaeian, A.H.; et al. Phosphorylation of PDHA by AMPK Drives TCA Cycle to Promote Cancer Metastasis. Mol. Cell 2020, 80, 263–278.e7. [Google Scholar] [CrossRef]
- Wang, H.; Sun, J.; Sun, H.; Wang, Y.; Lin, B.; Wu, L.; Qin, W.; Zhu, Q.; Yi, W. The OGT-c-Myc-PDK2 axis rewires the TCA cycle and promotes colorectal tumor growth. Cell Death Differ. 2024, 31, 1157–1169. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef]
- Tong, Y.; Qi, Y.; Xiong, G.; Li, J.; Scott, T.L.; Chen, J.; He, D.; Li, L.; Wang, C.; Lane, A.N.; et al. The PLOD2/succinate axis regulates the epithelial-mesenchymal plasticity and cancer cell stemness. Proc. Natl. Acad. Sci. USA 2023, 120, e2214942120. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Matsuura, Y.; Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 2025, 53, D672–D677. [Google Scholar] [CrossRef]
- Awad, H.; Allen, K.J.H.; Adamko, D.J.; El-Aneed, A. Development of a new quantification method for organic acids in urine as potential biomarkers for respiratory illness. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1122–1123, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Willacey, C.C.W.; Naaktgeboren, M.; Lucumi Moreno, E.; Wegrzyn, A.B.; van der Es, D.; Karu, N.; Fleming, R.M.T.; Harms, A.C.; Hankemeier, T. LC–MS/MS analysis of the central energy and carbon metabolites in biological samples following derivatization by dimethylaminophenacyl bromide. J. Chromatogr. A 2019, 1608, 460413. [Google Scholar] [CrossRef] [PubMed]
- Khamis, M.M.; Adamko, D.J.; Purves, R.W.; El-Aneed, A. Quantitative determination of potential urine biomarkers of respiratory illnesses using new targeted metabolomic approach. Anal. Chim. Acta 2019, 1047, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Khajavinia, A.; Michel, D.; Ezeaka, U.C.; Purves, R.W.; Laprairie, R.B.; El-Aneed, A. Addressing a major interference in the quantification of psilocin in mouse plasma: Development of a validated liquid chromatography tandem mass spectrometry method. J. Chromatogr. A 2024, 1730, 465123. [Google Scholar] [CrossRef]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef]
- Marcotte, R.; Brown, K.R.; Suarez, F.; Sayad, A.; Karamboulas, K.; Krzyzanowski, P.M.; Sircoulomb, F.; Medrano, M.; Fedyshyn, Y.; Koh, J.L.Y.; et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012, 2, 172–189. [Google Scholar] [CrossRef]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; East-Seletsky, A.; Ali, L.D.; Gerath, W.F.; Pantel, S.E.; et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 2014, 1, 140035. [Google Scholar] [CrossRef]
- McDonald, E.R., 3rd; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592.e10. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, X.; Chen, V.; Feng, Y.; Wang, L.; Croniger, C.; Conlon, R.A.; Markowitz, S.; Fearon, E.; Puchowicz, M.; et al. Colorectal cancers utilize glutamine as an anaplerotic substrate of the TCA cycle in vivo. Sci. Rep. 2019, 9, 19180. [Google Scholar] [CrossRef] [PubMed]
- Baksh, S.C.; Finley, L.W.S. Metabolic Coordination of Cell Fate by α-Ketoglutarate-Dependent Dioxygenases. Trends Cell Biol. 2021, 31, 24–36. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Dettmer, K.; Rümmele, P.; Schmidt, K.; Prehn, C.; Milenkovic, V.M.; Jagla, W.; Madej, G.M.; Lantow, M.; Schladt, M.; et al. Extracellular Citrate Affects Critical Elements of Cancer Cell Metabolism and Supports Cancer Development In Vivo. Cancer Res. 2018, 78, 2513–2523. [Google Scholar] [CrossRef]
- Icard, P.; Alifano, M.; Simula, L. The potential for citrate to reinforce epigenetic therapy by promoting apoptosis. Trends Endocrinol. Metab. 2023, 34, 586–589. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, X.; Si, F.; Huang, L.; Gao, A.; Lin, W.; Hoft, D.F.; Shao, Q.; Peng, G. Citrate Promotes Excessive Lipid Biosynthesis and Senescence in Tumor Cells for Tumor Therapy. Adv. Sci. 2022, 9, e2101553. [Google Scholar] [CrossRef]
- Ren, J.G.; Seth, P.; Ye, H.; Guo, K.; Hanai, J.I.; Husain, Z.; Sukhatme, V.P. Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci. Rep. 2017, 7, 4537. [Google Scholar] [CrossRef]
- Icard, P.; Simula, L.; Wu, Z.; Berzan, D.; Sogni, P.; Dohan, A.; Dautry, R.; Coquerel, A.; Lincet, H.; Loi, M.; et al. Why may citrate sodium significantly increase the effectiveness of transarterial chemoembolization in hepatocellular carcinoma? Drug Resist. Updat. 2021, 59, 100790. [Google Scholar] [CrossRef]
- Drexler, K.; Schmidt, K.M.; Jordan, K.; Federlin, M.; Milenkovic, V.M.; Liebisch, G.; Artati, A.; Schmidl, C.; Madej, G.; Tokarz, J.; et al. Cancer-associated cells release citrate to support tumour metastatic progression. Life Sci. Alliance 2021, 4, e202000903. [Google Scholar] [CrossRef]
- Cenigaonandia-Campillo, A.; Garcia-Bautista, A.; Rio-Vilariño, A.; Cebrian, A.; Del Puerto, L.; Pellicer, J.A.; Gabaldón, J.A.; Pérez-Sánchez, H.; Carmena-Bargueño, M.; Meroño, C.; et al. Vitamin-C-dependent downregulation of the citrate metabolism pathway potentiates pancreatic ductal adenocarcinoma growth arrest. Mol. Oncol. 2024, 18, 2212–2233. [Google Scholar] [CrossRef]
- Li, Z.; Li, D.; Choi, E.Y.; Lapidus, R.; Zhang, L.; Huang, S.M.; Shapiro, P.; Wang, H. Silencing of solute carrier family 13 member 5 disrupts energy homeostasis and inhibits proliferation of human hepatocarcinoma cells. J. Biol. Chem. 2017, 292, 13890–13901. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; He, C.; Li, Z.; Wang, Z.; Zhang, Q. FBP1 modulates cell metabolism of breast cancer cells by inhibiting the expression of HIF-1α. Neoplasma 2017, 64, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Tyrakis, P.A.; Yurkovich, M.E.; Sciacovelli, M.; Papachristou, E.K.; Bridges, H.R.; Gaude, E.; Schreiner, A.; D’Santos, C.; Hirst, J.; Hernandez-Fernaud, J.; et al. Fumarate Hydratase Loss Causes Combined Respiratory Chain Defects. Cell Rep. 2017, 21, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Ternette, N.; Yang, M.; Laroyia, M.; Kitagawa, M.; O’Flaherty, L.; Wolhulter, K.; Igarashi, K.; Saito, K.; Kato, K.; Fischer, R.; et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013, 3, 689–700. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Martinez-Garcia, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; Deberardinis, R.J.; Chandel, N.S. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 2013, 51, 236–248. [Google Scholar] [CrossRef]
- Broeks, M.H.; Meijer, N.W.F.; Westland, D.; Bosma, M.; Gerrits, J.; German, H.M.; Ciapaite, J.; van Karnebeek, C.D.M.; Wanders, R.J.A.; Zwartkruis, F.J.T.; et al. The malate-aspartate shuttle is important for de novo serine biosynthesis. Cell Rep. 2023, 42, 113043. [Google Scholar] [CrossRef]
- Lu, Y.X.; Ju, H.Q.; Liu, Z.X.; Chen, D.L.; Wang, Y.; Zhao, Q.; Wu, Q.N.; Zeng, Z.L.; Qiu, H.B.; Hu, P.S.; et al. ME1 Regulates NADPH Homeostasis to Promote Gastric Cancer Growth and Metastasis. Cancer Res. 2018, 78, 1972–1985. [Google Scholar] [CrossRef]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells—Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef]
- Tseng, C.W.; Kuo, W.H.; Chan, S.H.; Chan, H.L.; Chang, K.J.; Wang, L.H. Transketolase Regulates the Metabolic Switch to Control Breast Cancer Cell Metastasis via the α-Ketoglutarate Signaling Pathway. Cancer Res. 2018, 78, 2799–2812. [Google Scholar] [CrossRef]
- Tran, T.Q.; Hanse, E.A.; Habowski, A.N.; Li, H.; Ishak Gabra, M.B.; Yang, Y.; Lowman, X.H.; Ooi, A.M.; Liao, S.Y.; Edwards, R.A.; et al. α-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat. Cancer 2020, 1, 345–358. [Google Scholar] [CrossRef]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. 2014, 71, 2577–25604. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes. Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | p-Value | Distance Low–High | Protein Name | Dataset |
|---|---|---|---|---|
| ACLY | 0.0423 | 2.5934 | ATP Citrate Lyase | Marcotte (BRCA) 1 [45] |
| ACLY | 0.0079 | 1.4671 | ATP Citrate Lyase | Marcotte (All) 2 [46] |
| ACLY | 0.0228 | 0.3913 | ATP Citrate Lyase | Achilles (All) [47] |
| PCK1 | 0.0105 | 1.3177 | Phosphoenolpyruvate Carboxykinase 1 | Achilles (BRCA) |
| PCK2 | 0.0229 | 0.3120 | Phosphoenolpyruvate Carboxykinase 2 | DRIVE/Ataris (All) [48] |
| OGDH | 0.0382 | 1.0679 | Oxoglutarate Dehydrogenase | Marcotte (All) |
| OGDH | 0.0360 | 0.6191 | Oxoglutarate Dehydrogenase | Achilles (All) |
| OGDH | 0.0020 | 0.2184 | Oxoglutarate Dehydrogenase | CERES (All) [49] |
| MDH1 | 0.0416 | 0.7235 | Malate Dehydrogenase 1 | Achilles (BRCA) |
| ACO2 | 0.0319 | 0.6585 | Aconitase 2 | DRIVE/Ataris (All) |
| ACO2 | 0.0380 | 0.3233 | Aconitase 2 | CERES (All) |
| ACO1 | 0.0187 | 0.1600 | Aconitase 1 | DRIVE/Ataris (All) |
| SUCLA2 | 0.0423 | 0.5430 | Succinate-CoA Ligase ADP-Forming Beta Subunit | DRIVE/RSA (BRCA) |
| SUCLG1 | 0.0127 | 0.3360 | Succinate-CoA Ligase Alpha Subunit | DRIVE/RSA (BRCA) |
| PDHB | 0.0097 | 0.4362 | Pyruvate Dehydrogenase E1 Subunit Beta | Marcotte (All) |
| PDHA2 | 0.0410 | 0.0529 | Pyruvate Dehydrogenase E1 Subunit Alpha 2 | CERES (All) |
| DLAT | 0.0127 | 0.4250 | Dihydrolipoamide Acetyltransferase | DRIVE/RSA (BRCA) |
| IDH3B | 0.0486 | 0.2790 | Isocitrate Dehydrogenase 3 Beta | Marcotte (All) |
| IDH3B | 0.0161 | 0.1010 | Isocitrate Dehydrogenase 3 Beta | DRIVE/Ataris (All) |
| IDH3A | 0.0421 | 0.2750 | Isocitrate Dehydrogenase 3 Alpha | DRIVE/Ataris (All) |
| IDH1 | 0.0421 | 0.2380 | Isocitrate Dehydrogenase 1 | DRIVE/RSA (All) |
| SDHB | 0.0353 | 0.1842 | Succinate Dehydrogenase Iron-Sulfur Subunit | CERES (All) |
| SDHC | 0.0110 | 0.1763 | Succinate Dehydrogenase Complex Subunit C | CERES (All) |
| DLD | 0.0435 | 0.1330 | Dihydrolipoamide Dehydrogenase | Ataris (All) |
| DLD | 0.0353 | 0.1029 | Dihydrolipoamide Dehydrogenase | CERES (All) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghanbarian, M.; Dolgova, N.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Michel, D.; El-Aneed, A.; Dmitriev, O.Y. Metabolic Effects of the Cancer Metastasis Modulator MEMO1. Metabolites 2025, 15, 277. https://doi.org/10.3390/metabo15040277
Ghanbarian M, Dolgova N, Vizeacoumar FS, Vizeacoumar FJ, Michel D, El-Aneed A, Dmitriev OY. Metabolic Effects of the Cancer Metastasis Modulator MEMO1. Metabolites. 2025; 15(4):277. https://doi.org/10.3390/metabo15040277
Chicago/Turabian StyleGhanbarian, Marziyeh, Natalia Dolgova, Frederick S. Vizeacoumar, Franco J. Vizeacoumar, Deborah Michel, Anas El-Aneed, and Oleg Y. Dmitriev. 2025. "Metabolic Effects of the Cancer Metastasis Modulator MEMO1" Metabolites 15, no. 4: 277. https://doi.org/10.3390/metabo15040277
APA StyleGhanbarian, M., Dolgova, N., Vizeacoumar, F. S., Vizeacoumar, F. J., Michel, D., El-Aneed, A., & Dmitriev, O. Y. (2025). Metabolic Effects of the Cancer Metastasis Modulator MEMO1. Metabolites, 15(4), 277. https://doi.org/10.3390/metabo15040277