
Metabolic Plasticity of Glioblastoma Cells in Response to DHODH Inhibitor BAY2402234 Treatment


 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Cell Lysis, SDS PAGE and Western Blot Analysis
2.4. Metabolic Flux Analysis Using 13C-Fatty Acid Mix by Mass Spectrometry
2.5. Targeted Lipidomic Analysis
3. Results
3.1. Pharmacological Inhibition of DHODH Selectively Kills GBM Subtypes
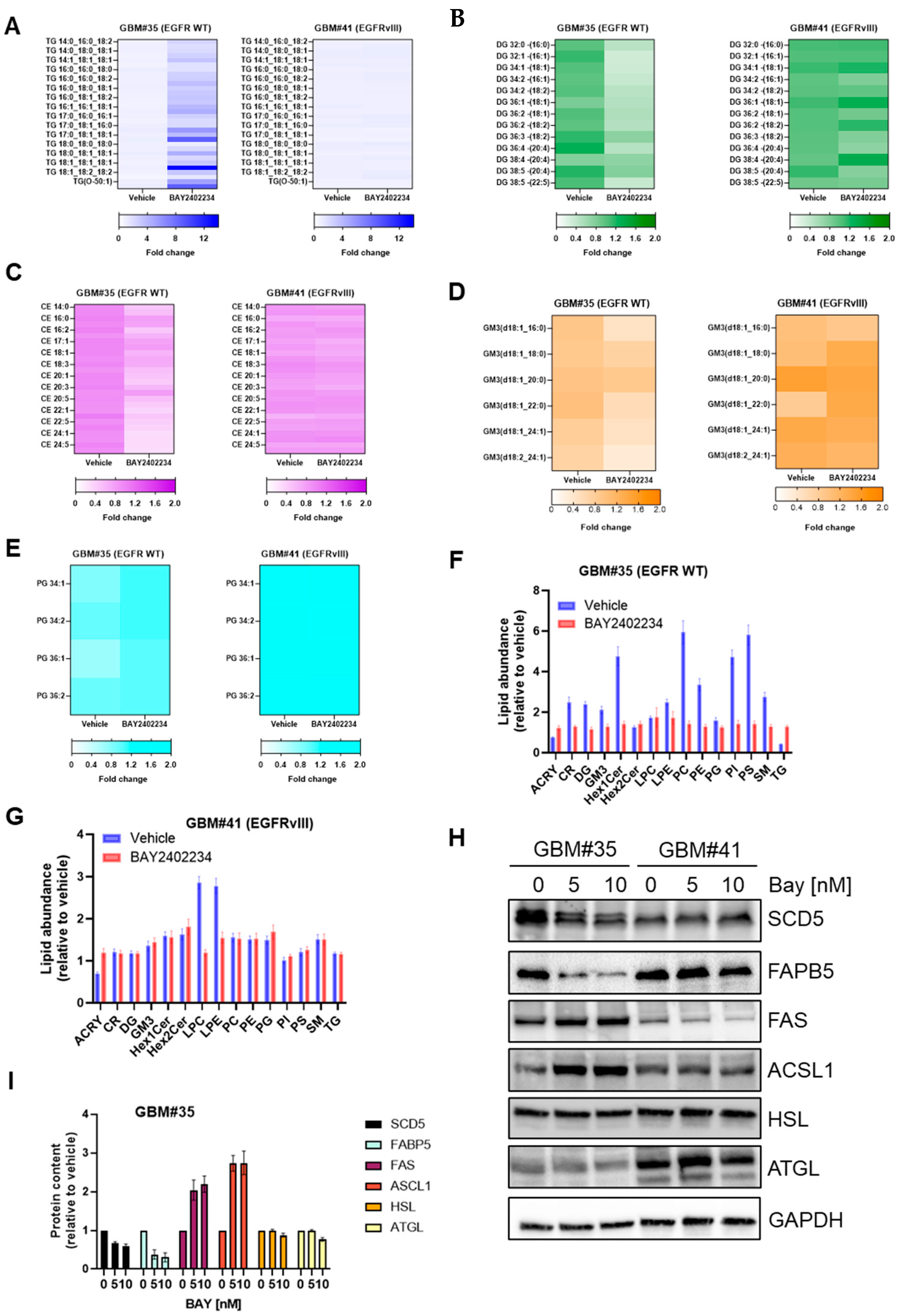
3.2. Treatment with the DHODH Inhibitor BAY2402234 Induces Triglyceride Accumulation at the Expense of Membrane Lipids in GBM Cells Harboring the Wild-Type EGFR Gene
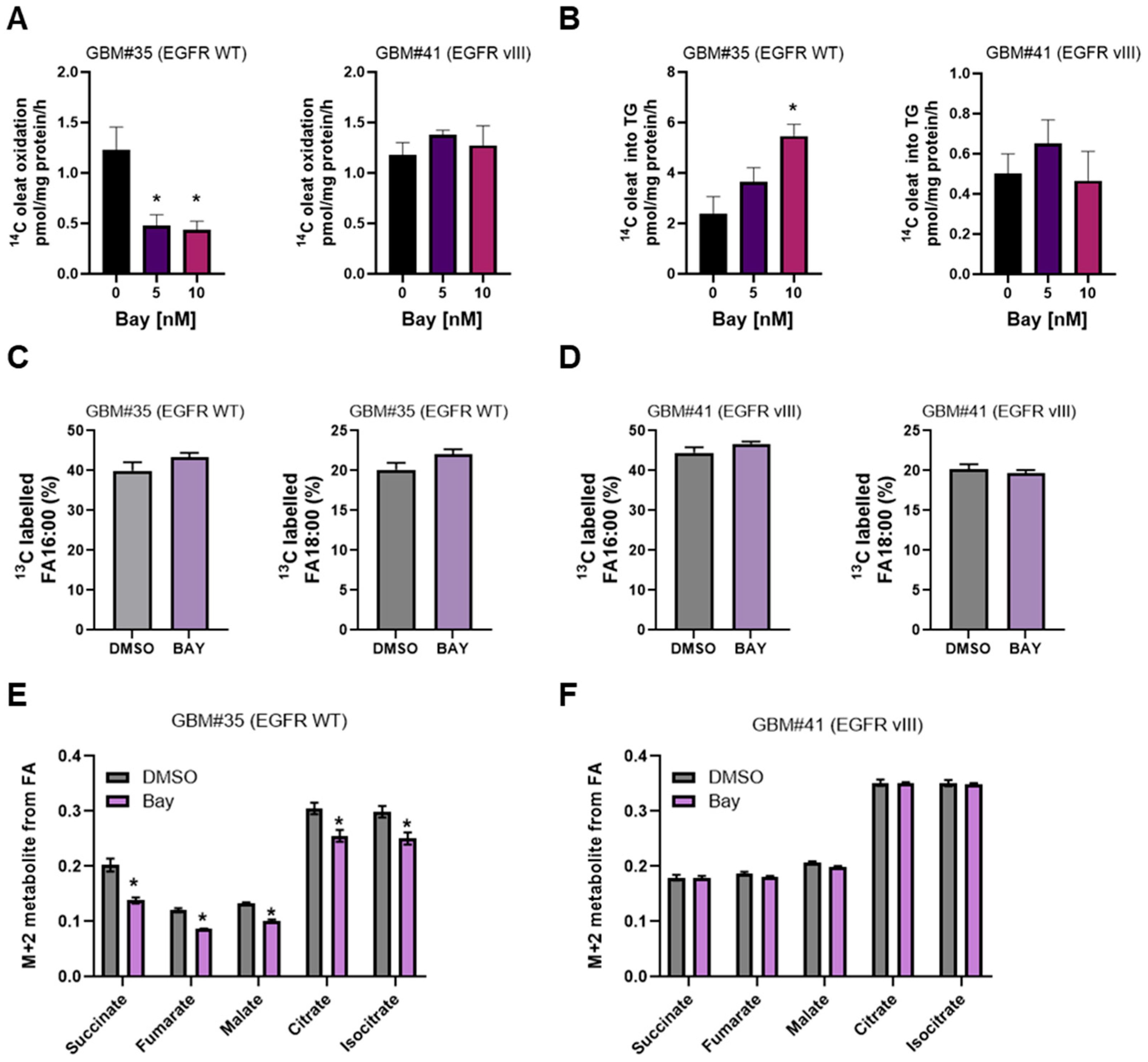
3.3. BAY2402234 Treatment Exerts Differential Regulation on Fatty Acid Metabolism and TCA Cycle Metabolic Flux in EGFR WT Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Reulen, H.J.; Meinel, T.; Pichlmeier, U.; Schumacher, W.; Tonn, J.C.; Rohde, V.; Oppel, F.; Turowski, B.; Woiciechowsky, C.; et al. Extent of resection and survival in glioblastoma multiforme: Identification of and adjustment for bias. Neurosurgery 2008, 62, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ferreira, W.A.; Pinheiro Ddo, R.; Costa Junior, C.A.; Rodrigues-Antunes, S.; Araujo, M.D.; Leao Barros, M.B.; Teixeira, A.C.; Faro, T.A.; Burbano, R.R.; Oliveira, E.H.; et al. An update on the epigenetics of glioblastomas. Epigenomics 2016, 8, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, G.; Wirsching, H.G.; Knobbe-Thomsen, C.B.; Weller, M. Advances in the molecular genetics of gliomas-implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Brennan, C.W.; DeAngelis, L.M.; Omuro, A.M. Emerging therapies for glioblastoma. JAMA Neurol. 2014, 71, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ning, S.; Ghandi, M.; Kryukov, G.V.; Gopal, S.; Deik, A.; Souza, A.; Pierce, K.; Keskula, P.; Hernandez, D.; et al. The landscape of cancer cell line metabolism. Nat. Med. 2019, 25, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.; Oudejans, L.L.; Geugien, M.; Pereira-Martins, D.A.; Wierenga, A.T.J.; Erdem, A.; Sternadt, D.; Huls, G.; Schuringa, J.J. The nonessential amino acid cysteine is required to prevent ferroptosis in acute myeloid leukemia. Blood Adv. 2024, 8, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Papalazarou, V.; Newman, A.C.; Huerta-Uribe, A.; Legrave, N.M.; Falcone, M.; Zhang, T.; McGarry, L.; Athineos, D.; Shanks, E.; Blyth, K.; et al. Phenotypic profiling of solute carriers characterizes serine transport in cancer. Nat. Metab. 2023, 5, 2148–2168. [Google Scholar] [CrossRef] [PubMed]
- Sannino, S.; Manuel, A.M.; Shang, C.; Wendell, S.G.; Wipf, P.; Brodsky, J.L. Non-Essential Amino Acid Availability Influences Proteostasis and Breast Cancer Cell Survival During Proteotoxic Stress. Mol. Cancer Res. 2023, 21, 675–690. [Google Scholar] [CrossRef]
- Yu, N.; Aboud, O. The Lipidomic Signature of Glioblastoma: A Promising Frontier in Cancer Research. Cancers 2024, 16, 1089. [Google Scholar] [CrossRef] [PubMed]
- Bezawork-Geleta, A.; Dimou, J.; Watt, M.J. Lipid droplets and ferroptosis as new players in brain cancer glioblastoma progression and therapeutic resistance. Front. Oncol. 2022, 12, 1085034. [Google Scholar] [CrossRef] [PubMed]
- Puca, F.; Yu, F.; Bartolacci, C.; Pettazzoni, P.; Carugo, A.; Huang-Hobbs, E.; Liu, J.; Zanca, C.; Carbone, F.; Del Poggetto, E.; et al. Medium-Chain Acyl-CoA Dehydrogenase Protects Mitochondria from Lipid Peroxidation in Glioblastoma. Cancer Discov. 2021, 11, 2904–2923. [Google Scholar] [CrossRef] [PubMed]
- Vogel, F.C.E.; Chaves-Filho, A.B.; Schulze, A. Lipids as mediators of cancer progression and metastasis. Nat. Cancer 2024, 5, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Geng, F.; Cheng, X.; Guo, Q.; Zhong, Y.; Cloughesy, T.F.; Yong, W.H.; Chakravarti, A.; Guo, D. Lipid Droplets Maintain Energy Homeostasis and Glioblastoma Growth via Autophagic Release of Stored Fatty Acids. Iscience 2020, 23, 101569. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Perkins, G. Challenges in targeting cancer metabolism for cancer therapy. EMBO Rep. 2012, 13, 1034–1035. [Google Scholar] [CrossRef] [PubMed]
- Heidelberger, C.; Leibman, K.C.; Harbers, E.; Bhargava, P.M. The comparative utilization of uracil-2-C14 by liver, intestinal mucosa, and Flexner-Jobling carcinoma in the rat. Cancer Res. 1957, 17, 399–404. [Google Scholar] [PubMed]
- Rutman, R.J.; Cantarow, A.; Paschkis, K.E. Studies in 2-acetylaminofluorene carcinogenesis. III. The utilization of uracil-2-C14 by preneoplastic rat liver and rat hepatoma. Cancer Res. 1954, 14, 119–123. [Google Scholar] [PubMed]
- White, J.E.; Ricketts, W.N.; Strudwick, W.J. A clinical study of 5-fluorouracil in a variety of far advanced human malignancies. J. Natl. Med. Assoc. 1962, 54, 315–317. [Google Scholar] [PubMed]
- Galmarini, C.M.; Thomas, X.; Calvo, F.; Rousselot, P.; Rabilloud, M.; El Jaffari, A.; Cros, E.; Dumontet, C. In vivo mechanisms of resistance to cytarabine in acute myeloid leukaemia. Br. J. Haematol. 2002, 117, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Gwynne, W.D.; Suk, Y.; Custers, S.; Mikolajewicz, N.; Chan, J.K.; Zador, Z.; Chafe, S.C.; Zhai, K.; Escudero, L.; Zhang, C.; et al. Cancer-selective metabolic vulnerabilities in MYC-amplified medulloblastoma. Cancer Cell 2022, 40, 1488–1502. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Kaplan, J.P.; Nguyen, H.; Stopka, S.A.; Savani, M.R.; Regan, M.S.; Nguyen, Q.D.; Jones, K.L.; Moreau, L.A.; Peng, J.; et al. A druggable addiction to de novo pyrimidine biosynthesis in diffuse midline glioma. Cancer Cell 2022, 40, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.D.; Savani, M.R.; Levitt, M.M.; Wang, A.C.; Endress, J.E.; Bird, C.E.; Buehler, J.; Stopka, S.A.; Regan, M.S.; Lin, Y.F.; et al. De novo pyrimidine synthesis is a targetable vulnerability in IDH mutant glioma. Cancer Cell 2022, 40, 939–956. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.F.; Bezawork-Geleta, A.; Areeb, Z.; Gomez, J.; Tsui, V.; Zulkifli, A.; Paradiso, L.; Jones, J.; Nguyen, H.P.T.; Putoczki, T.L.; et al. The Interleukin-11/IL-11 Receptor Promotes Glioblastoma Survival and Invasion under Glucose-Starved Conditions through Enhanced Glutaminolysis. Int. J. Mol. Sci. 2023, 24, 3356. [Google Scholar] [CrossRef] [PubMed]
- Dagley, M.J.; McConville, M.J. DExSI: A new tool for the rapid quantitation of 13C-labelled metabolites detected by GC-MS. Bioinformatics 2018, 34, 1957–1958. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Barlow, C.K.; Jayawardana, K.S.; Weir, J.M.; Mellett, N.A.; Cinel, M.; Magliano, D.J.; Shaw, J.E.; Drew, B.G.; Meikle, P.J. High-Throughput Plasma Lipidomics: Detailed Mapping of the Associations with Cardiometabolic Risk Factors. Cell Chem. Biol. 2019, 26, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Pernes, G.; Mellett, N.A.; Meikle, P.J.; Murphy, A.J.; Lancaster, G.I. Lipidomic Profiling of Murine Macrophages Treated with Fatty Acids of Varying Chain Length and Saturation Status. Metabolites 2018, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Bezawork-Geleta, A.; Wen, H.; Dong, L.; Yan, B.; Vider, J.; Boukalova, S.; Krobova, L.; Vanova, K.; Zobalova, R.; Sobol, M.; et al. Alternative assembly of respiratory complex II connects energy stress to metabolic checkpoints. Nat. Commun. 2018, 9, 2221. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Narita, Y.; Arakawa, Y.; Yamasaki, F.; Nishikawa, R.; Aoki, T.; Kanamori, M.; Nagane, M.; Kumabe, T.; Hirose, Y.; Ichikawa, T.; et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro Oncol. 2019, 21, 348–359. [Google Scholar] [CrossRef]
- Rouse, C.; Gittleman, H.; Ostrom, Q.T.; Kruchko, C.; Barnholtz-Sloan, J.S. Years of potential life lost for brain and CNS tumors relative to other cancers in adults in the United States, 2010. Neuro Oncol. 2016, 18, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.B. The emergence of dihydroorotate dehydrogenase (DHODH) as a therapeutic target in acute myeloid leukemia. Expert. Opin. Ther. Targets 2018, 22, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Wang, J.; Ren, C.; Tang, P.; Ouyang, L.; Wang, Y. Recent advances of human dihydroorotate dehydrogenase inhibitors for cancer therapy: Current development and future perspectives. Eur. J. Med. Chem. 2022, 232, 114176. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, T.; Mazzoni, M.; Greco, A.; Cassinelli, G. Non-oncogene dependencies: Novel opportunities for cancer therapy. Biochem. Pharmacol. 2024, 116254. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, L.; Zhou, X.; Zuo, Z.; Gong, J.; Liu, X.; Zhou, Y.; Liu, C.; Sang, N.; Liu, H.; et al. DHODH and cancer: Promising prospects to be explored. Cancer Metab. 2021, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.D.; Aziz, M.; Dyer, J.M.; Mullen, R.T. Mechanisms of lipid droplet biogenesis. Biochem. J. 2019, 476, 1929–1942. [Google Scholar] [CrossRef]
- Klemm, R.W.; Ikonen, E. The cell biology of lipid droplets: More than just a phase. Semin. Cell Dev. Biol. 2020, 108, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Piccolis, M.; Bond, L.M.; Kampmann, M.; Pulimeno, P.; Chitraju, C.; Jayson, C.B.K.; Vaites, L.P.; Boland, S.; Lai, Z.W.; Gabriel, K.R.; et al. Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids. Mol. Cell 2019, 74, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Rozeveld, C.N.; Johnson, K.M.; Zhang, L.; Razidlo, G.L. KRAS Controls Pancreatic Cancer Cell Lipid Metabolism and Invasive Potential through the Lipase HSL. Cancer Res. 2020, 80, 4932–4945. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Hagen, J.T.; Neufer, P.D.; Kassai, M.; Cabot, M.C. On the nature of ceramide-mitochondria interactions—Dissection using comprehensive mitochondrial phenotyping. Cell Signal. 2021, 78, 109838. [Google Scholar] [CrossRef] [PubMed]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines--old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezawork-Geleta, A.; Moujalled, D.; De Souza, D.P.; Narayana, V.K.; Dimou, J.; Luwor, R.; Watt, M.J. Metabolic Plasticity of Glioblastoma Cells in Response to DHODH Inhibitor BAY2402234 Treatment. Metabolites 2024, 14, 413. https://doi.org/10.3390/metabo14080413
Bezawork-Geleta A, Moujalled D, De Souza DP, Narayana VK, Dimou J, Luwor R, Watt MJ. Metabolic Plasticity of Glioblastoma Cells in Response to DHODH Inhibitor BAY2402234 Treatment. Metabolites. 2024; 14(8):413. https://doi.org/10.3390/metabo14080413
Chicago/Turabian StyleBezawork-Geleta, Ayenachew, Diane Moujalled, David P. De Souza, Vinod K. Narayana, James Dimou, Rodney Luwor, and Matthew J. Watt. 2024. "Metabolic Plasticity of Glioblastoma Cells in Response to DHODH Inhibitor BAY2402234 Treatment" Metabolites 14, no. 8: 413. https://doi.org/10.3390/metabo14080413
APA StyleBezawork-Geleta, A., Moujalled, D., De Souza, D. P., Narayana, V. K., Dimou, J., Luwor, R., & Watt, M. J. (2024). Metabolic Plasticity of Glioblastoma Cells in Response to DHODH Inhibitor BAY2402234 Treatment. Metabolites, 14(8), 413. https://doi.org/10.3390/metabo14080413