
Automated Liquid Handling Extraction and Rapid Quantification of Underivatized Amino Acids and Tryptophan Metabolites from Human Serum and Plasma Using Dual-Column U(H)PLC-MRM-MS and Its Application to Prostate Cancer Study

 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Metabolite Standards
2.3. U(H)PLC-MRM-MS
2.4. Method Validation
2.5. Manual Extraction from Serum
2.6. Semi-Automated Extraction from Serum and Plasma
3. Results
3.1. U(H)PLC-MRM-MS Method Development
3.2. Linearity, Lower Limit of Detection (Llod), and Lower Limit of Quantification (Lloq)
3.3. Within-Run and between-Run Accuracy and Precision
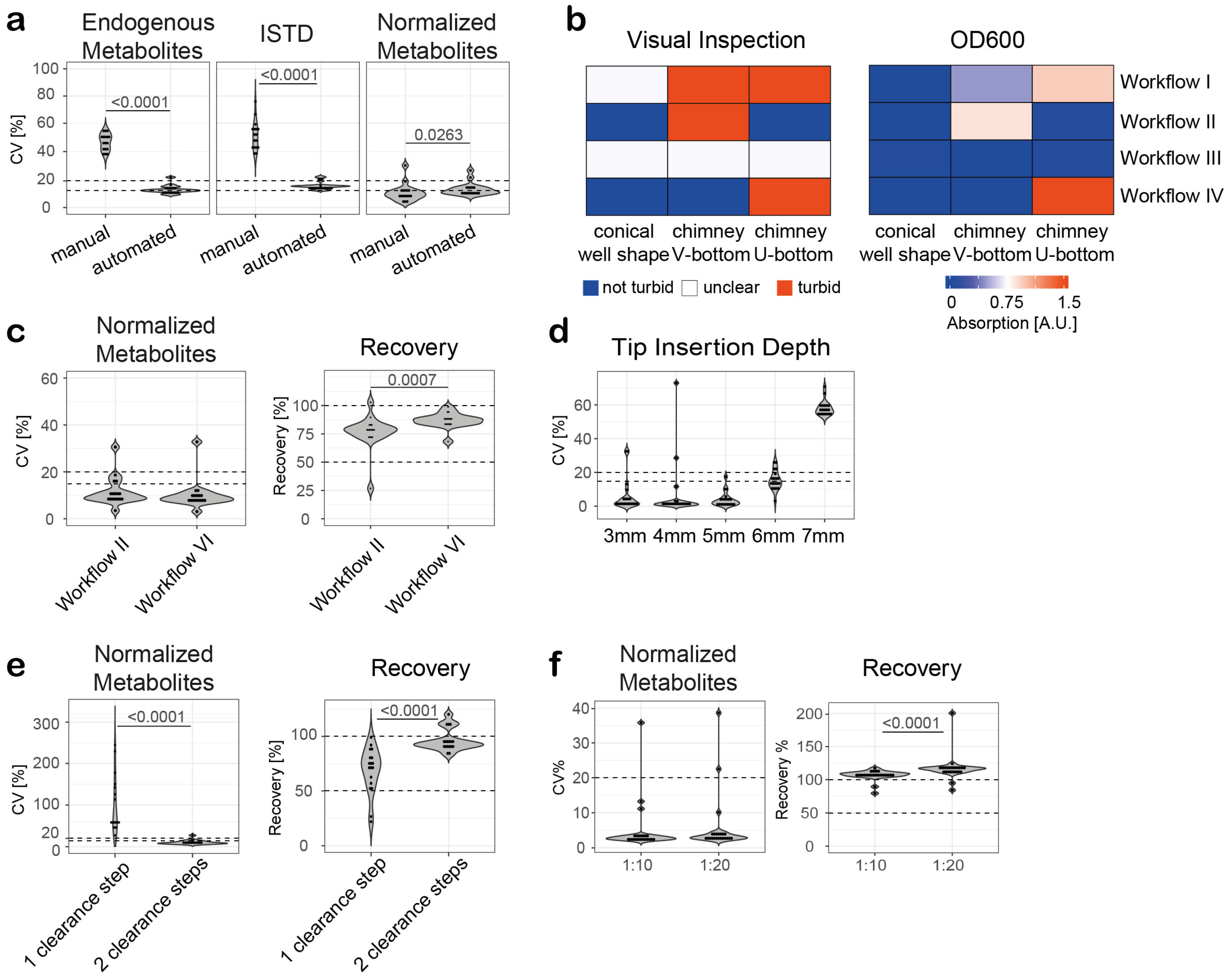
3.4. Semi-Automated Extraction and Sample Preparation of Human Serum and Plasma
3.5. Manual Extraction and Sample Preparation Versus Semi-Automated Workflow
3.6. Optimization of the Semi-Automated Extraction for Serum Samples
3.7. Optimization of the Semi-Automated Extraction for Plasma Samples
3.8. Recovery
3.9. Intra-Assay Variability and Inter-Assay Precision
3.10. Autosampler Stability
3.11. Analysis of NIST SRM 1950 Reference Plasma Sample
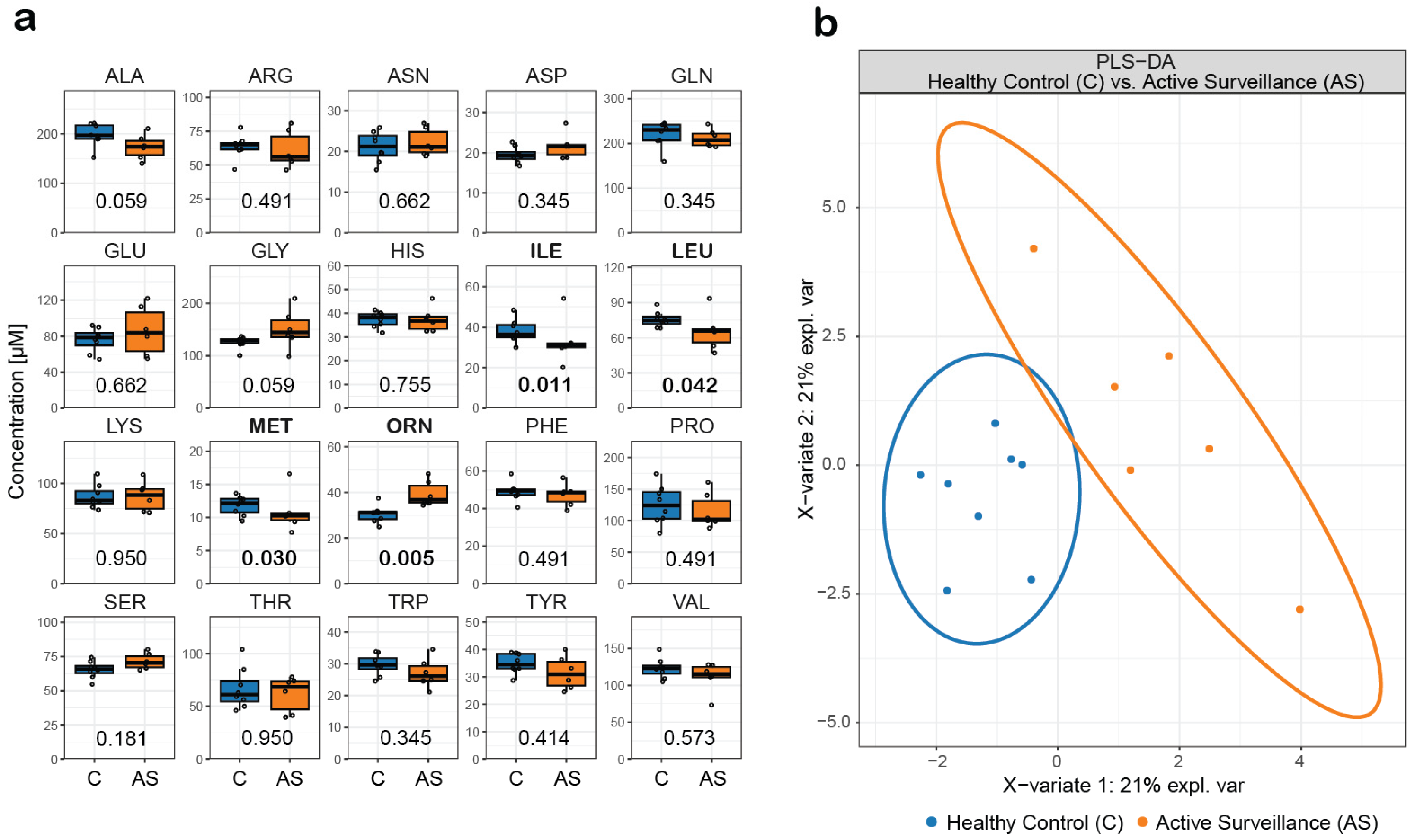
3.12. Automated Extraction and Analysis of Serum Samples from a Prostate Cancer Study
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Sadik, A.; Somarribas Patterson, L.F.; Ozturk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfander, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270.e34. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Li, G.; Zheng, Q.; Gu, X.; Shi, Q.; Su, Y.; Chu, Q.; Yuan, X.; Bao, Z.; Lu, J.; et al. Tryptophan metabolism in health and disease. Cell Metab. 2023, 35, 1304–1326. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef] [PubMed]
- Aliu, E.; Kanungo, S.; Arnold, G.L. Amino acid disorders. Ann. Transl. Med. 2018, 6, 471. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zheng, W.; Locasale, J.W. Amino acid variability, tradeoffs and optimality in human diet. Nat. Commun. 2022, 13, 6683. [Google Scholar] [CrossRef] [PubMed]
- La Du, B.N.; Zannoni, V.G.; Laster, L.; Seegmiller, J.E. The nature of the defect in tyrosine metabolism in alcaptonuria. J. Biol. Chem. 1958, 230, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Akter, S.; Kuwahara, K.; Matsushita, Y.; Nakagawa, T.; Konishi, M.; Honda, T.; Yamamoto, S.; Hayashi, T.; Noda, M.; et al. Serum amino acid profiles and risk of type 2 diabetes among Japanese adults in the Hitachi Health Study. Sci. Rep. 2019, 9, 7010. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef]
- Ling, Z.N.; Jiang, Y.F.; Ru, J.N.; Lu, J.H.; Ding, B.; Wu, J. Amino acid metabolism in health and disease. Signal Transduct. Target. Ther. 2023, 8, 345. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, C.; Zhang, Q.; Liu, Z. Metabolomic profiling of amino acids study reveals a distinct diagnostic model for diabetic kidney disease. Amino Acids 2023, 55, 1563–1572. [Google Scholar] [CrossRef]
- Kong, F.; Yuan, L.; Zheng, Y.F.; Chen, W. Automatic liquid handling for life science: A critical review of the current state of the art. J. Lab. Autom. 2012, 17, 169–185. [Google Scholar] [CrossRef]
- Ellinger, J.J.; Miller, D.C.; Lewis, I.A.; Markley, J.L. Semiautomated device for batch extraction of metabolites from tissue samples. Anal. Chem. 2012, 84, 1809–1812. [Google Scholar] [CrossRef]
- Malinowska, J.M.; Palosaari, T.; Sund, J.; Carpi, D.; Lloyd, G.R.; Weber, R.J.M.; Whelan, M.; Viant, M.R. Automated Sample Preparation and Data Collection Workflow for High-Throughput In Vitro Metabolomics. Metabolites 2022, 12, 52. [Google Scholar] [CrossRef]
- Sadok, I.; Gamian, A.; Staniszewska, M.M. Chromatographic analysis of tryptophan metabolites. J. Sep. Sci. 2017, 40, 3020–3045. [Google Scholar] [CrossRef]
- Whiley, L.; Nye, L.C.; Grant, I.; Andreas, N.; Chappell, K.E.; Sarafian, M.H.; Misra, R.; Plumb, R.S.; Lewis, M.R.; Nicholson, J.K.; et al. Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry with Electrospray Ionization Quantification of Tryptophan Metabolites and Markers of Gut Health in Serum and Plasma-Application to Clinical and Epidemiology Cohorts. Anal. Chem. 2019, 91, 5207–5216. [Google Scholar] [CrossRef]
- Panitz, V.; Koncarevic, S.; Sadik, A.; Friedel, D.; Bausbacher, T.; Trump, S.; Farztdinov, V.; Schulz, S.; Sievers, P.; Schmidt, S.; et al. Tryptophan metabolism is inversely regulated in the tumor and blood of patients with glioblastoma. Theranostics 2021, 11, 9217–9233. [Google Scholar] [CrossRef]
- Desmons, A.; Humbert, L.; Eguether, T.; Krasniqi, P.; Rainteau, D.; Mahdi, T.; Kapel, N.; Lamaziere, A. High performance liquid chromatography-tandem mass spectrometry quantification of tryptophan metabolites in human serum and stool—Application to clinical cohorts in Inflammatory Bowel Diseases. J. Chromatogr. A 2022, 1685, 463602. [Google Scholar] [CrossRef]
- Betts, M.; Russell, R. Amino Acid Properties and Consequences of Substitutions. In Bioinformatics for Geneticists; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2003. [Google Scholar]
- Furst, P.; Pollack, L.; Graser, T.A.; Godel, H.; Stehle, P. Appraisal of four pre-column derivatization methods for the high-performance liquid chromatographic determination of free amino acids in biological materials. J. Chromatogr. 1990, 499, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Fiechter, G.; Mayer, H.K. Characterization of amino acid profiles of culture media via pre-column 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate derivatization and ultra performance liquid chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 1353–1360. [Google Scholar] [CrossRef]
- Ferre, S.; Gonzalez-Ruiz, V.; Guillarme, D.; Rudaz, S. Analytical strategies for the determination of amino acids: Past, present and future trends. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1132, 121819. [Google Scholar] [CrossRef]
- Smon, A.; Cuk, V.; Brecelj, J.; Murko, S.; Groselj, U.; Zerjav Tansek, M.; Battelino, T.; Repic Lampret, B. Comparison of liquid chromatography with tandem mass spectrometry and ion-exchange chromatography by post-column ninhydrin derivatization for amino acid monitoring. Clin. Chim. Acta 2019, 495, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Armenta, J.M.; Cortes, D.F.; Pisciotta, J.M.; Shuman, J.L.; Blakeslee, K.; Rasoloson, D.; Ogunbiyi, O.; Sullivan, D.J., Jr.; Shulaev, V. Sensitive and rapid method for amino acid quantitation in malaria biological samples using AccQ.Tag ultra performance liquid chromatography-electrospray ionization-MS/MS with multiple reaction monitoring. Anal. Chem. 2010, 82, 548–558. [Google Scholar] [CrossRef]
- Ubhi, B.K.; Davenport, P.W.; Welch, M.; Riley, J.; Griffin, J.L.; Connor, S.C. Analysis of chloroformate-derivatised amino acids, dipeptides and polyamines by LC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 934, 79–88. [Google Scholar] [CrossRef]
- Piraud, M.; Vianey-Saban, C.; Petritis, K.; Elfakir, C.; Steghens, J.P.; Morla, A.; Bouchu, D. ESI-MS/MS analysis of underivatised amino acids: A new tool for the diagnosis of inherited disorders of amino acid metabolism. Fragmentation study of 79 molecules of biological interest in positive and negative ionisation mode. Rapid Commun. Mass. Spectrom. 2003, 17, 1297–1311. [Google Scholar] [CrossRef]
- Piraud, M.; Vianey-Saban, C.; Petritis, K.; Elfakir, C.; Steghens, J.P.; Bouchu, D. Ion-pairing reversed-phase liquid chromatography/electrospray ionization mass spectrometric analysis of 76 underivatized amino acids of biological interest: A new tool for the diagnosis of inherited disorders of amino acid metabolism. Rapid Commun. Mass. Spectrom. 2005, 19, 1587–1602. [Google Scholar] [CrossRef] [PubMed]
- de Person, M.; Chaimbault, P.; Elfakir, C. Analysis of native amino acids by liquid chromatography/electrospray ionization mass spectrometry: Comparative study between two sources and interfaces. J. Mass. Spectrom. 2008, 43, 204–215. [Google Scholar] [CrossRef]
- Krumpochova, P.; Bruyneel, B.; Molenaar, D.; Koukou, A.; Wuhrer, M.; Niessen, W.M.; Giera, M. Amino acid analysis using chromatography-mass spectrometry: An inter platform comparison study. J. Pharm. Biomed. Anal. 2015, 114, 398–407. [Google Scholar] [CrossRef]
- Zhou, G.; Pang, H.; Tang, Y.; Yao, X.; Mo, X.; Zhu, S.; Guo, S.; Qian, D.; Qian, Y.; Su, S.; et al. Hydrophilic interaction ultra-performance liquid chromatography coupled with triple-quadrupole tandem mass spectrometry for highly rapid and sensitive analysis of underivatized amino acids in functional foods. Amino Acids 2013, 44, 1293–1305. [Google Scholar] [CrossRef]
- Ammann, A.A.; Suter, M.J. Multimode gradient high performance liquid chromatography mass spectrometry method applicable to metabolomics and environmental monitoring. J. Chromatogr. A 2016, 1456, 145–151. [Google Scholar] [CrossRef]
- Choi, M.S.; Rehman, S.U.; Kim, I.S.; Park, H.J.; Song, M.Y.; Yoo, H.H. Development of a mixed-mode chromatography with tandem mass spectrometry method for the quantitative analysis of 23 underivatized amino acids in human serum. J. Pharm. Biomed. Anal. 2017, 145, 52–58. [Google Scholar] [CrossRef]
- DeArmond, P.D.; Bunch, D.R. Quantitation of non-derivatized free amino acids for detecting inborn errors of metabolism by incorporating mixed-mode chromatography with tandem mass spectrometry. J. Mass. Spectrom. Adv. Clin. Lab. 2022, 25, 1–11. [Google Scholar] [CrossRef]
- Ghosson, H.; Schwarzenberg, A.; Jamois, F.; Yvin, J.C. Simultaneous untargeted and targeted metabolomics profiling of underivatized primary metabolites in sulfur-deficient barley by ultra-high performance liquid chromatography-quadrupole/time-of-flight mass spectrometry. Plant Methods 2018, 14, 62. [Google Scholar] [CrossRef]
- Vilches, A.P.; Norstrom, S.H.; Bylund, D. Direct analysis of free amino acids by mixed-mode chromatography with tandem mass spectrometry. J. Sep. Sci. 2017, 40, 1482–1492. [Google Scholar] [CrossRef]
- Munjoma, N.; Isaac, G.; Muazzam, A.; Cexus, O.; Azhar, F.; Pandha, H.; Whetton, A.D.; Townsend, P.A.; Wilson, I.D.; Gethings, L.A.; et al. High Throughput LC-MS Platform for Large Scale Screening of Bioactive Polar Lipids in Human Plasma and Serum. J. Proteome Res. 2022, 21, 2596–2608. [Google Scholar] [CrossRef]
- van Pijkeren, A.; Egger, A.S.; Hotze, M.; Zimmermann, E.; Kipura, T.; Grander, J.; Gollowitzer, A.; Koeberle, A.; Bischoff, R.; Thedieck, K.; et al. Proteome Coverage after Simultaneous Proteo-Metabolome Liquid-Liquid Extraction. J. Proteome Res. 2023, 22, 951–966. [Google Scholar] [CrossRef]
- Anderson, D.J. Determination of the lower limit of detection. Clin. Chem. 1989, 35, 2152–2153. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, H.; Shi, G.; Yang, M.; Zheng, F.; Zheng, Z.; Zhang, S.; Zhong, S. Ultra-performance liquid chromatography-tandem mass spectrometry quantitative profiling of tryptophan metabolites in human plasma and its application to clinical study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1128, 121745. [Google Scholar] [CrossRef]
- Lai, Y.; Liu, C.W.; Chi, L.; Ru, H.; Lu, K. High-Resolution Metabolomics of 50 Neurotransmitters and Tryptophan Metabolites in Feces, Serum, and Brain Tissues Using UHPLC-ESI-Q Exactive Mass Spectrometry. ACS Omega 2021, 6, 8094–8103. [Google Scholar] [CrossRef]
- Hylen, U.; Sarndahl, E.; Bejerot, S.; Humble, M.B.; Hyotylainen, T.; Salihovic, S.; Eklund, D. Alterations in inflammasome-related immunometabolites in individuals with severe psychiatric disorders. BMC Psychiatry 2023, 23, 268. [Google Scholar] [CrossRef] [PubMed]
- Bobaly, B.; Beck, A.; Fekete, J.; Guillarme, D.; Fekete, S. Systematic evaluation of mobile phase additives for the LC-MS characterization of therapeutic proteins. Talanta 2015, 136, 60–67. [Google Scholar] [CrossRef]
- Khalikova, M.A.; Skarbalius, L.; Naplekov, D.K.; Jadeja, S.; Svec, F.; Lenco, J. Evaluation of strategies for overcoming trifluoroacetic acid ionization suppression resulted in single-column intact level, middle-up, and bottom-up reversed-phase LC-MS analyses of antibody biopharmaceuticals. Talanta 2021, 233, 122512. [Google Scholar] [CrossRef]
- Yamamoto, E.; Ishihama, Y.; Asakawa, N. Application of partially fluorinated carboxylic acids as ion-pairing reagents in LC/ESI-MS. Talanta 2014, 127, 219–224. [Google Scholar] [CrossRef]
- Nguyen, J.M.; Smith, J.; Rzewuski, S.; Legido-Quigley, C.; Lauber, M.A. High sensitivity LC-MS profiling of antibody-drug conjugates with difluoroacetic acid ion pairing. mAbs 2019, 11, 1358–1366. [Google Scholar] [CrossRef]
- Michel, M.; Salvador, C.; Wiedemair, V.; Adam, M.G.; Laser, K.T.; Dubowy, K.O.; Entenmann, A.; Karall, D.; Geiger, R.; Zlamy, M.; et al. Method comparison of HPLC-ninhydrin-photometry and UHPLC-PITC-tandem mass spectrometry for serum amino acid analyses in patients with complex congenital heart disease and controls. Metabolomics 2020, 16, 128. [Google Scholar] [CrossRef]
- Liu, Z.; Tu, M.J.; Zhang, C.; Jilek, J.L.; Zhang, Q.Y.; Yu, A.M. A reliable LC-MS/MS method for the quantification of natural amino acids in mouse plasma: Method validation and application to a study on amino acid dynamics during hepatocellular carcinoma progression. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1124, 72–81. [Google Scholar] [CrossRef]
- Schafheimer, N.; King, J. Tryptophan cluster protects human gammaD-crystallin from ultraviolet radiation-induced photoaggregation in vitro. Photochem. Photobiol. 2013, 89, 1106–1115. [Google Scholar] [CrossRef]
- Gray, N.; Zia, R.; King, A.; Patel, V.C.; Wendon, J.; McPhail, M.J.; Coen, M.; Plumb, R.S.; Wilson, I.D.; Nicholson, J.K. High-Speed Quantitative UPLC-MS Analysis of Multiple Amines in Human Plasma and Serum via Precolumn Derivatization with 6-Aminoquinolyl-N-hydroxysuccinimidyl Carbamate: Application to Acetaminophen-Induced Liver Failure. Anal. Chem. 2017, 89, 2478–2487. [Google Scholar] [CrossRef]
- Gray, N.; Lawler, N.G.; Yang, R.; Morillon, A.C.; Gay, M.C.L.; Bong, S.H.; Holmes, E.; Nicholson, J.K.; Whiley, L. A simultaneous exploratory and quantitative amino acid and biogenic amine metabolic profiling platform for rapid disease phenotyping via UPLC-QToF-MS. Talanta 2021, 223, 121872. [Google Scholar] [CrossRef]
- Thompson, J.W.; Adams, K.J.; Adamski, J.; Asad, Y.; Borts, D.; Bowden, J.A.; Byram, G.; Dang, V.; Dunn, W.B.; Fernandez, F.; et al. International Ring Trial of a High Resolution Targeted Metabolomics and Lipidomics Platform for Serum and Plasma Analysis. Anal. Chem. 2019, 91, 14407–14416. [Google Scholar] [CrossRef]
- Belkahla, S.; Nahvi, I.; Biswas, S.; Nahvi, I.; Ben Amor, N. Advances and development of prostate cancer, treatment, and strategies: A systemic review. Front. Cell Dev. Biol. 2022, 10, 991330. [Google Scholar] [CrossRef]
- Bergengren, O.; Pekala, K.R.; Matsoukas, K.; Fainberg, J.; Mungovan, S.F.; Bratt, O.; Bray, F.; Brawley, O.; Luckenbaugh, A.N.; Mucci, L.; et al. 2022 Update on Prostate Cancer Epidemiology and Risk Factors—A Systematic Review. Eur. Urol. 2023, 84, 191–206. [Google Scholar] [CrossRef]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef]
- Martin, R.M.; Donovan, J.L.; Turner, E.L.; Metcalfe, C.; Young, G.J.; Walsh, E.I.; Lane, J.A.; Noble, S.; Oliver, S.E.; Evans, S.; et al. Effect of a Low-Intensity PSA-Based Screening Intervention on Prostate Cancer Mortality: The CAP Randomized Clinical Trial. JAMA 2018, 319, 883–895. [Google Scholar] [CrossRef]
- Ahmad, F.; Cherukuri, M.K.; Choyke, P.L. Metabolic reprogramming in prostate cancer. Br. J. Cancer 2021, 125, 1185–1196. [Google Scholar] [CrossRef]
- Strmiska, V.; Michalek, P.; Eckschlager, T.; Stiborova, M.; Adam, V.; Krizkova, S.; Heger, Z. Prostate cancer-specific hallmarks of amino acids metabolism: Towards a paradigm of precision medicine. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 248–258. [Google Scholar] [CrossRef]
- Derezinski, P.; Klupczynska, A.; Sawicki, W.; Palka, J.A.; Kokot, Z.J. Amino Acid Profiles of Serum and Urine in Search for Prostate Cancer Biomarkers: A Pilot Study. Int. J. Med. Sci. 2017, 14, 1–12. [Google Scholar] [CrossRef]
- Miyagi, Y.; Higashiyama, M.; Gochi, A.; Akaike, M.; Ishikawa, T.; Miura, T.; Saruki, N.; Bando, E.; Kimura, H.; Imamura, F.; et al. Plasma free amino acid profiling of five types of cancer patients and its application for early detection. PLoS ONE 2011, 6, e24143. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Metabolite | LLOD [µM] | LLOQ [µM] |
|---|---|---|
| 3-hydroxykynurenine | 0.097 | 0.292 |
| Alanine | 0.140 | 0.420 |
| Arginine | 0.147 | 0.441 |
| Asparagine | 0.179 | 0.536 |
| Aspartic acid | 0.141 | 0.424 |
| Glutamine | 0.111 | 0.334 |
| Glutamic acid | 0.280 | 0.841 |
| Glycine | 0.822 | 2.467 |
| Histidine | 0.184 | 0.552 |
| Indole-3-acetic acid | 0.185 | 0.555 |
| Isoleucine | 0.124 | 0.372 |
| Kynurenine | 0.128 | 0.385 |
| Kynurenic acid | 0.053 | 0.158 |
| Leucine | 0.142 | 0.425 |
| Lysine | 0.155 | 0.465 |
| Methionine | 0.114 | 0.342 |
| Nicotinic acid | 0.082 | 0.245 |
| Nicotinamide | 0.075 | 0.226 |
| Ornithine | 0.121 | 0.363 |
| Phenylalanine | 0.178 | 0.534 |
| Proline | 0.086 | 0.257 |
| Serine | 0.198 | 0.594 |
| Threonine | 0.127 | 0.381 |
| Tryptophan | 0.097 | 0.292 |
| Tyrosine | 0.167 | 0.501 |
| Valine | 0.123 | 0.368 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kipura, T.; Hotze, M.; Hofer, A.; Egger, A.-S.; Timpen, L.E.; Opitz, C.A.; Townsend, P.A.; Gethings, L.A.; Thedieck, K.; Kwiatkowski, M. Automated Liquid Handling Extraction and Rapid Quantification of Underivatized Amino Acids and Tryptophan Metabolites from Human Serum and Plasma Using Dual-Column U(H)PLC-MRM-MS and Its Application to Prostate Cancer Study. Metabolites 2024, 14, 370. https://doi.org/10.3390/metabo14070370
Kipura T, Hotze M, Hofer A, Egger A-S, Timpen LE, Opitz CA, Townsend PA, Gethings LA, Thedieck K, Kwiatkowski M. Automated Liquid Handling Extraction and Rapid Quantification of Underivatized Amino Acids and Tryptophan Metabolites from Human Serum and Plasma Using Dual-Column U(H)PLC-MRM-MS and Its Application to Prostate Cancer Study. Metabolites. 2024; 14(7):370. https://doi.org/10.3390/metabo14070370
Chicago/Turabian StyleKipura, Tobias, Madlen Hotze, Alexa Hofer, Anna-Sophia Egger, Lea E. Timpen, Christiane A. Opitz, Paul A. Townsend, Lee A. Gethings, Kathrin Thedieck, and Marcel Kwiatkowski. 2024. "Automated Liquid Handling Extraction and Rapid Quantification of Underivatized Amino Acids and Tryptophan Metabolites from Human Serum and Plasma Using Dual-Column U(H)PLC-MRM-MS and Its Application to Prostate Cancer Study" Metabolites 14, no. 7: 370. https://doi.org/10.3390/metabo14070370
APA StyleKipura, T., Hotze, M., Hofer, A., Egger, A.-S., Timpen, L. E., Opitz, C. A., Townsend, P. A., Gethings, L. A., Thedieck, K., & Kwiatkowski, M. (2024). Automated Liquid Handling Extraction and Rapid Quantification of Underivatized Amino Acids and Tryptophan Metabolites from Human Serum and Plasma Using Dual-Column U(H)PLC-MRM-MS and Its Application to Prostate Cancer Study. Metabolites, 14(7), 370. https://doi.org/10.3390/metabo14070370