

In Vitro Astroglial Dysfunction Induced by Neurotoxins: Mimicking Astrocytic Metabolic Alterations of Alzheimer’s Disease

 ,
,  and
and
Abstract
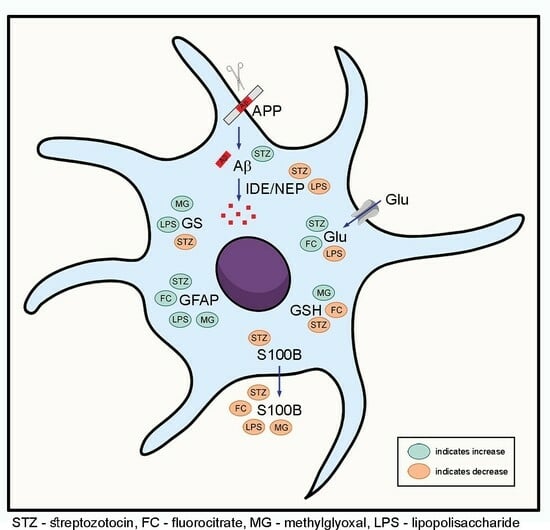
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Experimental Design
2.4. Methyl Thiazolyl Diphenyl-Tetrazolium Bromide Assay
2.5. Neutral Red Incorporation Assay
2.6. Propidium Iodide Uptake Assay
2.7. Assessment of Lactate Dehydrogenase Activity
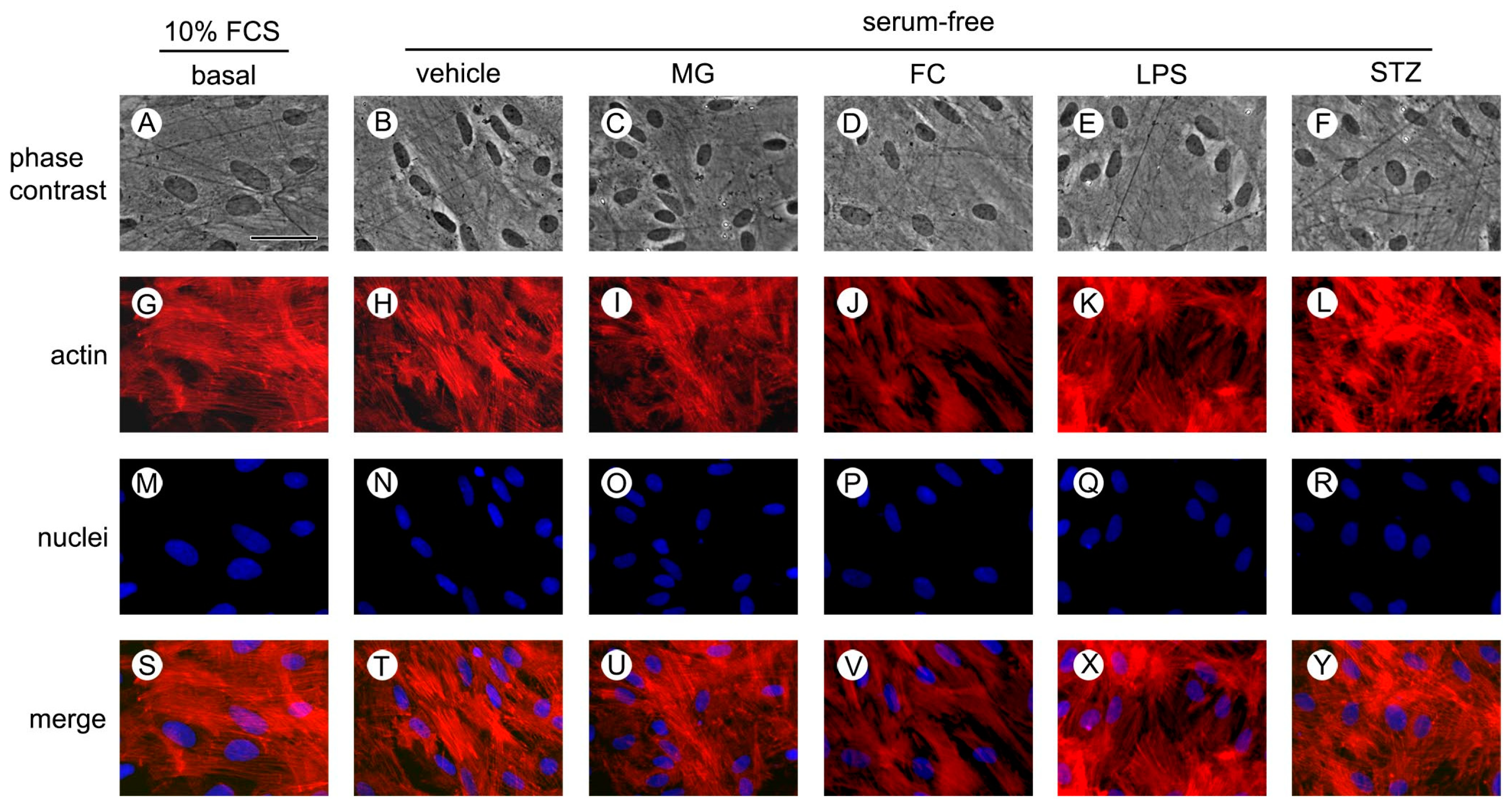
2.8. Cytochemistry for Actin and Nuclei
2.9. S100B Measurement
2.10. Glial Fibrillary Acidic Protein Measurement
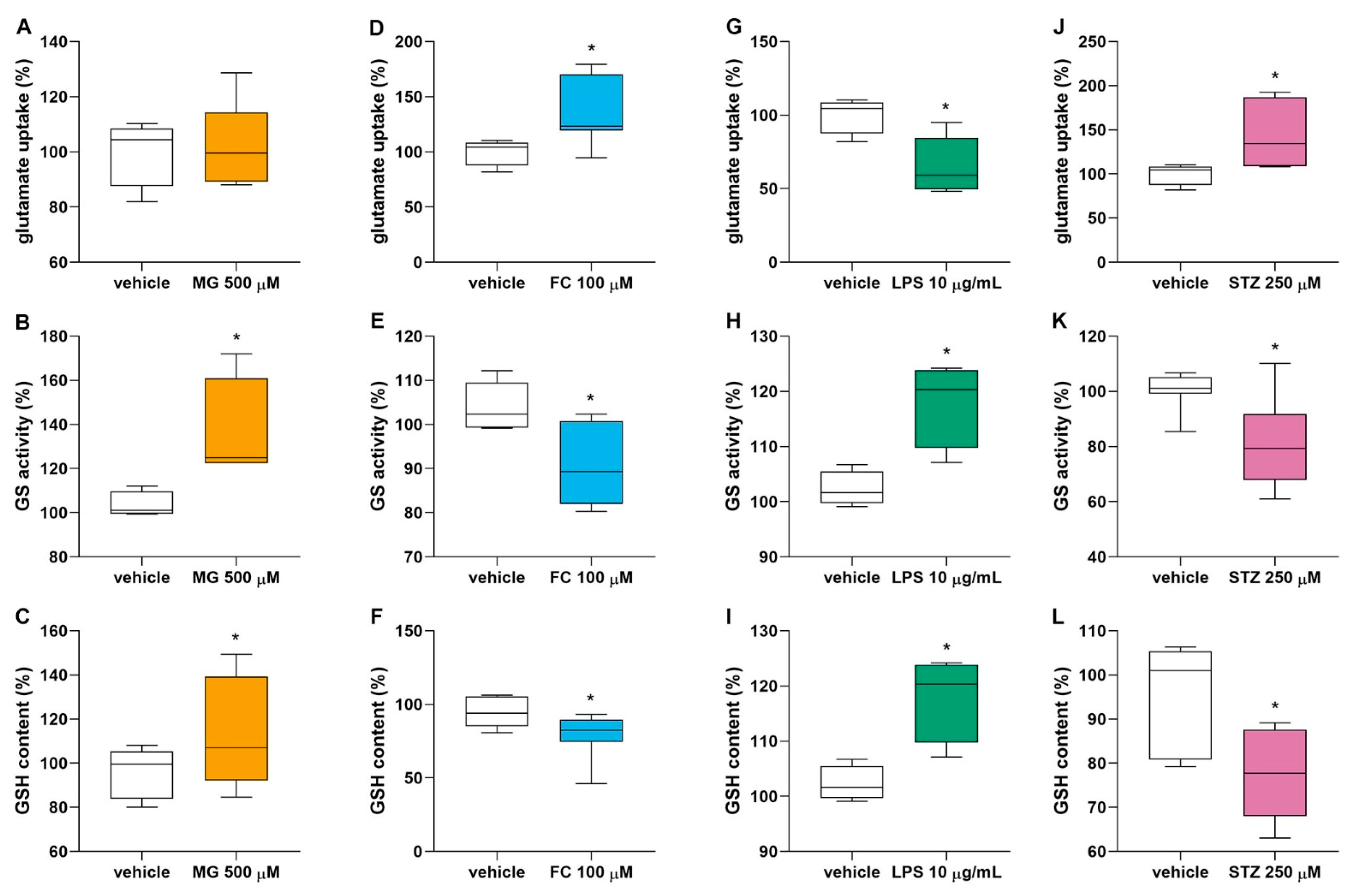
2.11. Glutamate Uptake Assay
2.12. Reduced Glutathione Content Assay
2.13. Glutamine Synthetase Activity
2.14. Western Blot Analysis
2.15. Protein Content
2.16. Statistical Analysis
3. Results
3.1. MG, FC, LPS and STZ Do Not Compromise Cell Viability, Integrity and Morphology
3.2. MG, FC, LPS and STZ Affect Astrocyte Protein Markers
3.3. MG, FC, LPS and STZ Affect the Glutamate Metabolism of Astrocytes
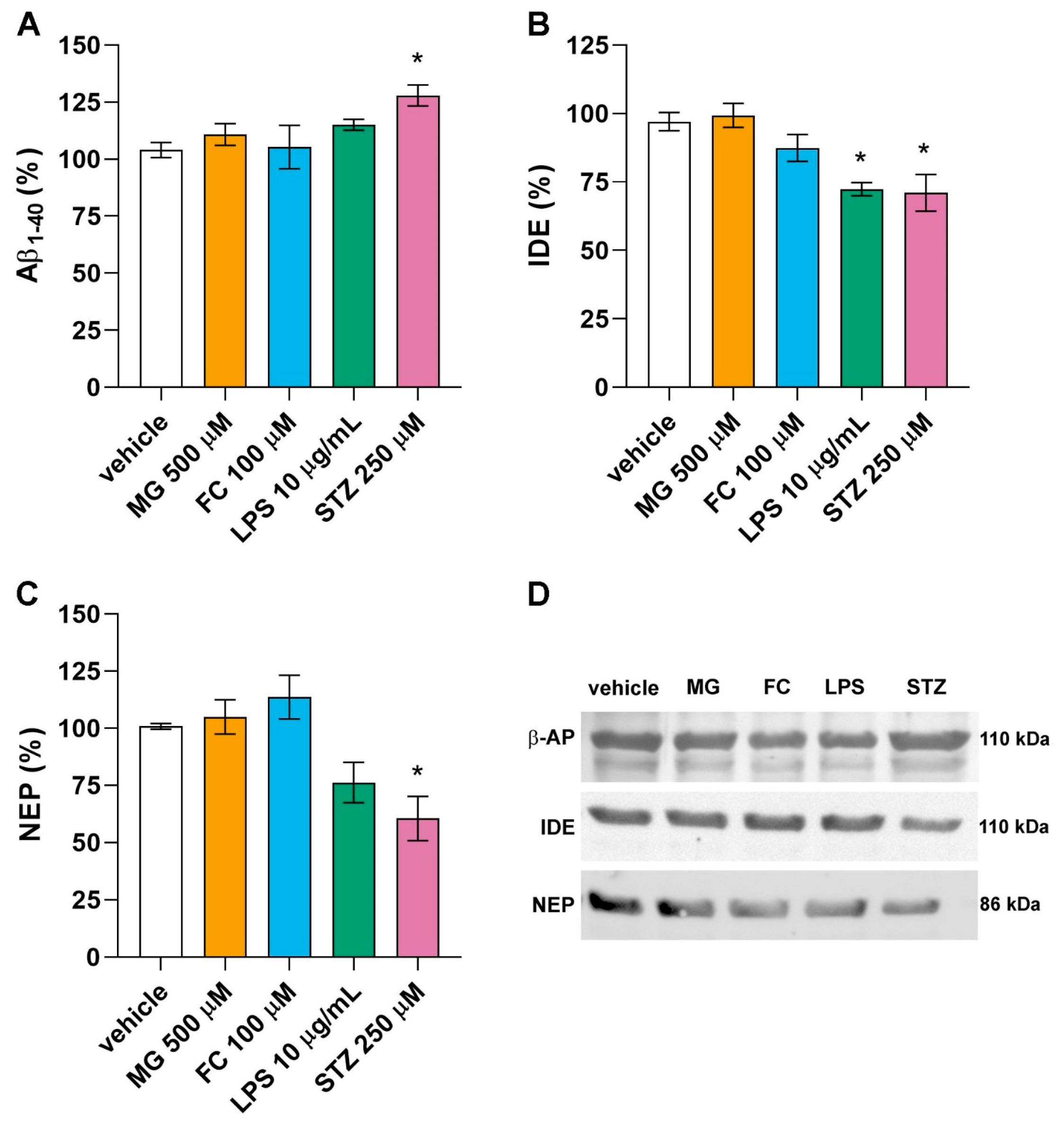
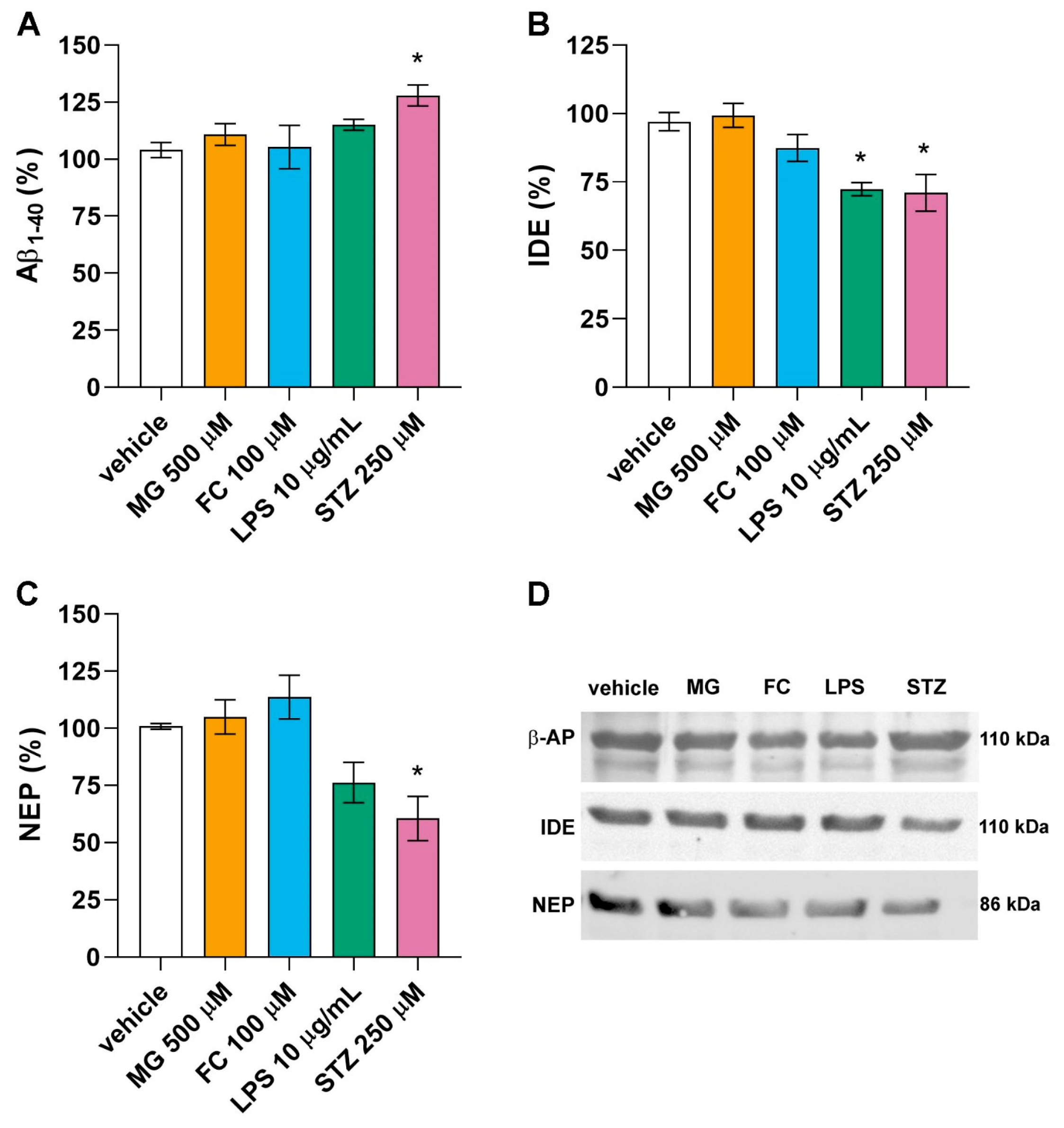
3.4. Only LPS and STZ Affect Proteins Involved in the Amyloid Cascade
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A Central Element in Neurological Diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef]
- Blanco-Suárez, E.; Caldwell, A.L.M.; Allen, N.J. Role of Astrocyte–Synapse Interactions in CNS Disorders. J. Physiol. 2017, 595, 1903–1916. [Google Scholar] [CrossRef]
- Chung, W.-S.; Welsh, C.A.; Barres, B.A.; Stevens, B. Do Glia Drive Synaptic and Cognitive Impairment in Disease? Nat. Neurosci. 2015, 18, 1539–1545. [Google Scholar] [CrossRef]
- Pérez Palmer, N.; Trejo Ortega, B.; Joshi, P. Cognitive Impairment in Older Adults: Epidemiology, Diagnosis, and Treatment. Psychiatr. Clin. N. Am. 2022, 45, 639–661. [Google Scholar] [CrossRef]
- Gauthier, S.; Webster, C.; Servaes, S.; Morais, J.A.; Rosa-Neto, P. World Alzheimer Report 2022: Life after Diagnosis: Navigating Treatment, Care and Support; Alzheimer’s Disease International: London, UK, 2022; pp. 1–414. [Google Scholar]
- Rae, C.D.; Baur, J.A.; Borges, K.; Dienel, G.; Díaz, C.M.; Starlette, G.; Kelly, R.D.; Duarte, J.M.N.; Liu, D.L.; Lindquist, B.E.; et al. Brain Energy Metabolism: A Roadmap for Future Research. J. Neurochem. 2024, 1–45. [Google Scholar] [CrossRef]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte Function from Information Processing to Cognition and Cognitive Impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef]
- Donato, R. S100: A Multigenic Family of Calcium-Modulated Proteins of the EF-Hand Type with Intracellular and Extracellular Functional Roles. Int. J. Biochem. Cell Biol. 2001, 33, 637–668. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate Metabolism and Recycling at the Excitatory Synapse in Health and Neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Palmer, A.L.; Ousman, S.S. Astrocytes and Aging. Front. Aging Neurosci. 2018, 10, 337. [Google Scholar] [CrossRef]
- Leclerc, B.; Abulrob, A. Perspectives in Molecular Imaging Using Staging Biomarkers and Immunotherapies in Alzheimer’s Disease. Sci. World J. 2013, 2013, 589308–589323. [Google Scholar] [CrossRef]
- Gonzales, M.M.; Garbarino, V.R.; Pollet, E.; Palavicini, J.P.; Kellogg, D.L.; Kraig, E.; Orr, M.E. Biological Aging Processes Underlying Cognitive Decline and Neurodegenerative Disease. J. Clin. Investig. 2022, 132, e158453. [Google Scholar] [CrossRef]
- Hassel, B.; Westergaard, N.; Schousboe, A.; Fonnum, F. Metabolic Differences between Primary Cultures of Astrocytes and Neurons from Cerebellum and Cerebral Cortex. Effects of Fluorocitrate. Neurochem. Res. 1995, 20, 413–420. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, X.; Dong, J.; Liu, W.C.; Song, M.; Sun, Y.; Shu, H.; Towse, C.L.; Liu, W.; Liu, C.F.; et al. Inhibition of Reactive Astrocytes with Fluorocitrate Ameliorates Learning and Memory Impairment Through Upregulating CRTC1 and Synaptophysin in Ischemic Stroke Rats. Cell. Mol. Neurobiol. 2019, 39, 1151–1163. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Wang, Y.; He, Y.; Wang, T.; Huang, X.H.; Zhao, C.M.; Zhang, L.; Li, S.W.; Wang, C.; Qu, Y.N.; et al. A1 Astrocytes Contribute to Murine Depression-like Behavior and Cognitive Dysfunction, Which Can Be Alleviated by IL-10 or Fluorocitrate Treatment. J. Neuroinflamm. 2020, 17, 200. [Google Scholar] [CrossRef]
- Rezagholizadeh, A.; Karimi, S.A.; Hosseinmardi, N.; Janahmadi, M.; Sayyah, M. The Effects of Glial Cells Inhibition on Spatial Reference, Reversal and Working Memory Deficits in a Rat Model of Traumatic Brain Injury (TBI). Int. J. Neurosci. 2022, 132, 226–236. [Google Scholar] [CrossRef]
- Rae, C.; Fekete, A.D.; Kashem, M.A.; Nasrallah, F.A.; Bröer, S. Metabolism, Compartmentation, Transport and Production of Acetate in the Cortical Brain Tissue Slice. Neurochem. Res. 2012, 37, 2541–2553. [Google Scholar] [CrossRef]
- Jash, K.; Gondaliya, P.; Kirave, P.; Kulkarni, B.; Sunkaria, A.; Kalia, K. Cognitive Dysfunction: A Growing Link between Diabetes and Alzheimer’s Disease. Drug Dev. Res. 2020, 81, 144–164. [Google Scholar] [CrossRef]
- Shopit, A.; Niu, M.; Wang, H.; Tang, Z.; Li, X.; Tesfaldet, T.; Ai, J.; Ahmad, N.; Al-Azab, M.; Tang, Z. Protection of Diabetes-Induced Kidney Injury by Phosphocreatine via the Regulation of ERK/Nrf2/HO-1 Signaling Pathway. Life Sci. 2020, 242, 117248. [Google Scholar] [CrossRef]
- Cho, C.H.; Lee, C.J.; Kim, M.G.; Ryu, B.; Je, J.G.; Kim, Y.; Lee, S.H. Therapeutic Potential of Phlorotannin-Rich Ecklonia Cava Extract on Methylglyoxal-Induced Diabetic Nephropathy in In Vitro Model. Mar. Drugs 2022, 20, 355. [Google Scholar] [CrossRef]
- Karumanchi, D.K.; Karunaratne, N.; Lurio, L.; Dillon, J.P.; Gaillard, E.R. Non-Enzymatic Glycation of α-Crystallin as an in Vitro Model for Aging, Diabetes and Degenerative Diseases. Amino Acids 2015, 47, 2601–2608. [Google Scholar] [CrossRef]
- Hansen, F.; Galland, F.; Lirio, F.; De Souza, D.F.; Da Ré, C.; Pacheco, R.F.; Vizuete, A.F.; Quincozes-Santos, A.; Leite, M.C.; Gonçalves, C.A. Methylglyoxal Induces Changes in the Glyoxalase System and Impairs Glutamate Uptake Activity in Primary Astrocytes. Oxid. Med. Cell Longev. 2017, 2017, 9574201–9574211. [Google Scholar] [CrossRef]
- Hansen, F.; Pandolfo, P.; Galland, F.; Torres, F.V.; Dutra, M.F.; Batassini, C.; Guerra, M.C.; Leite, M.C.; Gonçalves, C.A. Methylglyoxal Can Mediate Behavioral and Neurochemical Alterations in Rat Brain. Physiol. Behav. 2016, 164, 93–101. [Google Scholar] [CrossRef]
- Lissner, L.J.; Rodrigues, L.; Wartchow, K.M.; Borba, E.; Bobermin, L.D.; Fontella, F.U.; Hansen, F.; Quincozes-Santos, A.; Souza, D.O.G.; Gonçalves, C.A. Short-Term Alterations in Behavior and Astroglial Function after Intracerebroventricular Infusion of Methylglyoxal in Rats. Neurochem. Res. 2021, 46, 183–196. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the Dark Side of Glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of Advanced Glycation End Products in Cellular Signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Hansen, F.; Battú, C.E.; Dutra, M.F.; Galland, F.; Lirio, F.; Broetto, N.; Nardin, P.; Goncąlves, C.A. Methylglyoxal and Carboxyethyllysine Reduce Glutamate Uptake and S100B Secretion in the Hippocampus Independently of RAGE Activation. Amino Acids 2016, 48, 375–385. [Google Scholar] [CrossRef]
- Angeloni, C.; Zambonin, L.; Hrelia, S. Role of Methylglyoxal in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 238485. [Google Scholar] [CrossRef]
- Shi, D.Y.; Bierhaus, A.; Nawroth, P.P.; Stern, D.M. RAGE and Alzheimer’s Disease: A Progression Factor for Amyloid-β-Induced Cellular Perturbation? J. Alzheimer’s Dis. 2009, 16, 833–843. [Google Scholar] [CrossRef]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS Neurodegenerative Diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Singh, S.; Sahu, K.; Singh, C.; Singh, A. Lipopolysaccharide Induced Altered Signaling Pathways in Various Neurological Disorders. Naunyn. Schmiedebergs. Arch. Pharmacol. 2022, 395, 285–294. [Google Scholar] [CrossRef]
- Zakaria, R.; Wan Yaacob, W.M.; Othman, Z.; Long, I.; Ahmad, A.H.; Al-Rahbi, B. Lipopolysaccharide-Induced Memory Impairment in Rats: A Model of Alzheimer’s Disease. Physiol. Res. 2017, 66, 553–565. [Google Scholar] [CrossRef]
- Płóciennikowska, A.; Hromada-Judycka, A.; Borzęcka, K.; Kwiatkowska, K. Co-Operation of TLR4 and Raft Proteins in LPS-Induced pro-Inflammatory Signaling. Cell. Mol. Life Sci. 2015, 72, 557–581. [Google Scholar] [CrossRef]
- Trotta, T.; Porro, C.; Calvello, R.; Panaro, M.A. Biological Role of Toll-like Receptor-4 in the Brain. J. Neuroimmunol. 2014, 268, 1–12. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s Disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef]
- Mishra, S.K.; Singh, S.; Shukla, S.; Shukla, R. Intracerebroventricular Streptozotocin Impairs Adult Neurogenesis and Cognitive Functions via Regulating Neuroinflammation and Insulin Signaling in Adult Rats. Neurochem. Int. 2018, 113, 56–68. [Google Scholar] [CrossRef]
- Huf, F.; Gutierres, J.M.; da Silva, G.N.; Zago, A.M.; Koenig, L.F.C.; Fernandes, M.C. Neuroprotection Elicited by Taurine in Sporadic Alzheimer-like Disease: Benefits on Memory and Control of Neuroinflammation in the Hippocampus of Rats. Mol. Cell. Biochem. 2023, 2023, 1–16. [Google Scholar] [CrossRef]
- Rodrigues, L.; Wartchow, K.M.; Suardi, L.Z.; Federhen, B.C.; Selistre, N.G.; Gonçalves, C.A. Streptozotocin Causes Acute Responses on Hippocampal S100B and BDNF Proteins Linked to Glucose Metabolism Alterations. Neurochem. Int. 2019, 128, 85–93. [Google Scholar] [CrossRef]
- Akhtar, A.; Gupta, S.M.; Dwivedi, S.; Kumar, D.; Shaikh, M.F.; Negi, A. Preclinical Models for Alzheimer’s Disease: Past, Present, and Future Approaches. ACS Omega 2022, 7, 47504–47517. [Google Scholar] [CrossRef]
- Rodrigues, L.; Biasibetti, R.; Swarowsky, A.; Leite, M.C.; Quincozes-Santos, A.; Quilfeldt, J.A.; Achaval, M.; Gonalves, C.A. Hippocampal Alterations in Rats Submitted to Streptozotocin-Induced Dementia Model Are Prevented by Aminoguanidine. J. Alzheimer’s Dis. 2009, 17, 193–202. [Google Scholar] [CrossRef]
- Grieb, P. Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer’s Disease: In Search of a Relevant Mechanism. Mol. Neurobiol. 2016, 53, 1741–1752. [Google Scholar] [CrossRef]
- Davis, N.; Mota, B.C.; Stead, L.; Palmer, E.O.C.; Lombardero, L.; Rodríguez-Puertas, R.; de Paola, V.; Barnes, S.J.; Sastre, M. Pharmacological Ablation of Astrocytes Reduces Aβ Degradation and Synaptic Connectivity in an Ex Vivo Model of Alzheimer’s Disease. J. Neuroinflamm. 2021, 18, 73. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.M. The Role of Astrocytes in Amyloid Production and Alzheimer’s Disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult Mouse Astrocytes Degrade Amyloid-β In Vitro and In Situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Gottfried, C.; Valentim, L.; Salbego, C.; Karl, J.; Wofchuk, S.T.; Rodnight, R. Regulation of Protein Phosphorylation in Astrocyte Cultures by External Calcium Ions: Specific Effects on the Phosphorylation of Glial Fibrillary Acidic Protein (GFAP), Vimentin and Heat Shock Protein 27 (HSP27). Brain Res. 1999, 833, 142–149. [Google Scholar] [CrossRef]
- Guerra, M.C.; Tortorelli, L.S.; Galland, F.; Da Ré, C.; Negri, E.; Engelke, D.S.; Rodrigues, L.; Leite, M.C.; Gonçalves, C.A. Lipopolysaccharide Modulates Astrocytic S100B Secretion: A Study in Cerebrospinal Fluid and Astrocyte Cultures from Rats. J. Neuroinflamm. 2011, 8, 128. [Google Scholar] [CrossRef]
- Selistre, N.G.; Rodrigues, L.; Federhen, B.C.; Gayger-Dias, V.; Taday, J.; Wartchow, K.M.; Gonçalves, C.-A. S100B Secretion in Astrocytes, Unlike C6 Glioma Cells, Is Downregulated by Lactate. Metabolites 2023, 14, 7. [Google Scholar] [CrossRef]
- Plaschke, K.; Kopitz, J. In Vitro Streptozotocin Model for Modeling Alzheimer-like Changes: Effect on Amyloid Precursor Protein Secretases and Glycogen Synthase Kinase-3. J. Neural Transm. 2015, 122, 551–557. [Google Scholar] [CrossRef]
- Leite, M.C.; Galland, F.; Brolese, G.; Guerra, M.C.; Bortolotto, J.W.; Freitas, R.; de Almeida, L.M.V.; Gottfried, C.; Gonçalves, C.A. A Simple, Sensitive and Widely Applicable ELISA for S100B: Methodological Features of the Measurement of This Glial Protein. J. Neurosci. Methods 2008, 169, 93–99. [Google Scholar] [CrossRef]
- Tramontina, F.; Leite, M.C.; Cereser, K.; de Souza, D.F.; Tramontina, A.C.; Nardin, P.; Andreazza, A.C.; Gottfried, C.; Kapczinski, F.; Gonçalves, C.A. Immunoassay for Glial Fibrillary Acidic Protein: Antigen Recognition Is Affected by Its Phosphorylation State. J. Neurosci. Methods 2007, 162, 282–286. [Google Scholar] [CrossRef]
- Gottfried, C.; Tramontina, F.; Gonçalves, D.; Gonçalves, C.A.; Moriguchi, E.; Dias, R.D.; Wofchuk, S.T.; Souza, D.O. Glutamate Uptake in Cultured Astrocytes Depends on Age: A Study about the Effect of Guanosine and the Sensitivity to Oxidative Stress Induced by H2O2. Mech. Ageing Dev. 2002, 123, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Thomazi, A.P.; Godinho, G.F.R.S.; Rodrigues, J.M.; Schwalm, F.D.; Frizzo, M.E.S.; Moriguchi, E.; Souza, D.O.; Wofchuk, S.T. Ontogenetic Profile of Glutamate Uptake in Brain Structures Slices from Rats: Sensitivity to Guanosine. Mech. Ageing Dev. 2004, 125, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Browne, R.W.; Armstrong, D. Reduced Glutathione and Glutathione Disulfide. Methods Mol. Biol. 1998, 108, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Minet, R.; Villie, F.; Marcollet, M.; Meynial-Denis, D.; Cynober, L. Measurement of Glutamine Synthetase Activity in Rat Muscle by a Colorimetric Assay. Clin. Chim. Acta 1997, 268, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Hancock, K.; Tsang, V.C. India Ink Staining of Proteins on Nitrocellulose Paper. Anal. Biochem. 1983, 133, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Peterson, G.L. A Simplification of the Protein Assay Method of Lowry et al. Which Is More Generally Applicable. Anal. Biochem. 1977, 83, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ni, J.; Zhai, L.; Gao, C.; Xie, L.; Zhao, L.; Yin, X. Inhibition of Activated Astrocyte Ameliorates Lipopolysaccharide- Induced Depressive-like Behaviors. J. Affect. Disord. 2019, 242, 52–59. [Google Scholar] [CrossRef]
- Seminotti, B.; Amaral, A.U.; Grings, M.; Ribeiro, C.A.J.; Leipnitz, G.; Wajner, M. Lipopolysaccharide-Elicited Systemic Inflammation Induces Selective Vulnerability of Cerebral Cortex and Striatum of Developing Glutaryl-CoA Dehydrogenase Deficient (Gcdh−/−) Mice to Oxidative Stress. Neurotox. Res. 2020, 38, 1024–1036. [Google Scholar] [CrossRef]
- Goshi, N.; Morgan, R.K.; Lein, P.J.; Seker, E. A Primary Neural Cell Culture Model to Study Neuron, Astrocyte, and Microglia Interactions in Neuroinflammation. J. Neuroinflamm. 2020, 17, 155. [Google Scholar] [CrossRef]
- Dourado, N.S.; Souza, C.D.S.; de Almeida, M.M.A.; Bispo da Silva, A.; dos Santos, B.L.; Silva, V.D.A.; De Assis, A.M.; da Silva, J.S.; Souza, D.O.; Costa, M.d.F.D.; et al. Neuroimmunomodulatory and Neuroprotective Effects of the Flavonoid Apigenin in In Vitro Models of Neuroinflammation Associated with Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Junior, M.S.; Pereira, E.P.; de Amorim, V.C.M.; Reis, L.T.C.; do Nascimento, R.P.; da Silva, V.D.A.; Costa, S.L. Lupeol Inhibits LPS-Induced Neuroinflammation in Cerebellar Cultures and Induces Neuroprotection Associated to the Modulation of Astrocyte Response and Expression of Neurotrophic and Inflammatory Factors. Int. Immunopharmacol. 2019, 70, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Xiang, Y.; Qu, X.; Liu, H.; Liu, C.; Li, G.; Han, L.; Qin, X. Apelin-13 Suppresses Neuroinflammation against Cognitive Deficit in a Streptozotocin-Induced Rat Model of Alzheimer’s Disease through Activation of BDNF-TrkB Signaling Pathway. Front. Pharmacol. 2019, 10, 395. [Google Scholar] [CrossRef]
- Dos Santos, J.P.A.; Vizuete, A.; Hansen, F.; Biasibetti, R.; Gonçalves, C.A. Early and Persistent O-GlcNAc Protein Modification in the Streptozotocin Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 61, 237–249. [Google Scholar] [CrossRef]
- Gonçalves, C.A.; Concli Leite, M.; Nardin, P. Biological and Methodological Features of the Measurement of S100B, a Putative Marker of Brain Injury. Clin. Biochem. 2008, 41, 755–763. [Google Scholar] [CrossRef]
- Sood, A.; Preeti, K.; Fernandes, V.; Khatri, D.K.; Singh, S.B. Glia: A Major Player in Glutamate–GABA Dysregulation-Mediated Neurodegeneration. J. Neurosci. Res. 2021, 99, 3148–3189. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS β-Amyloid in Alzheimer’s Disease. Science (80-) 2010, 330, 1774. [Google Scholar] [CrossRef]
- Miners, J.S.; Baig, S.; Palmer, J.; Palmer, L.E.; Kehoe, P.G.; Love, S. Aβ-Degrading Enzymes in Alzheimer’s Disease. Brain Pathol. 2008, 18, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Patil, G.V.; Joshi, R.S.; Kazi, R.S.; Kulsange, S.E.; Kulkarni, M.J. A Possible Role of Glycation in the Regulation of Amyloid β Precursor Protein Processing Leading to Amyloid β Accumulation. Med. Hypotheses 2020, 142, 109799. [Google Scholar] [CrossRef]
- Yamamoto, N.; Ishikuro, R.; Tanida, M.; Suzuki, K.; Ikeda-Matsuo, Y.; Sobue, K. Insulin-Signaling Pathway Regulates the Degradation of Amyloid β-Protein via Astrocytes. Neuroscience 2018, 385, 227–236. [Google Scholar] [CrossRef]
- Adermark, L.; Lagström, O.; Loftén, A.; Licheri, V.; Havenäng, A.; Loi, E.A.; Stomberg, R.; Söderpalm, B.; Domi, A.; Ericson, M. Astrocytes Modulate Extracellular Neurotransmitter Levels and Excitatory Neurotransmission in Dorsolateral Striatum via Dopamine D2 Receptor Signaling. Neuropsychopharmacology 2022, 47, 1493–1502. [Google Scholar] [CrossRef]
- Swanson, R.A.; Graham, S.H. Fluorocitrate and Fluoroacetate Effects on Astrocyte Metabolism in Vitro. Brain Res. 1994, 664, 94–100. [Google Scholar] [CrossRef]
- Paulsen, R.E.; Contestabile, A.; Villani, L.; Fonnum, F. An In Vivo Model for Studying Function of Brain Tissue Temporarily Devoid of Glial Cell Metabolism: The Use of Fluorocitrate. J. Neurochem. 1987, 48, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.; Bhalerao, A.; Powers, M.; Gonzalez, M.; Mancuso, S.; Cucullo, L. Hypometabolism, Alzheimer’s Disease, and Possible Therapeutic Targets: An Overview. Cells 2023, 12, 2019. [Google Scholar] [CrossRef]
- Nikkar, R.; Esmaeili-bandboni, A.; Badrikoohi, M.; Babaei, P. Effects of Inhibiting Astrocytes and BET/BRD4 Chromatin Reader on Spatial Memory and Synaptic Proteins in Rats with Alzheimer’s Disease. Metab. Brain Dis. 2022, 37, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.L.; Wang, Q.B.; Liu, X.P.; Yao, X.Q.; Cao, F.Y.; Wang, Q.; Zhang, J.Y.; Wang, J.Z.; Liu, G.P. Fluorocitrate Induced the Alterations of Memory-Related Proteins and Tau Hyperphosphorylation in SD Rats. Neurosci. Lett. 2015, 584, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Bakaeva, Z.; Lizunova, N.; Tarzhanov, I.; Boyarkin, D.; Petrichuk, S.; Pinelis, V.; Fisenko, A.; Tuzikov, A.; Sharipov, R.; Surin, A. Lipopolysaccharide from E. Coli Increases Glutamate-Induced Disturbances of Calcium Homeostasis, the Functional State of Mitochondria, and the Death of Cultured Cortical Neurons. Front. Mol. Neurosci. 2022, 14, 811171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; White, T.D. The Bacterial Endotoxin Lipopolysaccharide Causes Rapid Inappropriate Excitation in Rat Cortex. J. Neurochem. 1999, 72, 652–660. [Google Scholar] [CrossRef]
- Vizuete, A.F.K.; Fróes, F.; Seady, M.; Zanotto, C.; Bobermin, L.D.; Roginski, A.C.; Wajner, M.; Quincozes-Santos, A.; Gonçalves, C.A. Early Effects of LPS-Induced Neuroinflammation on the Rat Hippocampal Glycolytic Pathway. J. Neuroinflamm. 2022, 19, 255. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment through Enhancement of Beta-Amyloid Generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Bhuvanendran, S.; Paudel, Y.N.; Kumari, Y.; Othman, I.; Shaikh, M.F. Embelin Prevents Amyloid-Beta Accumulation via Modulation of SOD1 in a Streptozotocin-Induced AD-like Condition: An Evidence from in Vitro Investigation. Curr. Res. Neurobiol. 2022, 3, 100032. [Google Scholar] [CrossRef] [PubMed]
- Bagaméry, F.; Varga, K.; Kecsmár, K.; Vincze, I.; Szökő, É.; Tábi, T. Lack of Insulin Resistance in Response to Streptozotocin Treatment in Neuronal SH-SY5Y Cell Line. J. Neural Transm. 2020, 127, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.; Biasibetti, R.; Swarowsky, A.; Leite, M.; Quincozes-Santos, A.; Achaval, M.; Gonçalves, C.A. Hippocampal Alterations in Rats Submitted to Streptozotocin-Induced Dementia Model: Neuroprotection with Aminoguanidine. Adv. Alzheimer’s Dis. 2011, 1, 215–227. [Google Scholar] [CrossRef]
- Tramontina, A.C.; Wartchow, K.M.; Rodrigues, L.; Biasibetti, R.; Quincozes-Santos, A.; Bobermin, L.; Tramontina, F.; Gonçalves, C.-A. The Neuroprotective Effect of Two Statins: Simvastatin and Pravastatin on a Streptozotocin-Induced Model of Alzheimer’s Disease in Rats. J. Neural Transm. 2011, 118, 1641–1649. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MG 500 μM | FC 100 μM | LPS 10 μg/mL | STZ 250 μM | |
|---|---|---|---|---|
| MTT | 102.5 ± 3.3 (p = 1) | 102.9 ± 3.3 (p = 0.645) | 99.4 ± 2.1 (p = 0.706) | 102.8 ± 2.0 (p = 1) |
| NR | 101.1 ± 2.7 (p = 1) | 107.1 ± 1.1 (p = 0.999) | 101.8 ± 3.6 (p = 1) | 100.9 ± 4.0 (p = 0.844) |
| PI | 103.5 ± 2.4 (p = 1) | 89.1 ± 3.7 (p = 0.074) | 97.5 ± 3.4 (p = 0.768) | 98.7 ± 2.2 (p = 0.875) |
| LDH | 101.0 ± 5.3 (p = 1) | 59.4 ± 5.6 (p < 0.001) * | 96.2 ± 4.5 (p = 0.819) | 97.1 ± 4.7 (p = 0.934) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taday, J.; Fróes, F.T.; Seady, M.; Gonçalves, C.A.; Leite, M.C. In Vitro Astroglial Dysfunction Induced by Neurotoxins: Mimicking Astrocytic Metabolic Alterations of Alzheimer’s Disease. Metabolites 2024, 14, 151. https://doi.org/10.3390/metabo14030151
Taday J, Fróes FT, Seady M, Gonçalves CA, Leite MC. In Vitro Astroglial Dysfunction Induced by Neurotoxins: Mimicking Astrocytic Metabolic Alterations of Alzheimer’s Disease. Metabolites. 2024; 14(3):151. https://doi.org/10.3390/metabo14030151
Chicago/Turabian StyleTaday, Jéssica, Fernanda Telles Fróes, Marina Seady, Carlos Alberto Gonçalves, and Marina Concli Leite. 2024. "In Vitro Astroglial Dysfunction Induced by Neurotoxins: Mimicking Astrocytic Metabolic Alterations of Alzheimer’s Disease" Metabolites 14, no. 3: 151. https://doi.org/10.3390/metabo14030151
APA StyleTaday, J., Fróes, F. T., Seady, M., Gonçalves, C. A., & Leite, M. C. (2024). In Vitro Astroglial Dysfunction Induced by Neurotoxins: Mimicking Astrocytic Metabolic Alterations of Alzheimer’s Disease. Metabolites, 14(3), 151. https://doi.org/10.3390/metabo14030151