
Integrating TCGA and Single-Cell Sequencing Data for Hepatocellular Carcinoma: A Novel Glycosylation (GLY)/Tumor Microenvironment (TME) Classifier to Predict Prognosis and Immunotherapy Response

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Preparation
2.2. CIBERSORT Analysis
2.3. Formation and Verification of the GLY/TME Scoring System
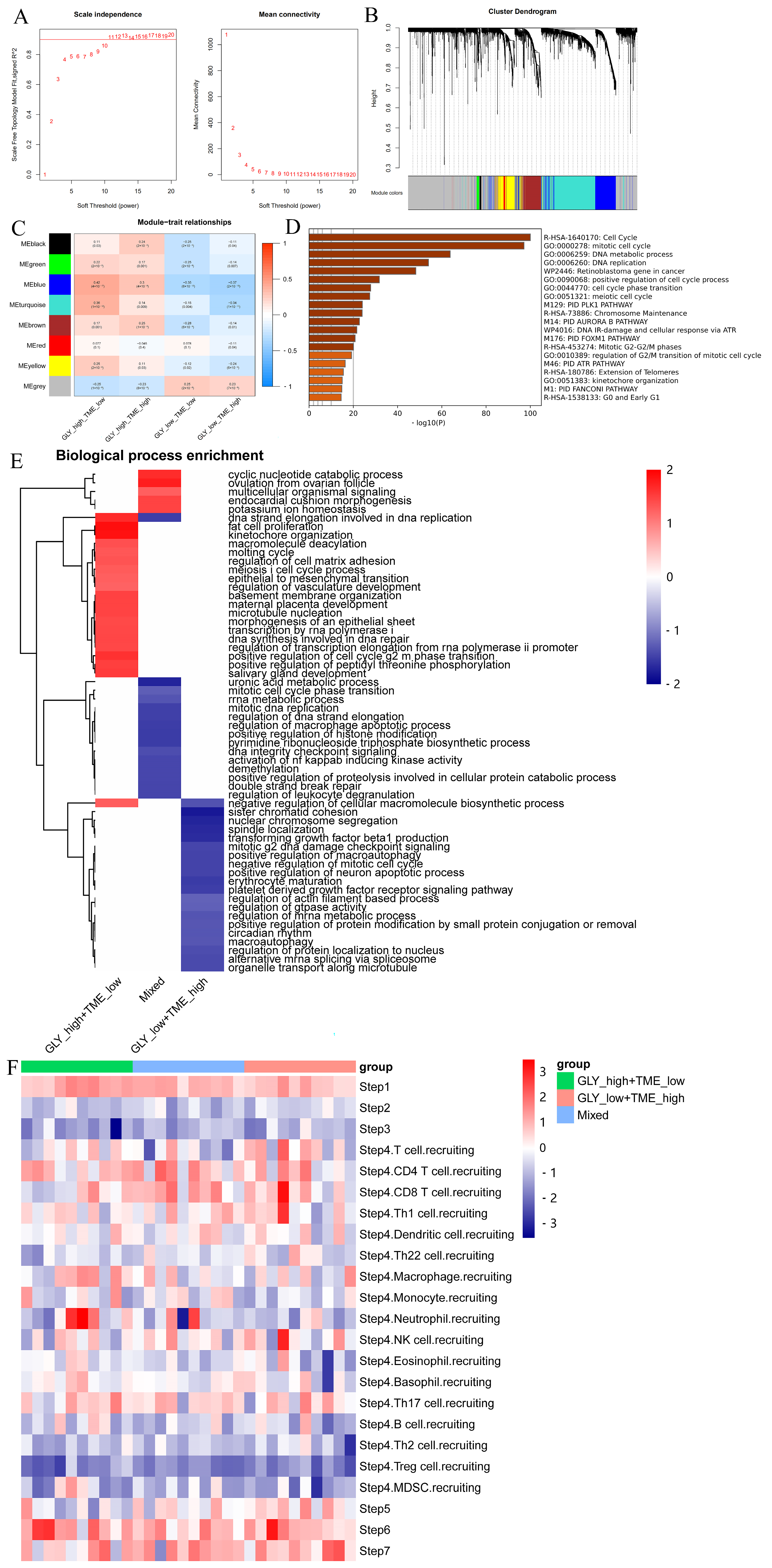
2.4. Weighted Gene Coexpression Network Analysis (WGCNA)
2.5. Functional Enrichment Analysis
2.6. TIP Analysis
2.7. Immunotherapy Response Prediction
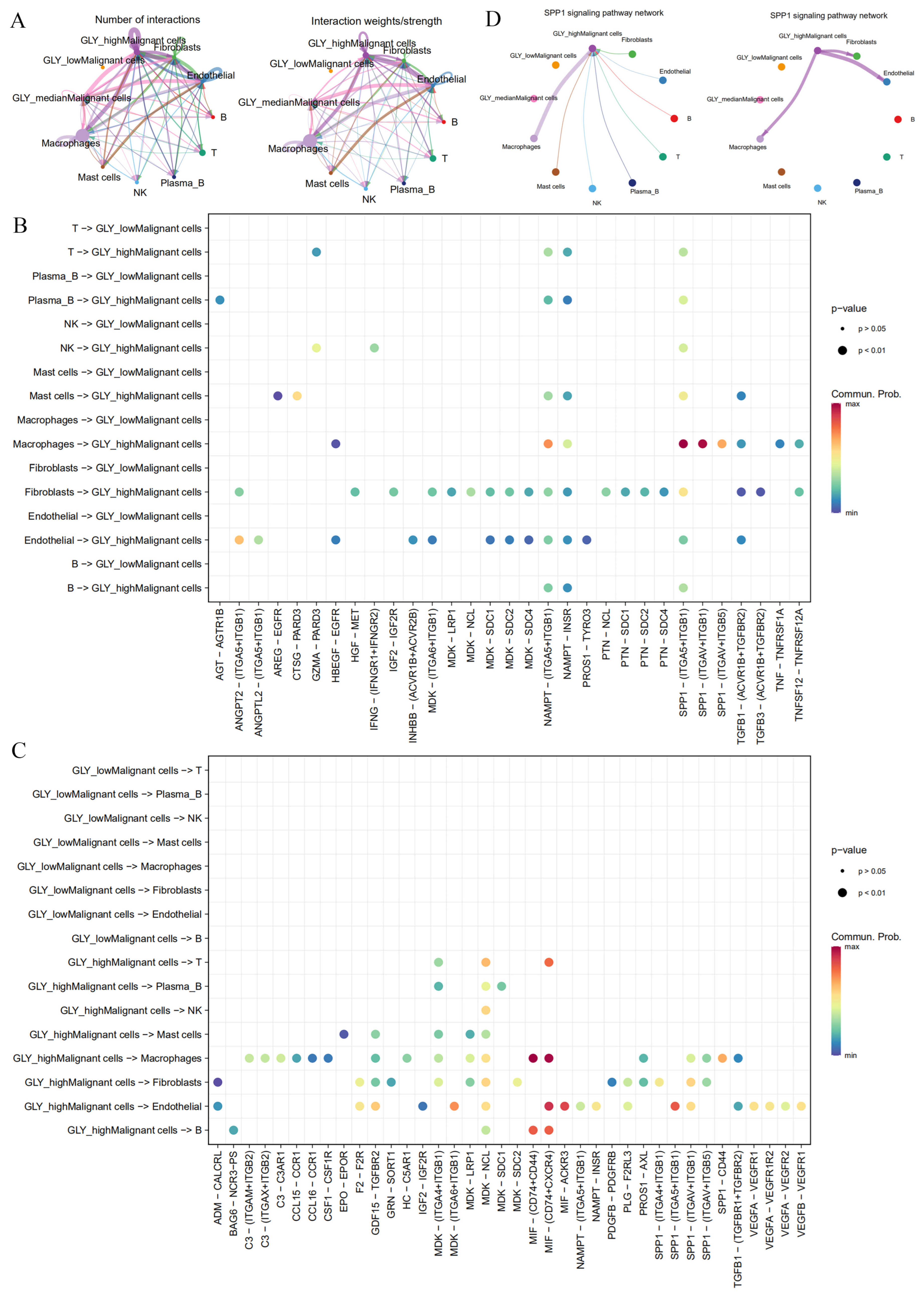
2.8. Intercellular Communications
2.9. Cell Culture and mRNA and Protein Level Analysis
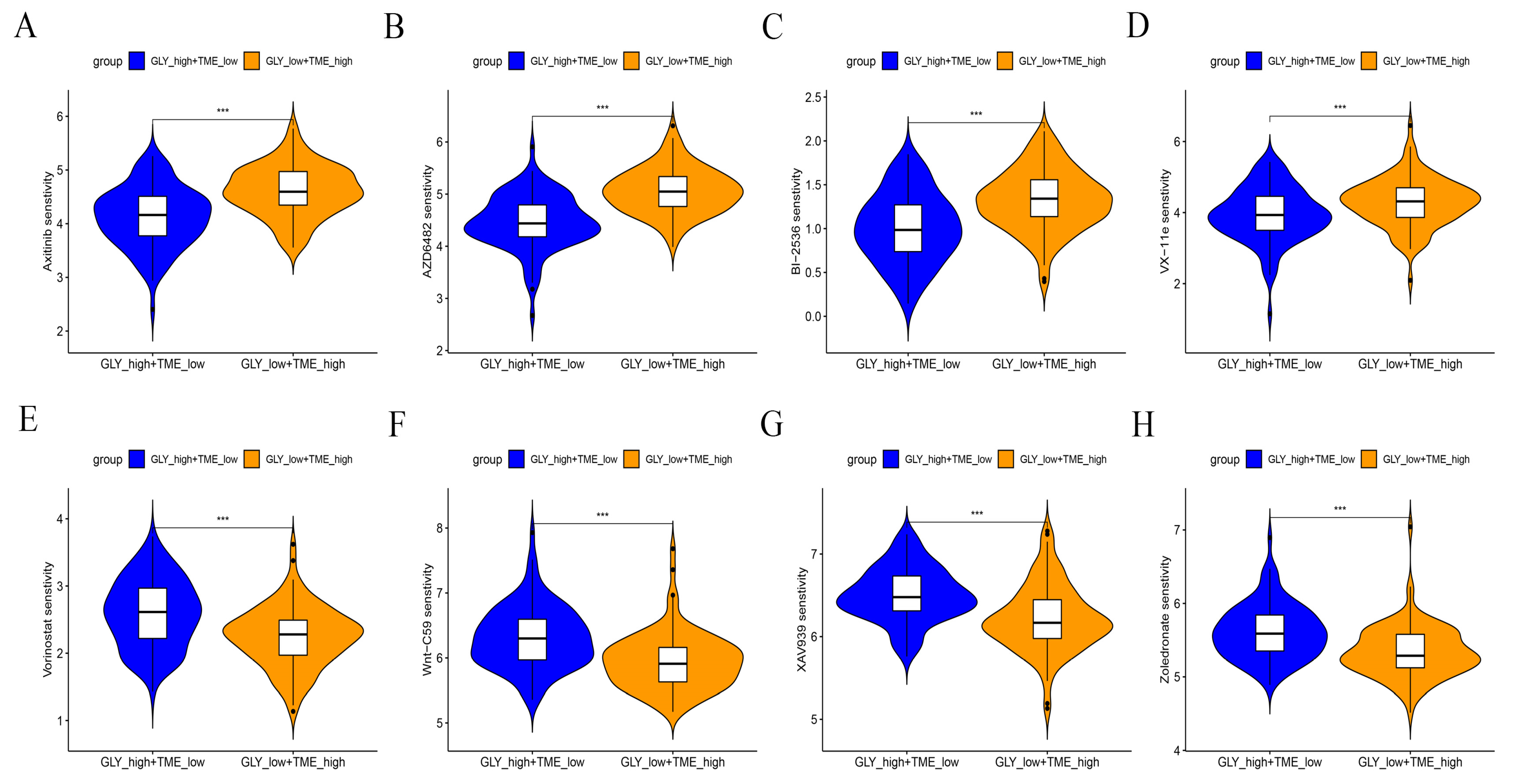
2.10. Prediction of Drug Response
2.11. Statistical Analysis
3. Results
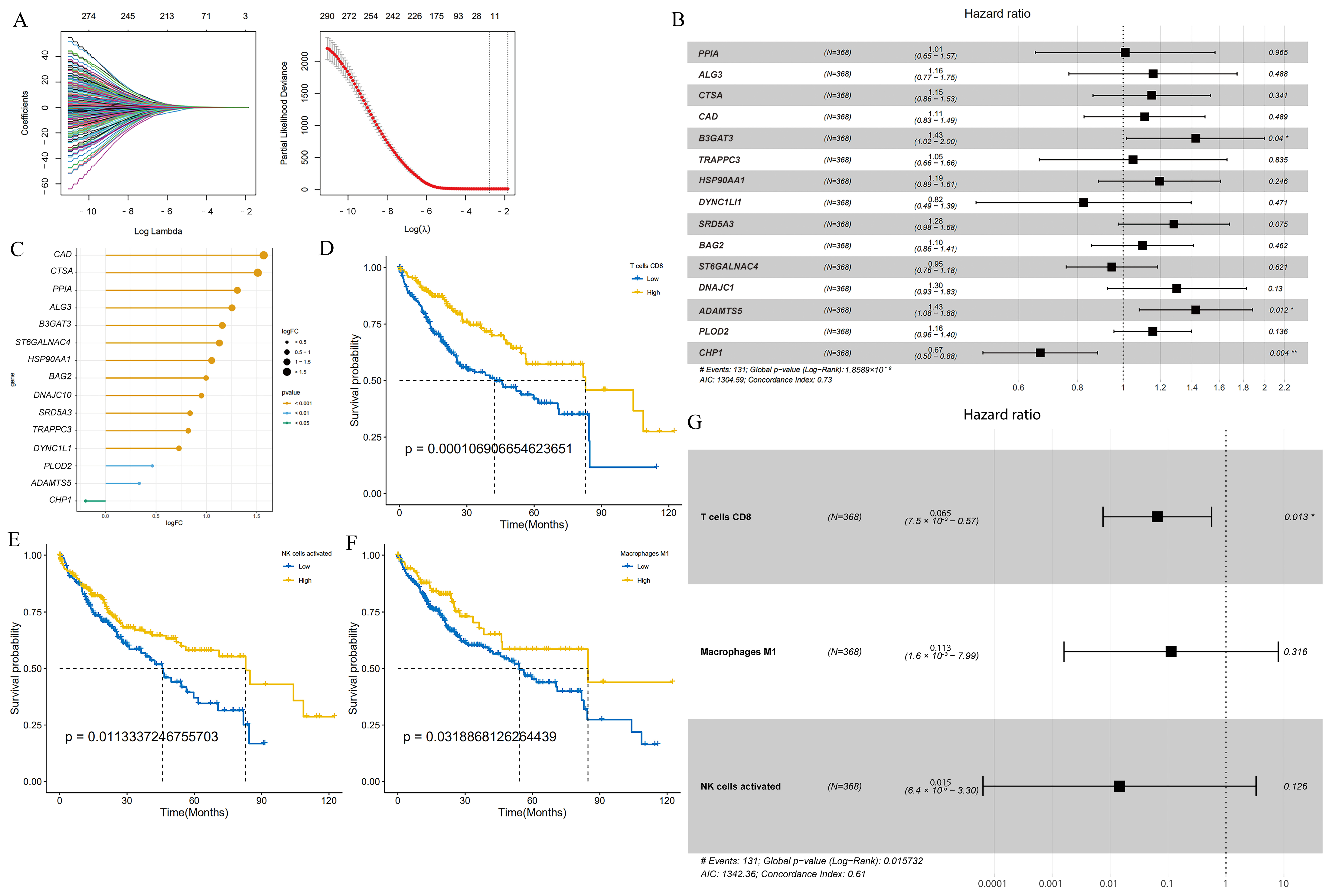
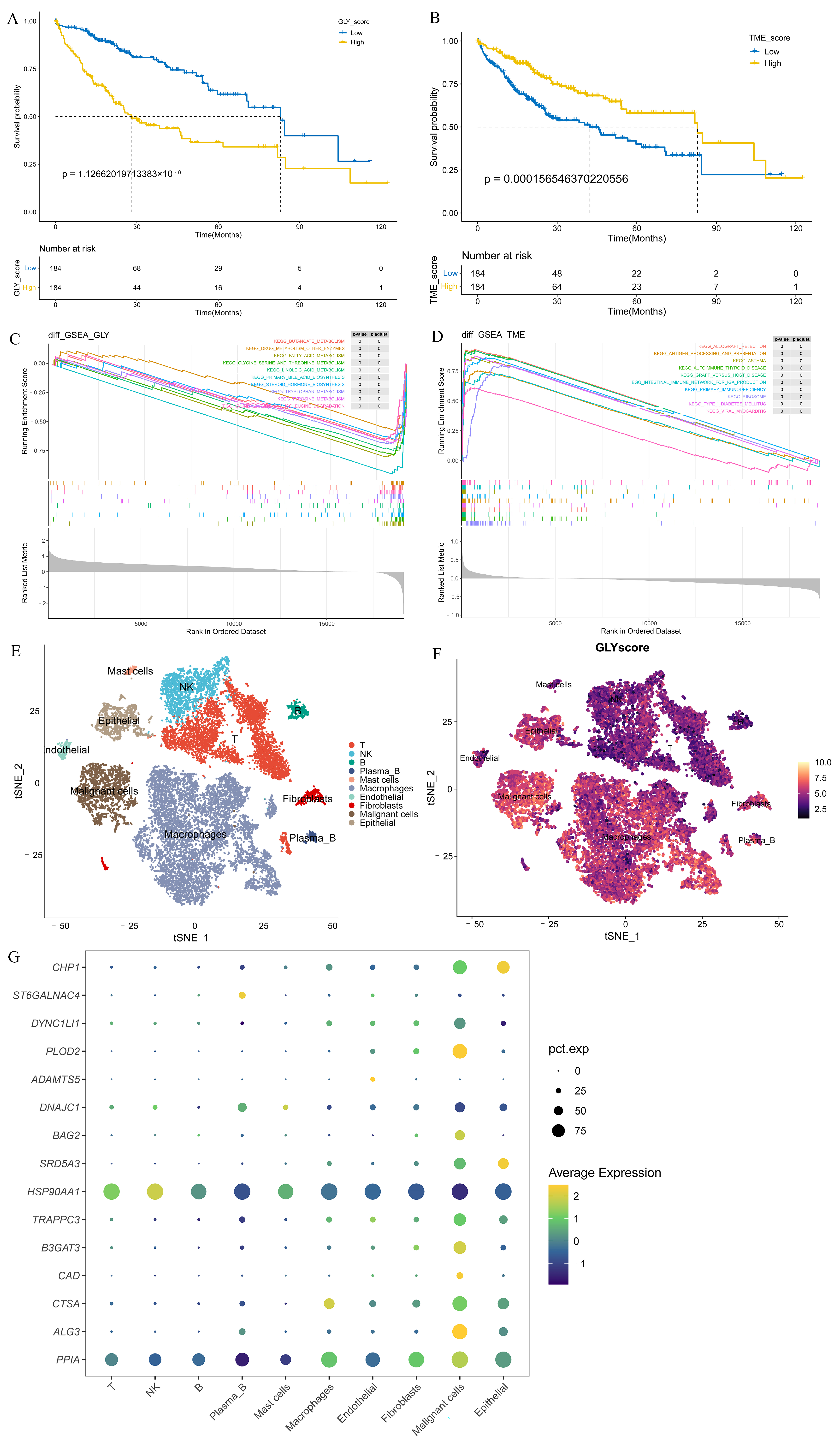
3.1. Construction of GLY Score and TME Score
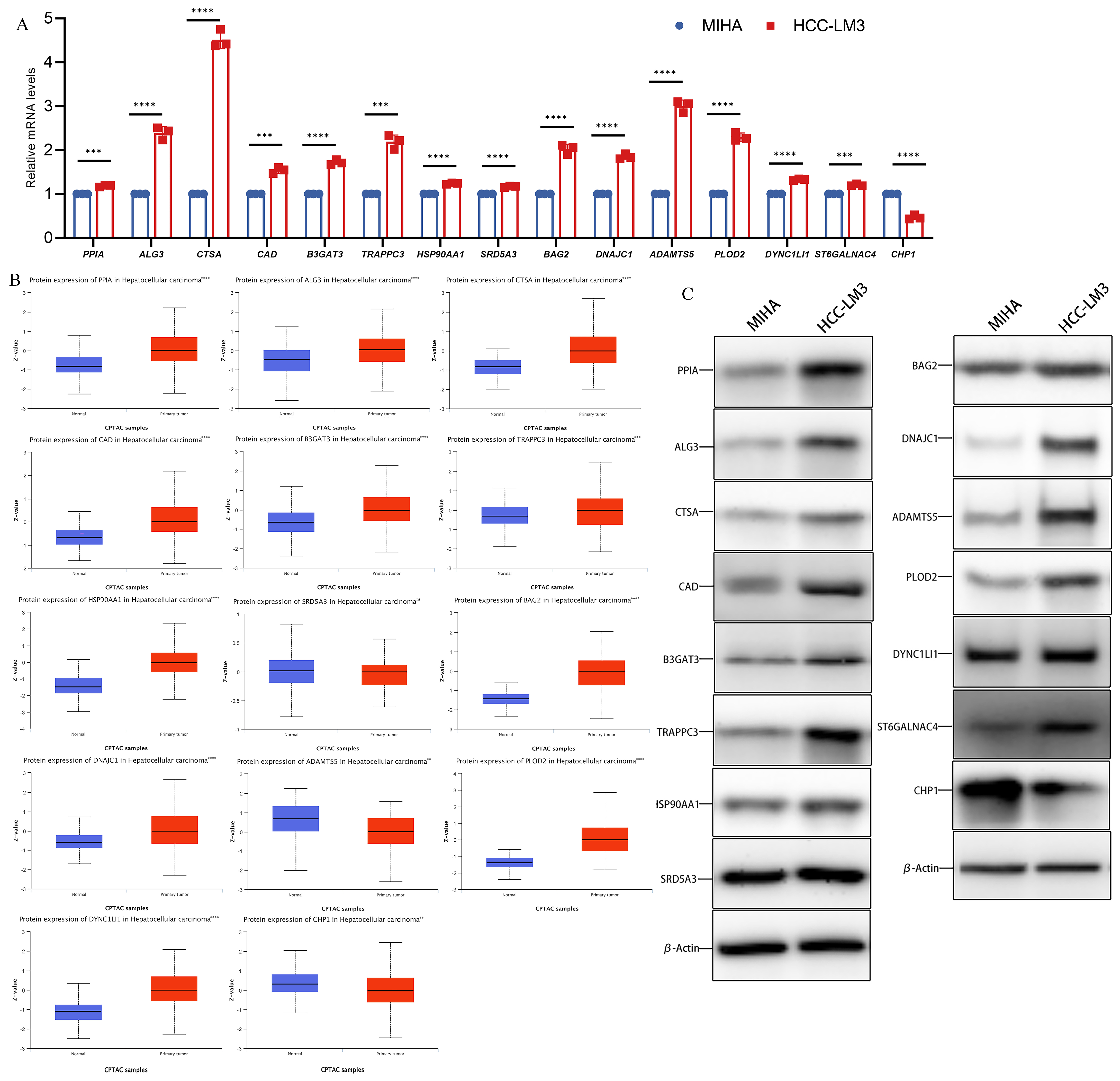
3.2. Differential mRNA and Protein Levels of Glycosylation-Related Prognostic Genes
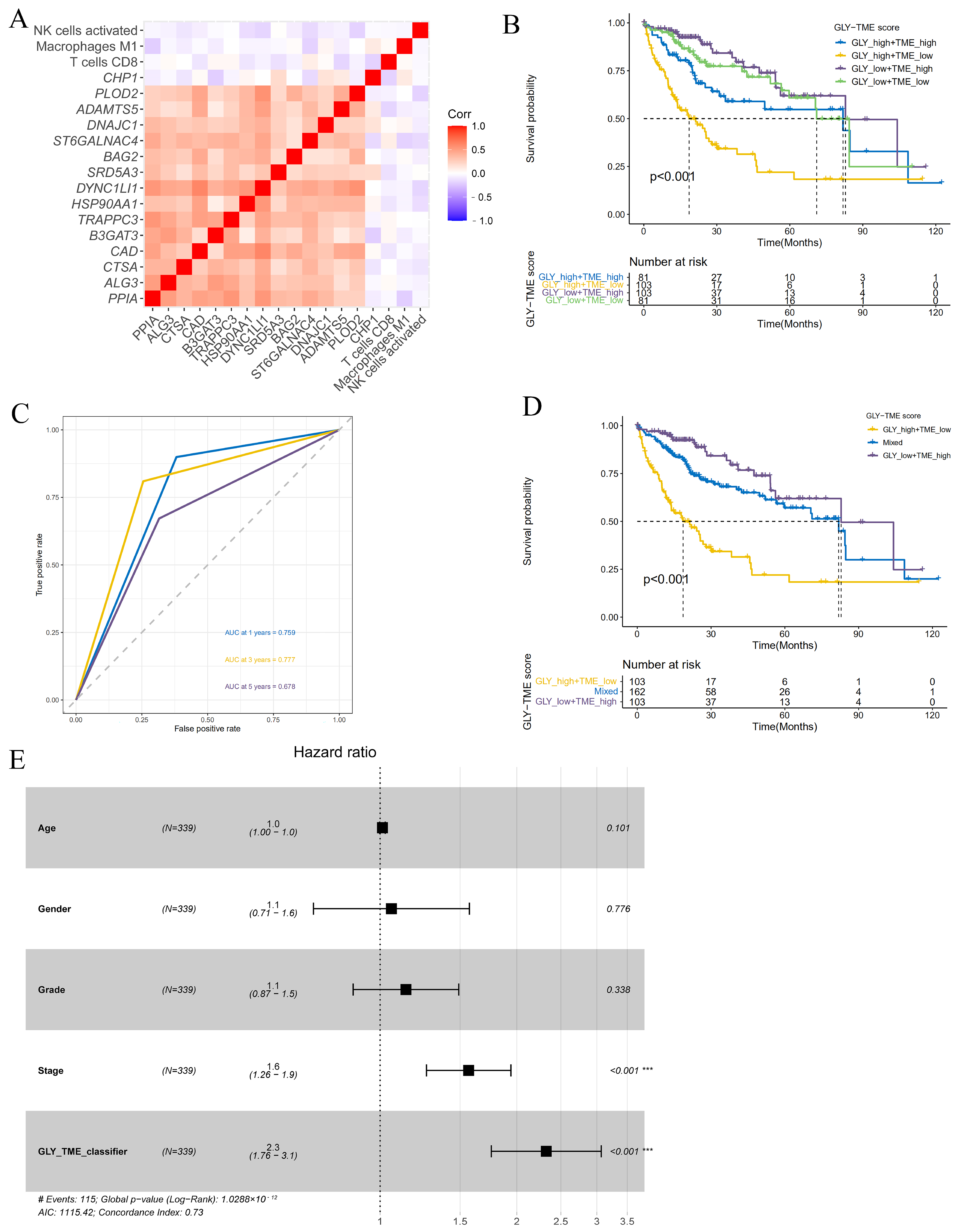
3.3. GLY/TME Score Performs Well in Predicting Prognosis
3.4. Construction and Prognostic Value Assessment of the GLY/TME Classifier
3.5. Different Underlying Molecular Mechanisms in the GLY/TME Classifier
3.6. High GLY Scores Were Associated with TME
3.7. Therapy Prediction Based on the GLY/TME Classifier
3.8. Benefit of Therapeutic Agents in GLYlow/TMEhigh and GLYhigh/TMElow Subgroups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
Abbreviations
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Anwanwan, D.; Singh, S.K.; Singh, S.; Saikam, V.; Singh, R. Challenges in liver cancer and possible treatment approaches. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188314. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Morizane, C.; Ueno, M.; Okusaka, T.; Ishii, H.; Furuse, J. Chemotherapy for hepatocellular carcinoma: Current status and future perspectives. Jpn. J. Clin. Oncol. 2018, 48, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Viatour, P. Hepatocellular carcinoma: Old friends and new tricks. Exp. Mol. Med. 2020, 52, 1898–1907. [Google Scholar] [CrossRef]
- Shokoohian, B.; Negahdari, B.; Aboulkheyr Es, H.; Abedi-Valugerdi, M.; Baghaei, K.; Agarwal, T.; Maiti, T.K.; Hassan, M.; Najimi, M.; Vosough, M. Advanced therapeutic modalities in hepatocellular carcinoma: Novel insights. J. Cell. Mol. Med. 2021, 25, 8602–8614. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, J.; Xu, L.; Du, Q.; Fang, N.; Wu, H.; Liu, F.; Hu, L.; Xu, J.; Hou, J.; et al. A theranostic metallodrug modulates immunovascular crosstalk to combat immunosuppressive liver cancer. Acta Biomater. 2022, 154, 478–496. [Google Scholar] [CrossRef]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef]
- Tiwari, A.; Trivedi, R.; Lin, S.Y. Tumor microenvironment: Barrier or opportunity towards effective cancer therapy. J. Biomed. Sci. 2022, 29, 83. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. From bench to bed: The tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 396. [Google Scholar] [CrossRef]
- Guizhen, Z.; Guanchang, J.; Liwen, L.; Huifen, W.; Zhigang, R.; Ranran, S.; Zujiang, Y. The tumor microenvironment of hepatocellular carcinoma and its targeting strategy by CAR-T cell immunotherapy. Front. Endocrinol. 2022, 13, 918869. [Google Scholar] [CrossRef]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef] [PubMed]
- Silsirivanit, A. Glycosylation markers in cancer. Adv. Clin. Chem. 2019, 89, 189–213. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, K.; Gao, Q.; Zhang, S. Glycoproteomic contributions to hepatocellular carcinoma research: A 2023 update. Expert Rev. Proteom. 2023, 20, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Gairing, S.J.; Ilyas, S.I.; Galle, P.R. Emerging immunotherapy for HCC: A guide for hepatologists. Hepatology 2022, 75, 1604–1626. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Pheatmap: Pretty Heatmaps, R Package Version 1.0.12. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 20 June 2023).
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Deng, C.; Pang, B.; Zhang, X.; Liu, W.; Liao, G.; Yuan, H.; Cheng, P.; Li, F.; Long, Z.; et al. TIP: A Web Server for Resolving Tumor Immunophenotype Profiling. Cancer Res. 2018, 78, 6575–6580. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Qi, J. The prognostic value and potential subtypes of immune activity scores in three major urological cancers. J. Cell. Physiol. 2021, 236, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Gillette, M.; Carr, S.A.; Paulovich, A.G.; Smith, R.D.; Rodland, K.K.; Townsend, R.R.; Kinsinger, C.; Mesri, M.; Rodriguez, H.; et al. Connecting genomic alterations to cancer biology with proteomics: The NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 2013, 3, 1108–1112. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Gowhari Shabgah, A.; Ezzatifar, F.; Aravindhan, S.; Olegovna Zekiy, A.; Ahmadi, M.; Gheibihayat, S.M.; Gholizadeh Navashenaq, J. Shedding more light on the role of Midkine in hepatocellular carcinoma: New perspectives on diagnosis and therapy. IUBMB Life 2021, 73, 659–669. [Google Scholar] [CrossRef]
- Liang, Y.; Fu, B.; Zhang, Y.; Lu, H. Progress of proteomics-driven precision medicine: From a glycosylation view. Rapid Commun. Mass Spectrom. 2022, 36, e9288. [Google Scholar] [CrossRef]
- Pandey, V.K.; Sharma, R.; Prajapati, G.K.; Mohanta, T.K.; Mishra, A.K. N-glycosylation, a leading role in viral infection and immunity development. Mol. Biol. Rep. 2022, 49, 8109–8120. [Google Scholar] [CrossRef] [PubMed]
- Leon, F.; Seshacharyulu, P.; Nimmakayala, R.K.; Chugh, S.; Karmakar, S.; Nallasamy, P.; Vengoji, R.; Rachagani, S.; Cox, J.L.; Mallya, K.; et al. Reduction in O-glycome induces differentially glycosylated CD44 to promote stemness and metastasis in pancreatic cancer. Oncogene 2022, 41, 57–71. [Google Scholar] [CrossRef]
- Rehman, S.; Aatif, M.; Rafi, Z.; Khan, M.Y.; Shahab, U.; Ahmad, S.; Farhan, M. Effect of non-enzymatic glycosylation in the epigenetics of cancer. Semin. Cancer Biol. 2022, 83, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Protein glycation—Biomarkers of metabolic dysfunction and early-stage decline in health in the era of precision medicine. Redox Biol. 2021, 42, 101920. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, T.; Du, Y.; Deng, J.; Gao, G.; Zhang, J. Identification of a Novel Glycosyltransferase Prognostic Signature in Hepatocellular Carcinoma Based on LASSO Algorithm. Front. Genet. 2022, 13, 823728. [Google Scholar] [CrossRef]
- Lv, W.; Yu, H.; Han, M.; Tan, Y.; Wu, M.; Zhang, J.; Wu, Y.; Zhang, Q. Analysis of Tumor Glycosylation Characteristics and Implications for Immune Checkpoint Inhibitor’s Efficacy for Breast Cancer. Front. Immunol. 2022, 13, 830158. [Google Scholar] [CrossRef]
- Sha, Y.; Han, L.; Sun, B.; Zhao, Q. Identification of a Glycosyltransferase Signature for Predicting Prognosis and Immune Microenvironment in Neuroblastoma. Front. Cell Dev. Biol. 2021, 9, 769580. [Google Scholar] [CrossRef]
- Munkley, J.; Elliott, D.J. Hallmarks of glycosylation in cancer. Oncotarget 2016, 7, 35478–35489. [Google Scholar] [CrossRef]
- Yu, D.M.; Li, X.H.; Mom, V.; Lu, Z.H.; Liao, X.W.; Han, Y.; Pichoud, C.; Gong, Q.M.; Zhang, D.H.; Zhang, Y.; et al. N-glycosylation mutations within hepatitis B virus surface major hydrophilic region contribute mostly to immune escape. J. Hepatol. 2014, 60, 515–522. [Google Scholar] [CrossRef]
- Verhelst, X.; Dias, A.M.; Colombel, J.F.; Vermeire, S.; Van Vlierberghe, H.; Callewaert, N.; Pinho, S.S. Protein Glycosylation as a Diagnostic and Prognostic Marker of Chronic Inflammatory Gastrointestinal and Liver Diseases. Gastroenterology 2020, 158, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Cortellino, S.; Longo, V.D. Metabolites and Immune Response in Tumor Microenvironments. Cancers 2023, 15, 3898. [Google Scholar] [CrossRef] [PubMed]
- Reinfeld, B.I.; Rathmell, W.K.; Kim, T.K.; Rathmell, J.C. The therapeutic implications of immunosuppressive tumor aerobic glycolysis. Cell. Mol. Immunol. 2022, 19, 46–58. [Google Scholar] [CrossRef]
- Shi, Q.; Shen, Q.; Liu, Y.; Shi, Y.; Huang, W.; Wang, X.; Li, Z.; Chai, Y.; Wang, H.; Hu, X.; et al. Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell 2022, 40, 1207–1222.e10. [Google Scholar] [CrossRef] [PubMed]
- DePeaux, K.; Delgoffe, G.M. Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. 2021, 21, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.; Khim, P.; Mkhikian, H.; Mortales, C.L.; Demetriou, M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife 2017, 6, e21330. [Google Scholar] [CrossRef]
- Karagiannis, F.; Peukert, K.; Surace, L.; Michla, M.; Nikolka, F.; Fox, M.; Weiss, P.; Feuerborn, C.; Maier, P.; Schulz, S.; et al. Impaired ketogenesis ties metabolism to T cell dysfunction in COVID-19. Nature 2022, 609, 801–807. [Google Scholar] [CrossRef]
- Wang, Y.N.; Lee, H.H.; Hsu, J.L.; Yu, D.; Hung, M.C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J. Biomed. Sci. 2020, 27, 77. [Google Scholar] [CrossRef]
- Cascio, S.; Finn, O.J. Intra- and Extra-Cellular Events Related to Altered Glycosylation of MUC1 Promote Chronic Inflammation, Tumor Progression, Invasion, and Metastasis. Biomolecules 2016, 6, 39. [Google Scholar] [CrossRef]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188464. [Google Scholar] [CrossRef]
- Huang, Y.L.; Liang, C.Y.; Labitzky, V.; Ritz, D.; Oliveira, T.; Cumin, C.; Estermann, M.; Lange, T.; Everest-Dass, A.V.; Jacob, F. Site-specific N-glycosylation of integrin alpha2 mediates collagen-dependent cell survival. iScience 2021, 24, 103168. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Yang, Q.; Tang, Q.; Li, X.; Ding, W.; Chen, W. Integrated transcriptomics unravels implications of glycosylation-regulating signature in diagnosis, prognosis and therapeutic benefits of hepatocellular carcinoma. Comput. Biol. Med. 2022, 148, 105886. [Google Scholar] [CrossRef] [PubMed]
- Caputo, W.L.; de Souza, M.C.; Basso, C.R.; Pedrosa, V.A.; Seiva, F.R.F. Comprehensive Profiling and Therapeutic Insights into Differentially Expressed Genes in Hepatocellular Carcinoma. Cancers 2023, 15, 5653. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Yang, S.; Tai, W.C.S.; Zhang, L.; Zhou, Y.; Cho, W.C.S.; Chan, L.W.C.; Wong, S.C.C. Bioinformatics Identification of Regulatory Genes and Mechanism Related to Hypoxia-Induced PD-L1 Inhibitor Resistance in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 8720. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Kong, N.; Zhao, Y.; Wu, Q.; Wang, X.; Xun, X.; Gao, P. Pan-Cancer Analyses Reveal Oncogenic and Immunological Role of PLOD2. Front. Genet. 2022, 13, 864655. [Google Scholar] [CrossRef]
- Zhu, X.G.; Nicholson Puthenveedu, S.; Shen, Y.; La, K.; Ozlu, C.; Wang, T.; Klompstra, D.; Gultekin, Y.; Chi, J.; Fidelin, J.; et al. CHP1 Regulates Compartmentalized Glycerolipid Synthesis by Activating GPAT4. Mol. Cell 2019, 74, 45–58.e7. [Google Scholar] [CrossRef]
- Liu, Y.; Zaun, H.C.; Orlowski, J.; Ackerman, S.L. CHP1-mediated NHE1 biosynthetic maturation is required for Purkinje cell axon homeostasis. J. Neurosci. 2013, 33, 12656–12669. [Google Scholar] [CrossRef]
- Xi, D.; Wang, J.; Yang, Y.; Ji, F.; Li, C.; Yan, X. A novel natural killer-related signature to effectively predict prognosis in hepatocellular carcinoma. BMC Med. Genom. 2023, 16, 211. [Google Scholar] [CrossRef]
- An, P.; Wang, L.H.; Hutcheson-Dilks, H.; Nelson, G.; Donfield, S.; Goedert, J.J.; Rinaldo, C.R.; Buchbinder, S.; Kirk, G.D.; O’Brien, S.J.; et al. Regulatory polymorphisms in the cyclophilin A gene, PPIA, accelerate progression to AIDS. PLoS Pathog. 2007, 3, e88. [Google Scholar] [CrossRef]
- Liu, P.; Lin, C.; Liu, Z.; Zhu, C.; Lin, Z.; Xu, D.; Chen, J.; Huang, Q.; Li, C.Y.; Hou, L.; et al. Inhibition of ALG3 stimulates cancer cell immunogenic ferroptosis to potentiate immunotherapy. Cell. Mol. Life Sci. 2022, 79, 352. [Google Scholar] [CrossRef]
- Wang, D.; Zaitsev, S.; Taylor, G.; D’azzo, A.; Bonten, E. Protective protein/cathepsin A rescues N-glycosylation defects in neuramini-dase-1. Biochim. Biophys. Acta 2009, 1790, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Mir, H.; Khurram, M.A.; Fujihara, K.M.; Dynlacht, B.D.; Cardozo, T.J.; Possemato, R. Allosteric regulation of CAD modulates de novo pyrimidine synthesis during the cell cycle. Nat. Metab. 2023, 5, 277–293. [Google Scholar] [CrossRef]
- Haltiwanger, R.S.; Lowe, J.B. Role of glycosylation in development. Annu. Rev. Biochem. 2004, 73, 491–537. [Google Scholar] [CrossRef] [PubMed]
- Barrowman, J.; Bhandari, D.; Reinisch, K.; Ferro-Novick, S. TRAPP complexes in membrane traffic: Convergence through a common Rab. Nat. Rev. Mol. Cell Biol. 2010, 11, 759–763. [Google Scholar] [CrossRef]
- Song, Q.; Wen, J.; Li, W.; Xue, J.; Zhang, Y.; Liu, H.; Han, J.; Ning, T.; Lu, Z. HSP90 promotes radioresistance of cervical cancer cells via reducing FBXO6-mediated CD147 polyubiquitination. Cancer Sci. 2022, 113, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, P.G.; Ng, B.G.; Sanford, L.; Sutton, V.R.; Bartholomew, D.W.; Pastore, M.T.; Bamshad, M.J.; Kircher, M.; Buckingham, K.J.; Nickerson, D.A. SRD5A3-CDG: Expanding the phenotype of a congenital disorder of glycosylation with emphasis on adult onset features. Am. J. Med. Genet. A 2016, 170, 3165–3171. [Google Scholar] [CrossRef]
- Pattingre, S.; Turtoi, A. BAG Family Members as Mitophagy Regulators in Mammals. Cells 2022, 11, 681. [Google Scholar] [CrossRef]
- Huang, L.; Yu, Z.; Zhang, T.; Zhao, X.; Huang, G. HSP40 interacts with pyruvate kinase M2 and regulates glycolysis and cell prolif-eration in tumor cells. PLoS ONE 2014, 9, e92949. [Google Scholar] [CrossRef]
- Clement-Lacroix, P.; Little, C.B.; Smith, M.M.; Cottereaux, C.; Merciris, D.; Meurisse, S.; Mollat, P.; Touitou, R.; Brebion, F.; Gosmini, R.; et al. Pharmacological characterization of GLPG1972/S201086, a potent and selective small-molecule inhibitor of ADAMTS5. Osteoarthr. Cartil. 2022, 30, 291–301. [Google Scholar] [CrossRef]
- Du, H.; Pang, M.; Hou, X.; Yuan, S.; Sun, L. PLOD2 in cancer research. Biomed. Pharmacother. 2017, 90, 670–676. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Zhou, H.; Ma, X.; Wu, L.; Tian, M.; Li, S.; Qian, X.; Gao, X.; Chai, R. Dync1li1 is required for the survival of mammalian cochlear hair cells by regulating the transportation of autophagosomes. PLoS Genet. 2022, 18, e1010232. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.Y.; Liang, S.Y.; Lu, S.C.; Tseng, H.C.; Tsai, H.Y.; Tang, C.J.; Sugata, M.; Chen, Y.J.; Chen, Y.J.; Wu, S.J.; et al. Molecular Basis and Role of Siglec-7 Ligand Expression on Chronic Lymphocytic Leukemia B Cells. Front. Immunol. 2022, 13, 840388. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Chen, J.; Zhu, R.; Huang, G.; Zeng, J.; Yu, H.; He, Z.; Han, C. Integrating TCGA and Single-Cell Sequencing Data for Hepatocellular Carcinoma: A Novel Glycosylation (GLY)/Tumor Microenvironment (TME) Classifier to Predict Prognosis and Immunotherapy Response. Metabolites 2024, 14, 51. https://doi.org/10.3390/metabo14010051
Wu Y, Chen J, Zhu R, Huang G, Zeng J, Yu H, He Z, Han C. Integrating TCGA and Single-Cell Sequencing Data for Hepatocellular Carcinoma: A Novel Glycosylation (GLY)/Tumor Microenvironment (TME) Classifier to Predict Prognosis and Immunotherapy Response. Metabolites. 2024; 14(1):51. https://doi.org/10.3390/metabo14010051
Chicago/Turabian StyleWu, Yun, Jiaru Chen, Riting Zhu, Guoliang Huang, Jincheng Zeng, Hongbing Yu, Zhiwei He, and Cuifang Han. 2024. "Integrating TCGA and Single-Cell Sequencing Data for Hepatocellular Carcinoma: A Novel Glycosylation (GLY)/Tumor Microenvironment (TME) Classifier to Predict Prognosis and Immunotherapy Response" Metabolites 14, no. 1: 51. https://doi.org/10.3390/metabo14010051
APA StyleWu, Y., Chen, J., Zhu, R., Huang, G., Zeng, J., Yu, H., He, Z., & Han, C. (2024). Integrating TCGA and Single-Cell Sequencing Data for Hepatocellular Carcinoma: A Novel Glycosylation (GLY)/Tumor Microenvironment (TME) Classifier to Predict Prognosis and Immunotherapy Response. Metabolites, 14(1), 51. https://doi.org/10.3390/metabo14010051