
Study on the Pharmacological Mechanism of Icariin for the Treatment of Alzheimer’s Disease Based on Network Pharmacology and Molecular Docking Techniques
Abstract
:1. Introduction
2. Materials and Methods
2.1. Prediction of Relevant Targets of AD and Epimedium Components
2.2. Protein–Protein Interaction (PPI) Data
2.3. GO and KEGG Pathway Enrichment Analysis
2.4. Molecular Docking
3. Results
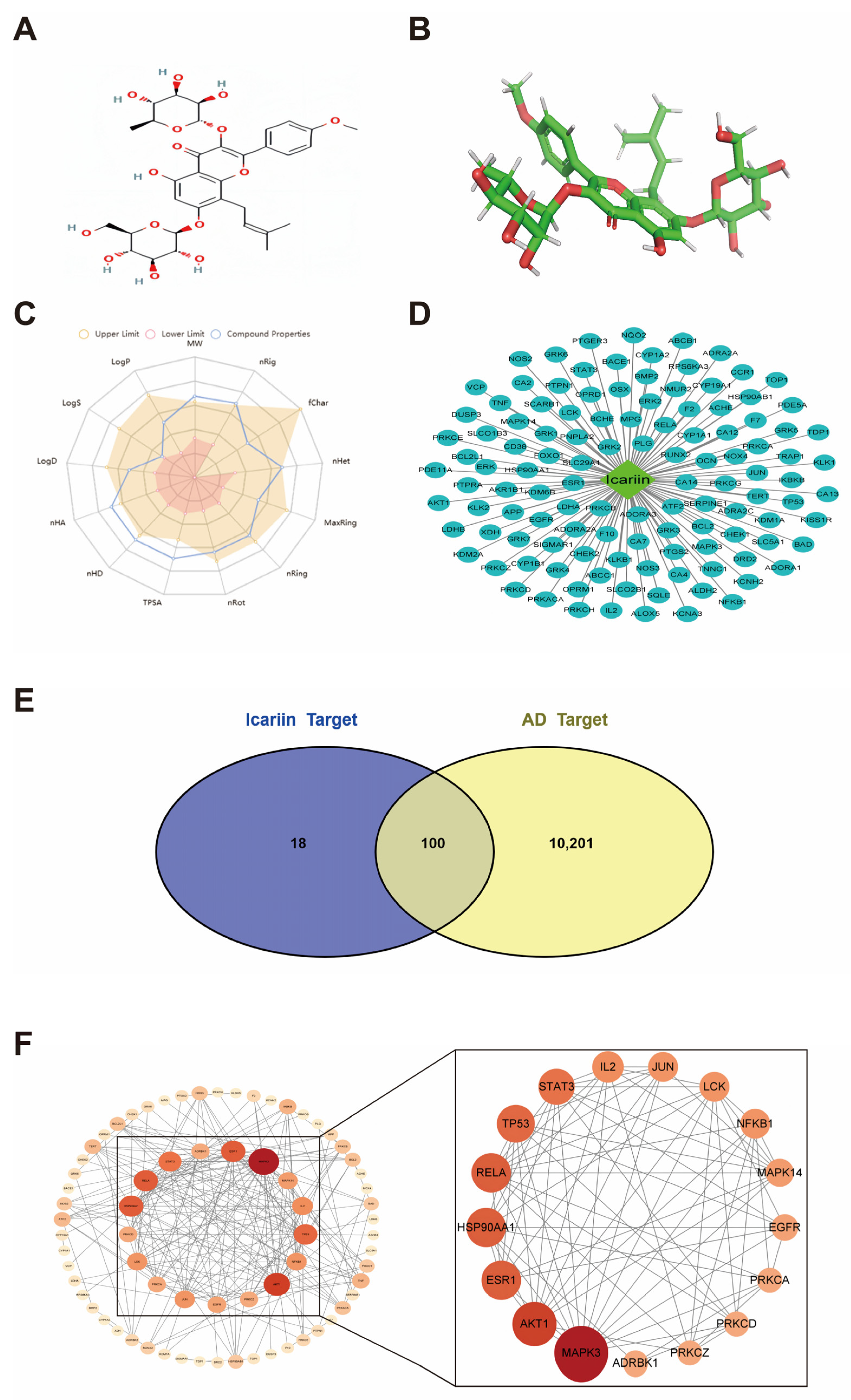
3.1. Physicochemical Properties and Pharmacokinetic Information of Icariin
3.2. Potential Targets of ICA for AD
3.3. Potential Target PPI Networks
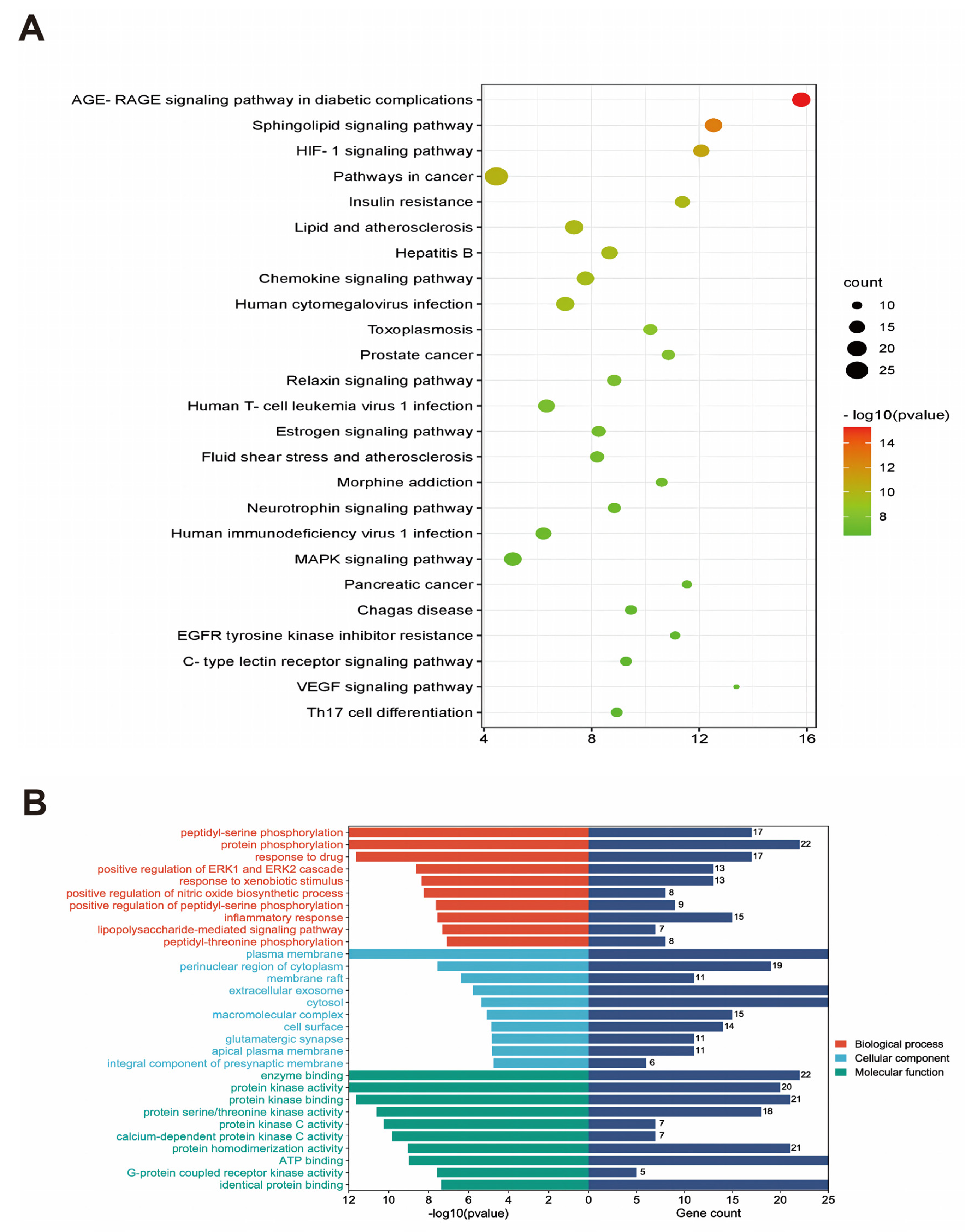
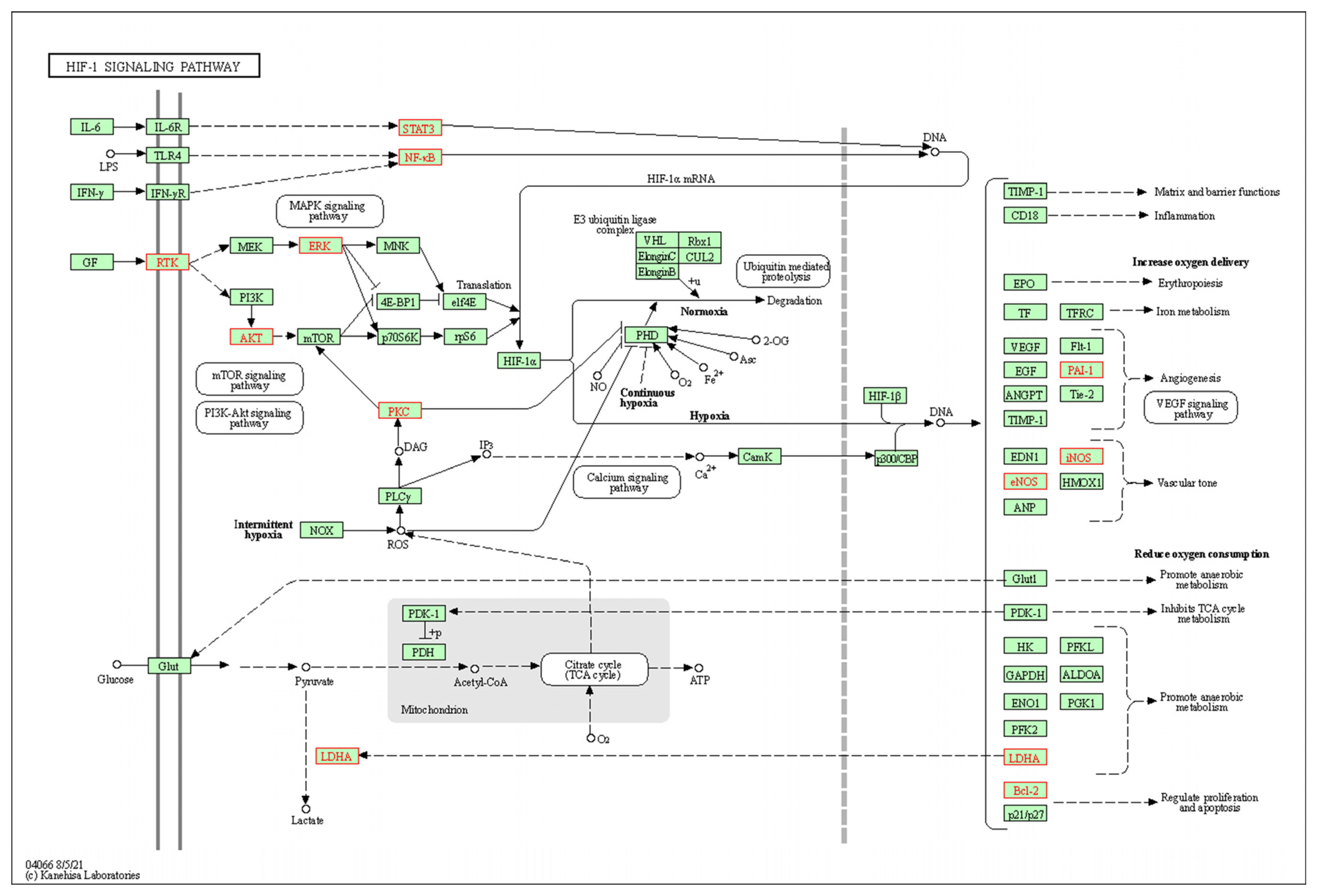
3.4. Results of GO Analysis and KEGG Metabolic Pathway Enrichment Analysis
3.5. Construction of the Compound-Target-Pathway-Disease Pharmacology Network for Compound ICA
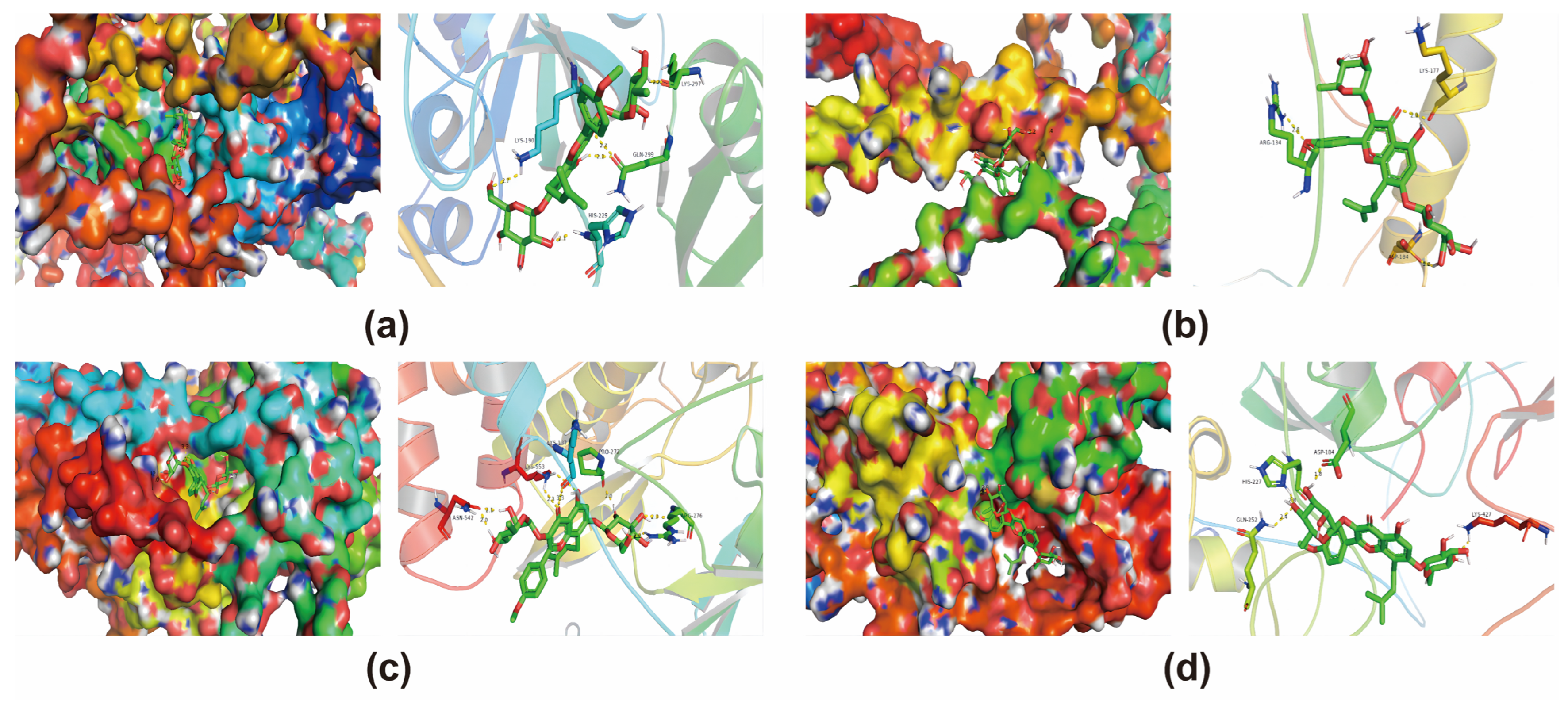
3.6. Molecular Docking Validation of Core Targets and Active Compounds
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laws, K.R.; Irvine, K.; Gale, T.M. Sex differences in cognitive impairment in Alzheimer’s disease. World J. Psychiatry 2016, 6, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.K.; Zhang, S.M.; Ni, J.J. Research progress of cathepsin B in brain aging and Alzheimer’s diseases. Yi Chuan 2023, 45, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Acosta, D.; Jiménez Velázquez, I.Z.; Llibre Guerra, J.J.; Peeters, G.; Rodriguez-Salgado, A.; de Jesús Llibre Rodriguez, J. 101—Alzheimer Disease and Dementia: Diagnostic challenges and future directions in Hispanic populations. Int. Psychogeriatr. 2021, 33, 2. [Google Scholar] [CrossRef]
- Courtney, C.; Farrell, D.; Gray, R.; Hills, R.; Lynch, L.; Sellwood, E.; Edwards, S.; Hardyman, W.; Raftery, J.; Crome, P.; et al. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): Randomised double-blind trial. Lancet 2004, 363, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Farlow, M. A clinical overview of cholinesterase inhibitors in Alzheimer’s disease. Int. Psychogeriatr. 2002, 14 (Suppl. S1), 93–126. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- Cochran, J.N.; Hall, A.M.; Roberson, E.D. The dendritic hypothesis for Alzheimer’s disease pathophysiology. Brain Res. Bull. 2014, 103, 18–28. [Google Scholar] [CrossRef]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Arai, R.; Takahashi, D.; Inoue, M.; Irie, T.; Asano, T.; Konno, T.; Terkawi, M.A.; Onodera, T.; Kondo, E.; Iwasaki, N. Efficacy of teriparatide in the treatment of nontraumatic osteonecrosis of the femoral head: A retrospective comparative study with alendronate. BMC Musculoskelet. Disord. 2017, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wei, J.; Xiao, H.; Zhu, L.; Weng, X.; Jin, J.; Zhang, J.; Wang, H.; Ge, J.; Tao, T.; et al. Bone-strengthening supplement (BSP) promotes bone and cartilage repair, for the treatment of Osteonecrosis of Femoral Head: An MRI-based study. Am. J. Transl. Res. 2019, 11, 7449–7455. [Google Scholar] [PubMed]
- Bi, Z.; Zhang, W.; Yan, X. Anti-inflammatory and immunoregulatory effects of icariin and icaritin. Biomed. Pharmacother. 2022, 151, 113180. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zhao, L.; Zhang, L.; Li, Y.; Zhang, L.; Li, L. Icariin Promotes Survival, Proliferation, and Differentiation of Neural Stem Cells In Vitro and in a Rat Model of Alzheimer’s Disease. Stem Cells Int. 2021, 2021, 9974625. [Google Scholar] [CrossRef]
- Qasim, M.; Abdullah, M.; Ali Ashfaq, U.; Noor, F.; Nahid, N.; Alzamami, A.; Alturki, N.A.; Khurshid, M. Molecular mechanism of Ferula asafoetida for the treatment of asthma: Network pharmacology and molecular docking approach. Saudi J. Biol. Sci. 2023, 30, 103527. [Google Scholar] [CrossRef]
- Zhai, Z.; Tao, X.; Alami, M.M.; Shu, S.; Wang, X. Network Pharmacology and Molecular Docking Combined to Analyze the Molecular and Pharmacological Mechanism of Pinellia ternata in the Treatment of Hypertension. Curr. Issues Mol. Biol. 2021, 43, 6. [Google Scholar] [CrossRef]
- Luo, W.; Deng, J.; He, J.; Yin, L.; You, R.; Zhang, L.; Shen, J.; Han, Z.; Xie, F.; He, J.; et al. Integration of molecular docking, molecular dynamics and network pharmacology to explore the multi-target pharmacology of fenugreek against diabetes. J. Cell Mol. Med. 2023, 27, 1959–1974. [Google Scholar] [CrossRef]
- Yi, P.; Zhang, Z.; Huang, S.; Huang, J.; Peng, W.; Yang, J. Integrated meta-analysis, network pharmacology, and molecular docking to investigate the efficacy and potential pharmacological mechanism of Kai-Xin-San on Alzheimer’s disease. Pharm. Biol. 2020, 58, 932–943. [Google Scholar] [CrossRef]
- Qiu, Z.-K.; Zhou, B.-X.; Pang, J.; Zeng, W.-Q.; Wu, H.-B.; Yang, F. The network pharmacology study and molecular docking to investigate the potential mechanism of Acoritataninowii Rhizoma against Alzheimer’s Disease. Metab. Brain Dis. 2023, 38, 1937–1962. [Google Scholar] [CrossRef]
- Evering, T.H.; Marston, J.L.; Gan, L.; Nixon, D.F. Transposable elements and Alzheimer’s disease pathogenesis. Trends Neurosci. 2023, 46, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Shi, J.; Lin, Y.; Zhang, Y.; Luo, Q.; Huang, S.; Wang, S.; Wei, J.; Huang, J.; Li, C.; et al. Icariin Ameliorates Alzheimer’s Disease Pathology by Alleviating Myelin Injury in 3 × Tg-AD Mice. Neurochem. Res. 2022, 47, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Khezri, M.R.; Ghasemnejad-Berenji, M. Icariin: A Potential Neuroprotective Agent in Alzheimer’s Disease and Parkinson’s Disease. Neurochem. Res. 2022, 47, 2954–2962. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ma, J.; Zeng, Y.; Zhou, G.; Wang, Y.; Zhou, W.; Sun, X.; Wu, M. Icariin, an Up-and-Coming Bioactive Compound Against Neurological Diseases: Network Pharmacology-Based Study and Literature Review. Drug Des. Dev. Ther. 2021, 15, 3619–3641. [Google Scholar] [CrossRef] [PubMed]
- Valle, M.L.; Zastre, J. Thiamine Insufficiency Induced Hypoxia-Inducible Factor 1-α (HIF-1α) Regulates Alzheimer’s Disease-Like Neuropathology. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Shang, C.; Ou, X.; Zhang, H.; Wei, D.; Wang, Q.; Li, G. Activation of PGRN/MAPK axis stimulated by the hypoxia-conditioned mesenchymal stem cell-derived HIF-1α facilitates osteosarcoma progression. Exp. Cell Res. 2022, 421, 113373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef]
- Zhang, X.; Zuo, L.; Geng, Z.; Song, X.; Li, J.; Ge, S.; Jiang, Y.; Yang, Z.; Liu, G.; Zhao, Y.; et al. Vindoline ameliorates intestinal barrier damage in Crohn’s disease mice through MAPK signaling pathway. FASEB J. 2022, 36, e22589. [Google Scholar] [CrossRef]
- Luo, Q.; Schnöder, L.; Hao, W.; Litzenburger, K.; Decker, Y.; Tomic, I.; Menger, M.D.; Liu, Y.; Fassbender, K. p38α-MAPK-deficient myeloid cells ameliorate symptoms and pathology of APP-transgenic Alzheimer’s disease mice. Aging Cell 2022, 21, e13679. [Google Scholar] [CrossRef]
- La Rosa, F.; Zoia, C.P.; Bazzini, C.; Bolognini, A.; Saresella, M.; Conti, E.; Ferrarese, C.; Piancone, F.; Marventano, I.; Galimberti, D.; et al. Modulation of MAPK- and PI3/AKT-Dependent Autophagy Signaling by Stavudine (D4T) in PBMC of Alzheimer’s Disease Patients. Cells 2022, 11, 2180. [Google Scholar] [CrossRef]
- Kang, K.; Bai, J.; Zhong, S.; Zhang, R.; Zhang, X.; Xu, Y.; Zhao, M.; Zhao, C.; Zhou, Z. Down-Regulation of Insulin Like Growth Factor 1 Involved in Alzheimer’s Disease via MAPK, Ras, and FoxO Signaling Pathways. Oxid. Med. Cell Longev. 2022, 2022, 8169981. [Google Scholar] [CrossRef] [PubMed]
- Ramasubbu, K.; Devi Rajeswari, V. Impairment of insulin signaling pathway PI3K/Akt/mTOR and insulin resistance induced AGEs on diabetes mellitus and neurodegenerative diseases: A perspective review. Mol. Cell Biochem. 2023, 478, 1307–1324. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chang, C.; Saliba, A.; Bhat, M.A. Microglial mTOR Activation Upregulates Trem2 and Enhances β-Amyloid Plaque Clearance in the 5XFAD Alzheimer’s Disease Model. J. Neurosci. 2022, 42, 5294–5313. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Liu, G.; Yang, Y.; Chen, L.; Shang, Y.; Hong, Q. Identifying mechanisms of Epimedii Folium against Alzheimer’s disease via a network pharmacology approach Epimedii Folium treats Alzheimer’s disease via PI3K-AKT. Eur. J. Inflamm. 2021, 19, 20587392211041435. [Google Scholar] [CrossRef]
- Kumar, M.; Bansal, N. Implications of Phosphoinositide 3-Kinase-Akt (PI3K-Akt) Pathway in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 354–385. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Yum, K.S.; Chang, J.Y.; Kim, M.; Ahn, J.Y.; Kim, S.; Lapchak, P.A.; Han, M.K. Dose-specific effect of simvastatin on hypoxia-induced HIF-1α and BACE expression in Alzheimer’s disease cybrid cells. BMC Neurol. 2015, 15, 127. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Wu, C.H.; Yan, Y.J.; Wang, D.H.; Wang, M.J.; Hou, Z.W. Effect of Biantie pretreatment on serum level of PHD2/HIF-1α and brain tissue damage in rats during acute hypobaric hypoxia exposure. Zhongguo Zhen Jiu 2022, 42, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Li, N.; Shu, J.; Yang, X.; Wei, W.; Yan, A. Exosomes Derived From M2 Microglia Cells Attenuates Neuronal Impairment and Mitochondrial Dysfunction in Alzheimer’s Disease Through the PINK1/Parkin Pathway. Front. Cell Neurosci. 2022, 16, 874102. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Kocki, J.; Januszewski, S.; Bogucki, J.; Bogucka-Kocka, A.; Pluta, R. Dysregulation of Autophagy, Mitophagy, and Apoptosis Genes in the CA3 Region of the Hippocampus in the Ischemic Model of Alzheimer’s Disease in the Rat. J. Alzheimer’s Dis. 2019, 72, 1279–1286. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.S. PINK1 Activation Attenuates Impaired Neuronal-Like Differentiation and Synaptogenesis and Mitochondrial Dysfunction in Alzheimer’s Disease Trans-Mitochondrial Cybrid Cells. J. Alzheimer’s Dis. 2021, 81, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Matenia, D.; Hempp, C.; Timm, T.; Eikhof, A.; Mandelkow, E.M. Microtubule affinity-regulating kinase 2 (MARK2) turns on phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) at Thr-313, a mutation site in Parkinson disease: Effects on mitochondrial transport. J. Biol. Chem. 2012, 287, 8174–8186. [Google Scholar] [CrossRef] [PubMed]
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free Radic. Biol. Med. 2021, 172, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xue, Y.; Yao, Y.; Li, Y.; Ji, X.; Chi, T.; Liu, P.; Zou, L. PINK1 regulates mitochondrial fission/fusion and neuroinflammation in β-amyloid-induced Alzheimer’s disease models. Neurochem. Int. 2022, 154, 105298. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.M.; Elnagar, M.R.; Abdelrazik, E.; Mahdi, M.R.; Hamza, E.; Elattar, E.M.; ElNashar, E.M.; Alghamdi, M.A.; Al-Qahtani, Z.; Al-Khater, K.M.; et al. Neuroprotective effect of naringin against cerebellar changes in Alzheimer’s disease through modulation of autophagy, oxidative stress and tau expression: An experimental study. Front. Neuroanat. 2022, 16, 1012422. [Google Scholar] [CrossRef]
- Chung, K.M.; Hernández, N.; Sproul, A.A.; Yu, W.H. Alzheimer’s disease and the autophagic-lysosomal system. Neurosci. Lett. 2019, 697, 49–58. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Wang, C.; Cai, X.; Hu, W.; Li, Z.; Kong, F.; Chen, X.; Wang, D. Investigation of the neuroprotective effects of crocin via antioxidant activities in HT22 cells and in mice with Alzheimer’s disease. Int. J. Mol. Med. 2019, 43, 956–966. [Google Scholar] [CrossRef]
- Begcevic, I.; Brinc, D.; Brown, M.; Martinez-Morillo, E.; Goldhardt, O.; Grimmer, T.; Magdolen, V.; Batruch, I.; Diamandis, E.P. Brain-related proteins as potential CSF biomarkers of Alzheimer’s disease: A targeted mass spectrometry approach. J. Proteom. 2018, 182, 12–20. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Alzheimer’s disease (AD) therapeutics—1: Repeated clinical failures continue to question the amyloid hypothesis of AD and the current understanding of AD causality. Biochem. Pharmacol. 2018, 158, 359–375. [Google Scholar] [CrossRef]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef]
- Huang, F.; Wang, M.; Liu, R.; Wang, J.Z.; Schadt, E.; Haroutunian, V.; Katsel, P.; Zhang, B.; Wang, X. CDT2-controlled cell cycle reentry regulates the pathogenesis of Alzheimer’s disease. Alzheimer’s Dement. 2019, 15, 217–231. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Leanza, G.; Gulino, R.; Zorec, R. Noradrenergic Hypothesis Linking Neurodegeneration-Based Cognitive Decline and Astroglia. Front. Mol. Neurosci. 2018, 11, 254. [Google Scholar] [CrossRef]
- Vakalopoulos, C. Alzheimer’s Disease: The Alternative Serotonergic Hypothesis of Cognitive Decline. J. Alzheimer’s Dis. 2017, 60, 859–866. [Google Scholar] [CrossRef]
- Guo, L.; Tian, J.; Du, H. Mitochondrial Dysfunction and Synaptic Transmission Failure in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1071–1086. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Property | Medicinal Chemistry | Absorption | Metabolism | ||||
|---|---|---|---|---|---|---|---|
| Molecular Weight (MW) | 676.240 | QED | 0.140 | Caco-2 Permeability | −6.178 | CYP1A2 inhibitor | --- |
| Volume | 644.667 | SAscore | 4.912 | MDCK Permeability | 7.7 × 10−5 | CYP1A2 substrate | --- |
| Density | 1.049 | Fsp3 | 0.485 | Pgp-inhibitor | --- | CYP2C19 inhibitor | --- |
| nHA | 15 | MCE-18 | 122.694 | Pgp-substrate | +++ | CYP2C19 substrate | -- |
| nHD | 8 | NPscore | 1.959 | HIA | ++ | CYP2C9 inhibitor | --- |
| nRot | 9 | Lipinski Rule | Rejected | F20% | --- | CYP2C9 substrate | + |
| nRing | 5 | Pfizer Rule | Accepted | F30% | +++ | CYP2D6 inhibitor | --- |
| MaxRing | 10 | GSK Rule | Rejected | CYP2D6 substrate | -- | ||
| nHet | 15 | Golden Triangle | Rejected | CYP3A4 inhibitor | --- | ||
| fChar | 0 | PAINS | 0 alert(s) | CYP3A4 substrate | --- | ||
| nRig | 31 | ALARM NMR Rule | 2 alert(s) | Distribution | Excretion | ||
| Flexibility | 0.290 | BMS Rule | 1 alert(s) | PPB | 64.816% | CL | 1.757 |
| Stereo Centers | 10 | Chelator Rule | 0 alert(s) | VD | 0.772 | T1/2 | 0.109 |
| TPSA | 238.200 | BBB Penetration | -- | ||||
| logS | −3.781 | Fu | 24.984% | ||||
| logP | 1.587 | ||||||
| logD | 2.066 | ||||||
| Classification | Target | Shorthand | Prediction | Probability |
|---|---|---|---|---|
| Organ toxicity | Hepatotoxicity | dili | Inactive | 0.74 |
| Toxicity end points | Carcinogenicity | carcino | Inactive | 0.83 |
| Toxicity end points | Immunotoxicity | immuno | Active | 0.98 |
| Toxicity end points | Mutagenicity | mutagen | Inactive | 0.70 |
| Toxicity end points | Cytotoxicity | cyto | Inactive | 0.61 |
| Tox21-Nuclear receptor signaling pathways | Aryl hydrocarbon Receptor (AhR) | nr_ahr | Inactive | 0.73 |
| Tox21-Nuclear receptor signaling pathways | Androgen Receptor (AR) | nr_ar | Inactive | 0.97 |
| Tox21-Nuclear receptor signaling pathways | Androgen Receptor Ligand Binding Domain (AR-LBD) | nr_ar_lbd | Inactive | 0.97 |
| Tox21-Nuclear receptor signaling pathways | Aromatase | nr_aromatase | Inactive | 0.85 |
| Tox21-Nuclear receptor signaling pathways | Estrogen Receptor Alpha (ER) | nr_er | Inactive | 0.88 |
| Tox21-Nuclear receptor signaling pathways | Estrogen Receptor Ligand Binding Domain (ER-LBD) | nr_er_lbd | Inactive | 0.99 |
| Tox21-Nuclear receptor signaling pathways | Peroxisome Proliferator Activated Receptor Gamma (PPAR-Gamma) | nr_ppar_gamma | Inactive | 0.95 |
| Tox21-Stress response pathways | Nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element (nrf2/ARE) | sr_are | Inactive | 0.89 |
| Tox21-Stress response pathways | Heat shock factor response element (HSE) | sr_hse | Inactive | 0.89 |
| Tox21-Stress response pathways | Mitochondrial Membrane Potential (MMP) | sr_mmp | Inactive | 0.84 |
| Tox21-Stress response pathways | Phosphoprotein (Tumor Suppressor) p53 | sr_p53 | Inactive | 0.73 |
| Tox21-Stress response pathways | ATPase family AAA domain-containing protein 5 (ATAD5) | sr_atad5 | Inactive | 0.97 |
| Gene Symbol | Degree | Betweenness | Closeness | Eigenvector | Information | LAC | Network |
|---|---|---|---|---|---|---|---|
| MAPK3 | 28 | 1213.18 | 0.26 | 0.34 | 3.35 | 6.71 | 18.45 |
| AKT1 | 22 | 371.72 | 0.25 | 0.29 | 3.26 | 6.36 | 14.02 |
| HSP90AA1 | 19 | 810.08 | 0.25 | 0.23 | 3.19 | 4.53 | 8.41 |
| ESR1 | 19 | 557.74 | 0.25 | 0.24 | 3.19 | 5.68 | 11.20 |
| RELA | 19 | 398.36 | 0.25 | 0.26 | 3.19 | 6.63 | 12.23 |
| TP53 | 18 | 776.05 | 0.25 | 0.21 | 3.16 | 3.89 | 8.48 |
| STAT3 | 17 | 229.70 | 0.24 | 0.25 | 3.14 | 6.35 | 9.86 |
| IL2 | 14 | 63.35 | 0.24 | 0.22 | 3.03 | 6.29 | 8.73 |
| NFKB1 | 13 | 165.17 | 0.23 | 0.18 | 2.99 | 4.92 | 6.70 |
| LCK | 13 | 48.24 | 0.24 | 0.21 | 2.99 | 6.31 | 8.17 |
| JUN | 13 | 309.10 | 0.24 | 0.20 | 2.99 | 6.00 | 7.71 |
| MAPK14 | 12 | 33.53 | 0.23 | 0.22 | 2.94 | 7.17 | 8.25 |
| EGFR | 11 | 252.93 | 0.24 | 0.15 | 2.89 | 3.45 | 4.26 |
| ADRBK1 | 10 | 923.55 | 0.23 | 0.05 | 2.83 | 1.00 | 4.22 |
| PRKCD | 10 | 248.02 | 0.24 | 0.15 | 2.83 | 3.00 | 3.42 |
| PRKCA | 10 | 202.93 | 0.24 | 0.14 | 2.83 | 3.60 | 4.21 |
| PRKCZ | 10 | 13.82 | 0.22 | 0.15 | 2.83 | 5.00 | 5.92 |
| HIF-1 | BNIP3 | PINK1 | Parkin | |
|---|---|---|---|---|
| Icariin | −8.0 | −7.6 | −8.1 | −8.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Zheng, J.; Sun, X.; Xie, L.; Yang, Y. Study on the Pharmacological Mechanism of Icariin for the Treatment of Alzheimer’s Disease Based on Network Pharmacology and Molecular Docking Techniques. Metabolites 2024, 14, 1. https://doi.org/10.3390/metabo14010001
Wang D, Zheng J, Sun X, Xie L, Yang Y. Study on the Pharmacological Mechanism of Icariin for the Treatment of Alzheimer’s Disease Based on Network Pharmacology and Molecular Docking Techniques. Metabolites. 2024; 14(1):1. https://doi.org/10.3390/metabo14010001
Chicago/Turabian StyleWang, Dongwei, Jilong Zheng, Xingsheng Sun, Liuwei Xie, and Yang Yang. 2024. "Study on the Pharmacological Mechanism of Icariin for the Treatment of Alzheimer’s Disease Based on Network Pharmacology and Molecular Docking Techniques" Metabolites 14, no. 1: 1. https://doi.org/10.3390/metabo14010001
APA StyleWang, D., Zheng, J., Sun, X., Xie, L., & Yang, Y. (2024). Study on the Pharmacological Mechanism of Icariin for the Treatment of Alzheimer’s Disease Based on Network Pharmacology and Molecular Docking Techniques. Metabolites, 14(1), 1. https://doi.org/10.3390/metabo14010001