

Mucosal Metabolomic Signatures in Chronic Colitis: Novel Insights into the Pathophysiology of Inflammatory Bowel Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Metabolomic Analysis
2.2.1. Sample Preparation
2.2.2. Ultrahigh Performance Liquid Chromatography–Tandem Mass Spectroscopy (UPLC-MS/MS)
2.2.3. Compound Identification and Metabolite Quantification
2.3. Bioinformatic Analysis
2.4. Myeloperoxidase Activity
2.5. Hematoxylin and Eosin Staining
2.6. Microbial Sampling and 16S rRNA Sequencing
2.7. Statistical Analysis
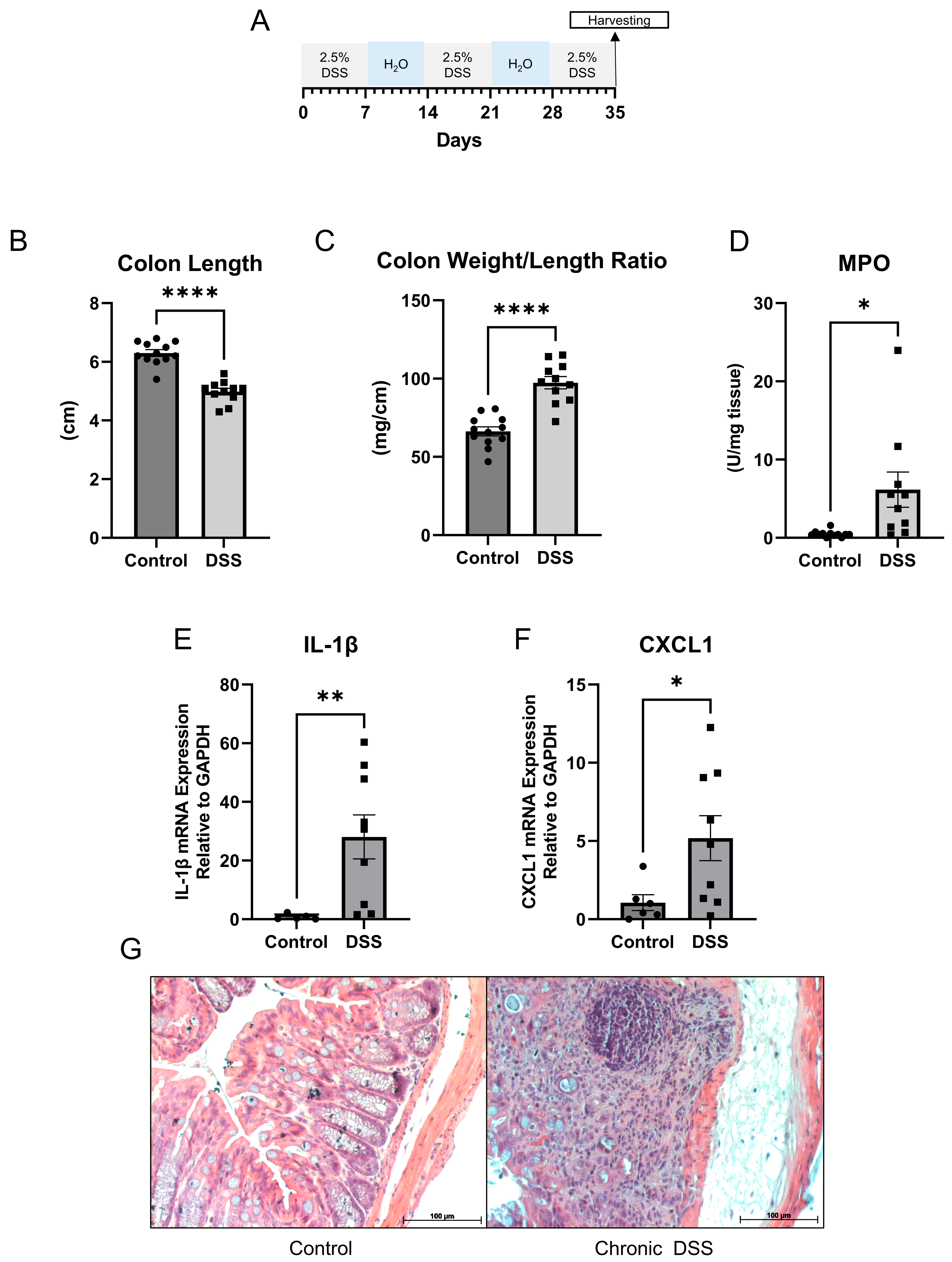
3. Results
3.1. Gut Microbial Dysbiosis Is Associated with Chronic DSS Treatment
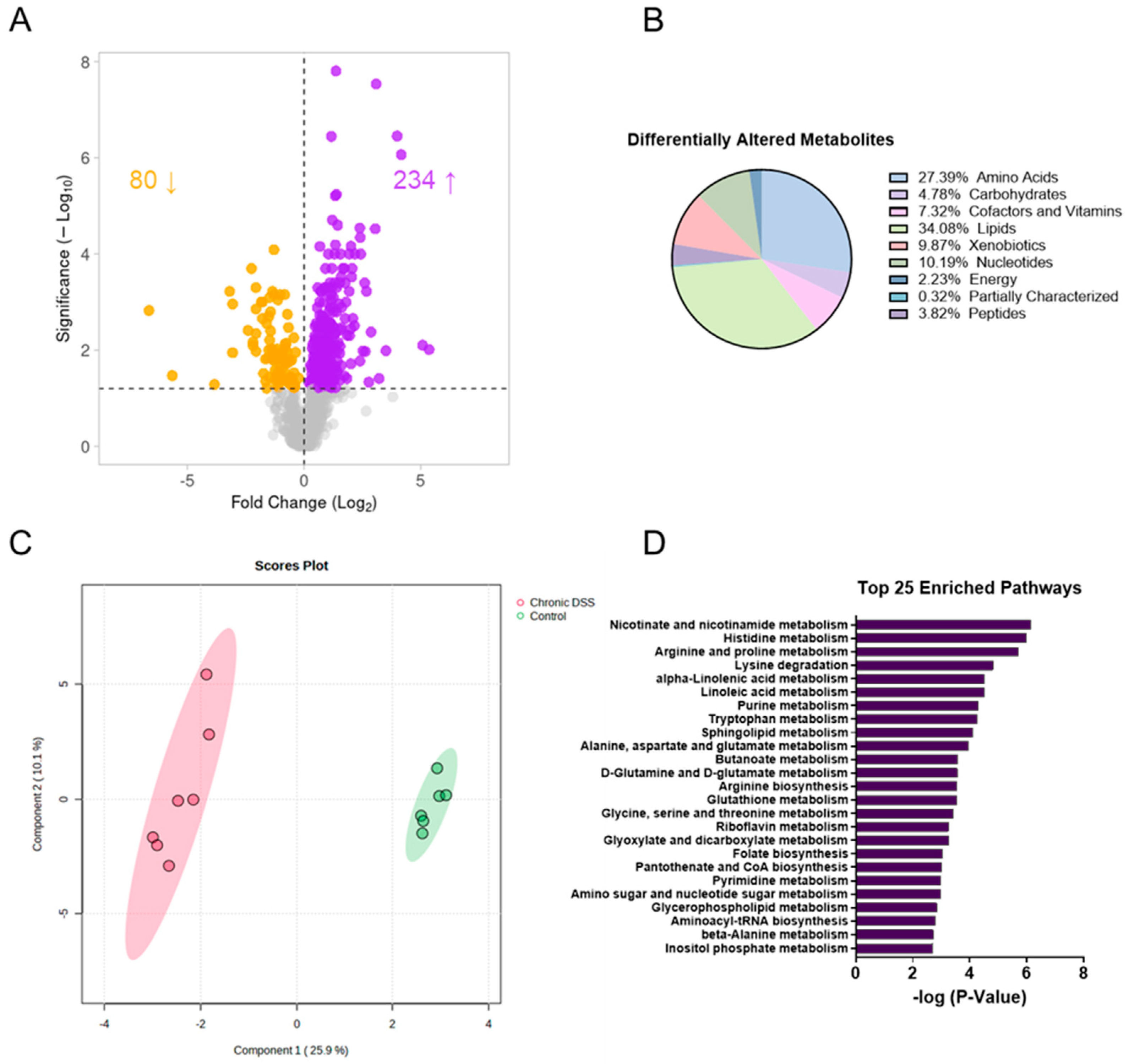
3.2. Chronic Colitis Leads to Distinct Metabolomic Profile in the Colonic Mucosa
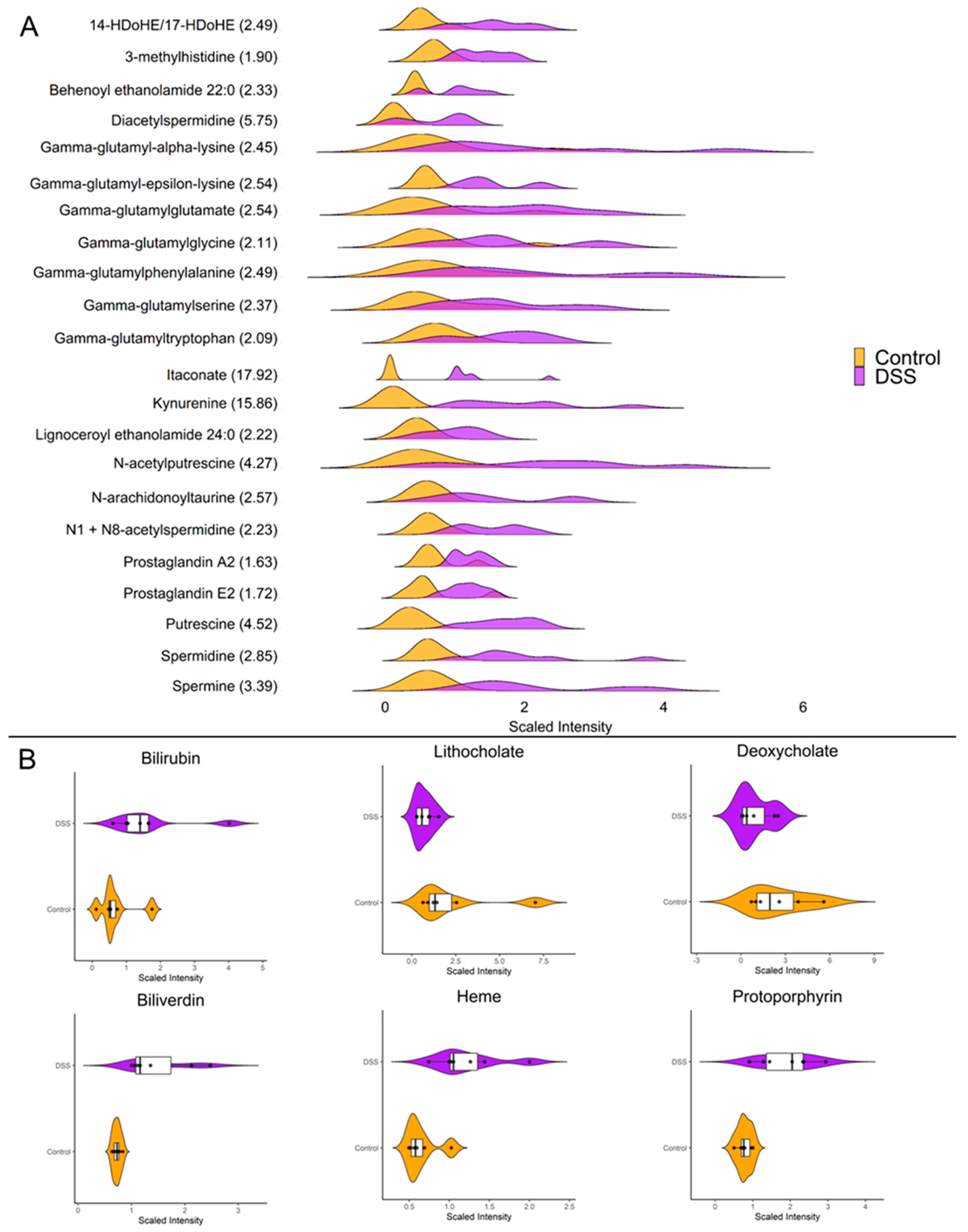
3.3. Chronic Colitis Leads to Increased Mucosal Localization of Inflammatory Metabolites and Impaired Enterohepatic Circulation
3.4. Contribution of Microbial Mucosal Metabolomic Profile during Chronic Colitis
3.5. Chronic Colitis Alters Mucosal Dietary, Nucleotide, Amino Acid, and Lipid Metabolism
4. Discussion
4.1. Facts and Perspectives
4.2. Strengths of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321.e2. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [PubMed]
- Gohil, K.; Carramusa, B. Ulcerative Colitis and Crohn’s Disease. P T 2014, 39, 576–577. [Google Scholar] [PubMed]
- Al-Bawardy, B.; Shivashankar, R.; Proctor, D.D. Novel and Emerging Therapies for Inflammatory Bowel Disease. Front. Pharmacol. 2021, 12, 651415. [Google Scholar] [CrossRef]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut Microbiome Structure and Metabolic Activity in Inflammatory Bowel Disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Zheng, L.; Wen, X.-L.; Duan, S.-L. Role of Metabolites Derived from Gut Microbiota in Inflammatory Bowel Disease. World J. Clin. Cases 2022, 10, 2660–2677. [Google Scholar] [CrossRef]
- Aldars-García, L.; Gisbert, J.P.; Chaparro, M. Metabolomics Insights into Inflammatory Bowel Disease: A Comprehensive Review. Pharmaceuticals 2021, 14, 1190. [Google Scholar] [CrossRef]
- Kolho, K.-L.; Pessia, A.; Jaakkola, T.; de Vos, W.M.; Velagapudi, V. Faecal and Serum Metabolomics in Paediatric Inflammatory Bowel Disease. J. Crohn’s Colitis 2017, 11, 321–334. [Google Scholar] [CrossRef]
- Le Gall, G.; Noor, S.O.; Ridgway, K.; Scovell, L.; Jamieson, C.; Johnson, I.T.; Colquhoun, I.J.; Kemsley, E.K.; Narbad, A. Metabolomics of Fecal Extracts Detects Altered Metabolic Activity of Gut Microbiota in Ulcerative Colitis and Irritable Bowel Syndrome. J. Proteome Res. 2011, 10, 4208–4218. [Google Scholar] [CrossRef]
- Bjerrum, J.T.; Wang, Y.; Hao, F.; Coskun, M.; Ludwig, C.; Günther, U.; Nielsen, O.H. Metabonomics of Human Fecal Extracts Characterize Ulcerative Colitis, Crohn’s Disease and Healthy Individuals. Metabolomics 2015, 11, 122–133. [Google Scholar] [CrossRef]
- Manzella, C.R.; Jayawardena, D.; Pagani, W.; Li, Y.; Alrefai, W.A.; Bauer, J.; Jung, B.; Weber, C.R.; Gill, R.K. Serum Serotonin Differentiates Between Disease Activity States in Crohn’s Patients. Inflamm. Bowel Dis. 2020, 26, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- De Preter, V.; Machiels, K.; Joossens, M.; Arijs, I.; Matthys, C.; Vermeire, S.; Rutgeerts, P.; Verbeke, K. Faecal Metabolite Profiling Identifies Medium-Chain Fatty Acids as Discriminating Compounds in IBD. Gut 2015, 64, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Jansson, J.; Willing, B.; Lucio, M.; Fekete, A.; Dicksved, J.; Halfvarson, J.; Tysk, C.; Schmitt-Kopplin, P. Metabolomics Reveals Metabolic Biomarkers of Crohn’s Disease. PLoS ONE 2009, 4, e6386. [Google Scholar] [CrossRef] [PubMed]
- Perez, K.; Ngollo, M.; Rabinowitz, K.; Hammoudi, N.; Seksik, P.; Xavier, R.J.; Daly, M.J.; Dotan, I.; Le Bourhis, L.; Allez, M. Meta-Analysis of IBD Gut Samples Gene Expression Identifies Specific Markers of Ileal and Colonic Diseases. Inflamm. Bowel Dis. 2022, 28, 775–782. [Google Scholar] [CrossRef]
- Abreu, M.T.; Davies, J.M.; Quintero, M.A.; Delmas, A.; Diaz, S.; Martinez, C.D.; Venables, T.; Reich, A.; Crynen, G.; Deshpande, A.R.; et al. Transcriptional Behavior of Regulatory T Cells Predicts IBD Patient Responses to Vedolizumab Therapy. Inflamm. Bowel Dis. 2022, 28, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.; Catesson, A.; Griffin, J.L.; Holmes, E.; Williams, H.R.T. Metabolomic Analysis in Inflammatory Bowel Disease: A Systematic Review. J. Crohn’s Colitis 2021, 15, 813–826. [Google Scholar] [CrossRef]
- Aldars-García, L.; Chaparro, M.; Gisbert, J.P. Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease. Microorganisms 2021, 9, 977. [Google Scholar] [CrossRef]
- Baydi, Z.; Limami, Y.; Khalki, L.; Zaid, N.; Naya, A.; Mtairag, E.M.; Oudghiri, M.; Zaid, Y. An Update of Research Animal Models of Inflammatory Bowel Disease. Sci. World J. 2021, 2021, 7479540. [Google Scholar] [CrossRef]
- Lanis, J.M.; Alexeev, E.E.; Curtis, V.F.; Kitzenberg, D.A.; Kao, D.J.; Battista, K.D.; Gerich, M.E.; Glover, L.E.; Kominsky, D.J.; Colgan, S.P. Tryptophan Metabolite Activation of the Aryl Hydrocarbon Receptor Regulates IL-10 Receptor Expression on Intestinal Epithelia. Mucosal. Immunol. 2017, 10, 1133–1144. [Google Scholar] [CrossRef]
- Chen, Y.; Mai, Q.; Chen, Z.; Lin, T.; Cai, Y.; Han, J.; Wang, Y.; Zhang, M.; Tan, S.; Wu, Z.; et al. Dietary Palmitoleic Acid Reprograms Gut Microbiota and Improves Biological Therapy against Colitis. Gut Microbes 2023, 15, 2211501. [Google Scholar] [CrossRef]
- Kang, S.; Kim, J.; Park, A.; Koh, M.; Shin, W.; Park, G.; Lee, T.A.; Kim, H.J.; Han, H.; Kim, Y.; et al. TRIM40 Is a Pathogenic Driver of Inflammatory Bowel Disease Subverting Intestinal Barrier Integrity. Nat. Commun. 2023, 14, 700. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef]
- Guo, L.; Milburn, M.V.; Ryals, J.A.; Lonergan, S.C.; Mitchell, M.W.; Wulff, J.E.; Alexander, D.C.; Evans, A.M.; Bridgewater, B.; Miller, L.; et al. Plasma Metabolomic Profiles Enhance Precision Medicine for Volunteers of Normal Health. Proc. Natl. Acad. Sci. USA 2015, 112, E4901–E4910. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z. Complex Heatmap Visualization. iMeta 2022, 1, e43. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Xu, C.; Zhang, D.; Ju, F.; Ni, Y. MetOrigin: Discriminating the Origins of Microbial Metabolites for Integrative Analysis of the Gut Microbiome and Metabolome. iMeta 2022, 1, e10. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A Web Server for Metabolomic Data Analysis and Interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef]
- Krawisz, J.E.; Sharon, P.; Stenson, W.F. Quantitative Assay for Acute Intestinal Inflammation Based on Myeloperoxidase Activity. Assessment of Inflammation in Rat and Hamster Models. Gastroenterology 1984, 87, 1344–1350. [Google Scholar] [CrossRef]
- Kumar, A.; Anbazhagan, A.N.; Coffing, H.; Chatterjee, I.; Priyamvada, S.; Gujral, T.; Saksena, S.; Gill, R.K.; Alrefai, W.A.; Borthakur, A.; et al. Lactobacillus Acidophilus Counteracts Inhibition of NHE3 and DRA Expression and Alleviates Diarrheal Phenotype in Mice Infected with Citrobacter Rodentium. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G817–G826. [Google Scholar] [CrossRef] [PubMed]
- McIver, L.J.; Abu-Ali, G.; Franzosa, E.A.; Schwager, R.; Morgan, X.C.; Waldron, L.; Segata, N.; Huttenhower, C. BioBakery: A Meta’omic Analysis Environment. Bioinformatics 2018, 34, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating Taxonomic, Functional, and Strain-Level Profiling of Diverse Microbial Communities with BioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Sorrell, M.F.; Batra, S.K.; Dhawan, P.; Singh, A.B. Gut Permeability and Mucosal Inflammation: Bad, Good or Context Dependent. Mucosal. Immunol. 2017, 10, 307–317. [Google Scholar] [CrossRef]
- Santana, P.T.; Rosas, S.L.B.; Ribeiro, B.E.; Marinho, Y.; de Souza, H.S.P. Dysbiosis in Inflammatory Bowel Disease: Pathogenic Role and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 3464. [Google Scholar] [CrossRef]
- Qiu, Z.; Yang, H.; Rong, L.; Ding, W.; Chen, J.; Zhong, L. Targeted Metagenome Based Analyses Show Gut Microbial Diversity of Inflammatory Bowel Disease Patients. Indian J. Microbiol. 2017, 57, 307–315. [Google Scholar] [CrossRef]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased Proportions of Bifidobacterium and the Lactobacillus Group and Loss of Butyrate-Producing Bacteria in Inflammatory Bowel Disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef]
- Glover, J.S.; Browning, B.D.; Ticer, T.D.; Engevik, A.C.; Engevik, M.A. Acinetobacter Calcoaceticus Is Well Adapted to Withstand Intestinal Stressors and Modulate the Gut Epithelium. Front. Physiol. 2022, 13, 880024. [Google Scholar] [CrossRef]
- Facchin, S.; Vitulo, N.; Calgaro, M.; Buda, A.; Romualdi, C.; Pohl, D.; Perini, B.; Lorenzon, G.; Marinelli, C.; D’Incà, R.; et al. Microbiota Changes Induced by Microencapsulated Sodium Butyrate in Patients with Inflammatory Bowel Disease. Neurogastroenterol. Motil. 2020, 32, e13914. [Google Scholar] [CrossRef]
- Dahal, R.H.; Kim, S.; Kim, Y.K.; Kim, E.S.; Kim, J. Insight into Gut Dysbiosis of Patients with Inflammatory Bowel Disease and Ischemic Colitis. Front. Microbiol. 2023, 14, 1174832. [Google Scholar] [CrossRef]
- Sasaki, K.; Inoue, J.; Sasaki, D.; Hoshi, N.; Shirai, T.; Fukuda, I.; Azuma, T.; Kondo, A.; Osawa, R. Construction of a Model Culture System of Human Colonic Microbiota to Detect Decreased Lachnospiraceae Abundance and Butyrogenesis in the Feces of Ulcerative Colitis Patients. Biotechnol. J. 2019, 14, e1800555. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhao, W.; Lan, P.; Mou, X. The Microbiome in Inflammatory Bowel Diseases: From Pathogenesis to Therapy. Protein Cell 2021, 12, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Luan, Y.; Sun, F. Variance Adjusted Weighted UniFrac: A Powerful Beta Diversity Measure for Comparing Communities Based on Phylogeny. BMC Bioinform. 2011, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Peace, C.G.; O’Neill, L.A. The Role of Itaconate in Host Defense and Inflammation. J. Clin. Investig. 2022, 132, e148548. [Google Scholar] [CrossRef]
- Clooney, A.G.; Eckenberger, J.; Laserna-Mendieta, E.; Sexton, K.A.; Bernstein, M.T.; Vagianos, K.; Sargent, M.; Ryan, F.J.; Moran, C.; Sheehan, D.; et al. Ranking Microbiome Variance in Inflammatory Bowel Disease: A Large Longitudinal Intercontinental Study. Gut 2021, 70, 499–510. [Google Scholar] [CrossRef]
- Abdel-Rahman, L.I.H.; Morgan, X.C. Searching for a Consensus Among Inflammatory Bowel Disease Studies: A Systematic Meta-Analysis. Inflamm. Bowel Dis. 2023, 29, 125–139. [Google Scholar] [CrossRef]
- Nie, K.; Ma, K.; Luo, W.; Shen, Z.; Yang, Z.; Xiao, M.; Tong, T.; Yang, Y.; Wang, X. Roseburia Intestinalis: A Beneficial Gut Organism from the Discoveries in Genus and Species. Front. Cell. Infect. Microbiol. 2021, 11, 757718. [Google Scholar] [CrossRef]
- Luo, W.; Shen, Z.; Deng, M.; Li, X.; Tan, B.; Xiao, M.; Wu, S.; Yang, Z.; Zhu, C.; Tian, L.; et al. Roseburia Intestinalis Supernatant Ameliorates Colitis Induced in Mice by Regulating the Immune Response. Mol. Med. Rep. 2019, 20, 1007–1016. [Google Scholar] [CrossRef]
- Tilg, H.; Danese, S. Roseburia Hominis: A Novel Guilty Player in Ulcerative Colitis Pathogenesis? Gut 2014, 63, 1204–1205. [Google Scholar] [CrossRef]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, M.; Li, W.; Zhu, S.; Zhang, D. Stachydrine Attenuates IL-1β-Induced Inflammatory Response in Osteoarthritis Chondrocytes through the NF-ΚB Signaling Pathway. Chem. Biol. Interact. 2020, 326, 109136. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, B.; Zhang, H.; Chen, L.; Wang, X.; Chen, H.; Zhou, L. L-Ergothioneine Exhibits Protective Effects against Dextran Sulfate Sodium-Induced Colitis in Mice. ACS Omega 2022, 7, 21554–21565. [Google Scholar] [CrossRef]
- Gründemann, D.; Harlfinger, S.; Golz, S.; Geerts, A.; Lazar, A.; Berkels, R.; Jung, N.; Rubbert, A.; Schömig, E. Discovery of the Ergothioneine Transporter. Proc. Natl. Acad. Sci. USA 2005, 102, 5256–5261. [Google Scholar] [CrossRef] [PubMed]
- Bryan, P.-F.; Karla, C.; Edgar Alejandro, M.-T.; Sara Elva, E.-P.; Gemma, F.; Luz, C. Sphingolipids as Mediators in the Crosstalk between Microbiota and Intestinal Cells: Implications for Inflammatory Bowel Disease. Mediat. Inflamm. 2016, 2016, 9890141. [Google Scholar] [CrossRef]
- Hu, D.; Zhang, D.; Zheng, S.; Guo, M.; Lin, X.; Jiang, Y. Association of Ulcerative Colitis with FUT2 and FUT3 Polymorphisms in Patients from Southeast China. PLoS ONE 2016, 11, e0146557. [Google Scholar] [CrossRef]
- Kappler, K.; Lasanajak, Y.; Smith, D.F.; Opitz, L.; Hennet, T. Increased Antibody Response to Fucosylated Oligosaccharides and Fucose-Carrying Bacteroides Species in Crohn’s Disease. Front. Microbiol. 2020, 11, 1553. [Google Scholar] [CrossRef]
- Vermeulen, N.; Vermeire, S.; Arijs, I.; Michiels, G.; Ballet, V.; Derua, R.; Waelkens, E.; Van Lommel, L.; Schuit, F.; Rutgeerts, P.; et al. Seroreactivity against Glycolytic Enzymes in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2011, 17, 557–564. [Google Scholar] [CrossRef]
- Hryhorowicz, S.; Kaczmarek-Ryś, M.; Zielińska, A.; Scott, R.J.; Słomski, R.; Pławski, A. Endocannabinoid System as a Promising Therapeutic Target in Inflammatory Bowel Disease—A Systematic Review. Front. Immunol. 2021, 12, 790803. [Google Scholar] [CrossRef]
- Maki, J.J.; Nielsen, D.W.; Looft, T. Complete Genome Sequence and Annotation for Turicibacter Sanguinis MOL361T (DSM 14220). Microbiol Resour Announc 2020, 9, e00475-20. [Google Scholar] [CrossRef]
- Lim, S.; Rhee, M.-S.; Chang, D.-H.; Kim, B.-C. Whole-Genome Sequence of Erysipelothrix Larvae LV19T (=KCTC 33523T), a Useful Strain for Arsenic Detoxification, from the Larval Gut of the Rhinoceros Beetle, Trypoxylus Dichotomus. J. Biotechnol. 2016, 223, 40–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-M.; Bao, C.-H.; Wu, Y.; Liang, S.-H.; Wang, D.; Wu, L.-Y.; Huang, Y.; Liu, H.-R.; Wu, H.-G. Tryptophan-Kynurenine Metabolism: A Link between the Gut and Brain for Depression in Inflammatory Bowel Disease. J. Neuroinflamm. 2021, 18, 135. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, S.D.; Kayal, M.; Shah, S.C. Sex-Based Differences in Inflammatory Bowel Diseases: A Review. Therap. Adv. Gastroenterol. 2020, 13, 1756284820915043. [Google Scholar] [CrossRef]
- Sochal, M.; Ditmer, M.; Binienda, A.; Gabryelska, A.; Białasiewicz, P.; Talar-Wojnarowska, R.; Fichna, J.; Małecka-Wojciesko, E. Relation between Selected Sleep Parameters, Depression, Anti-Tumor Necrosis Factor Therapy, and the Brain-Derived Neurotrophic Factor Pathway in Inflammatory Bowel Disease. Metabolites 2023, 13, 450. [Google Scholar] [CrossRef]
- Jo, J.-K.; Seo, S.-H.; Park, S.-E.; Kim, H.-W.; Kim, E.-J.; Kim, J.-S.; Pyo, J.-Y.; Cho, K.-M.; Kwon, S.-J.; Park, D.-H.; et al. Gut Microbiome and Metabolome Profiles Associated with High-Fat Diet in Mice. Metabolites 2021, 11, 482. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calzadilla, N.; Qazi, A.; Sharma, A.; Mongan, K.; Comiskey, S.; Manne, J.; Youkhana, A.G.; Khanna, S.; Saksena, S.; Dudeja, P.K.; et al. Mucosal Metabolomic Signatures in Chronic Colitis: Novel Insights into the Pathophysiology of Inflammatory Bowel Disease. Metabolites 2023, 13, 873. https://doi.org/10.3390/metabo13070873
Calzadilla N, Qazi A, Sharma A, Mongan K, Comiskey S, Manne J, Youkhana AG, Khanna S, Saksena S, Dudeja PK, et al. Mucosal Metabolomic Signatures in Chronic Colitis: Novel Insights into the Pathophysiology of Inflammatory Bowel Disease. Metabolites. 2023; 13(7):873. https://doi.org/10.3390/metabo13070873
Chicago/Turabian StyleCalzadilla, Nathan, Aisha Qazi, Anchal Sharma, Kai Mongan, Shane Comiskey, Jahnavi Manne, Alvin G. Youkhana, Sonam Khanna, Seema Saksena, Pradeep K. Dudeja, and et al. 2023. "Mucosal Metabolomic Signatures in Chronic Colitis: Novel Insights into the Pathophysiology of Inflammatory Bowel Disease" Metabolites 13, no. 7: 873. https://doi.org/10.3390/metabo13070873
APA StyleCalzadilla, N., Qazi, A., Sharma, A., Mongan, K., Comiskey, S., Manne, J., Youkhana, A. G., Khanna, S., Saksena, S., Dudeja, P. K., Alrefai, W. A., & Gill, R. K. (2023). Mucosal Metabolomic Signatures in Chronic Colitis: Novel Insights into the Pathophysiology of Inflammatory Bowel Disease. Metabolites, 13(7), 873. https://doi.org/10.3390/metabo13070873