

Metabolic Inheritance and the Competition for Calories between Mother and Fetus

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Evolution of the Competition for Calories
2.1. The Mammalian ‘Ecosystem’
2.2. Organismal Behavior: Consumption and ‘Effective Caloric Intake’
2.3. Cell-Type-Specific Behavior
2.4. Skeletal Muscle-Cells
2.5. Fat-Cells
3. The Inheritance of Metabolic Phenotypes
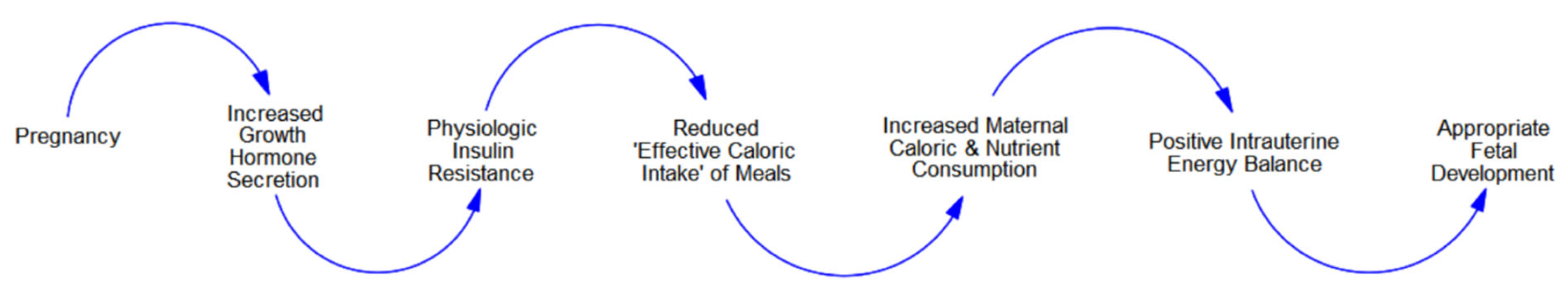
3.1. ‘Normal’ Pregnancy and the ‘Competition for Calories’
3.2. Pathologic Pregnancies
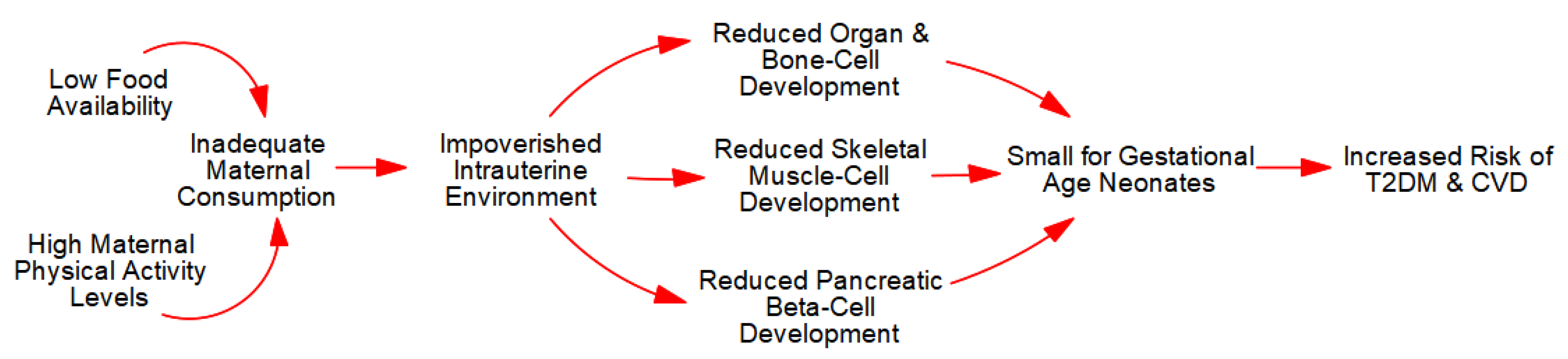
3.2.1. Intrauterine Growth Restriction
3.2.2. Atypical IUGR: ‘Big’ Does Not Always Beget ‘Big’
3.2.3. Maternal Fat-Cells Outcompete Fetal-Cells
3.2.4. Sitting-Induced IUGR
4. Inherited Obesity
4.1. Nongenetic versus Genetic Inheritance
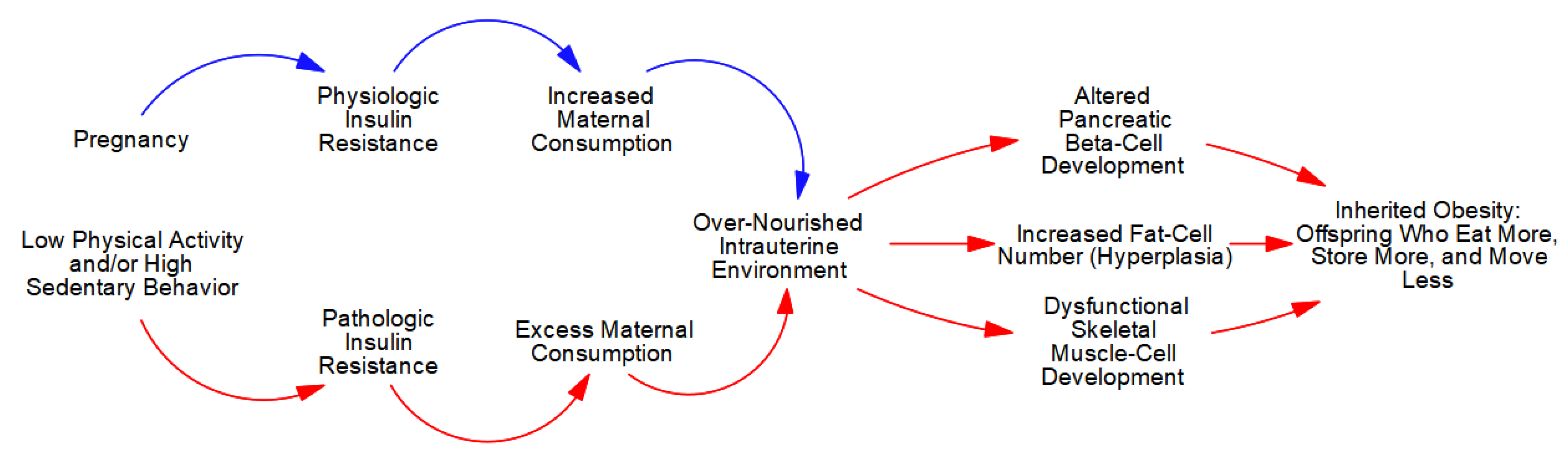
4.2. The Etiology of Inherited (Pediatric/Childhood) Obesity
4.3. ‘Grand-Maternal Effects’ and Trends in Obesity and T2DM
5. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Archer, E.; Pavela, G.; McDonald, S.; Lavie, C.J.; Hill, J.O. Cell-Specific “Competition for Calories” Drives Asymmetric Nutrient-Energy Partitioning, Obesity, and Metabolic Diseases in Human and Non-human Animals. Front. Physiol. 2018, 9, 1053. [Google Scholar] [CrossRef] [PubMed]
- Archer, E.; Hill, J.O. Body and fat mass are not regulated, controlled, or defended: An introduction to the ‘invisible Hand’ and ‘competition’ models of metabolism. Prog. Cardiovasc. Dis. 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Dobersek, U.; Stallings, B.; Wy, G.C.; Case, C.R.; Maner, J.K. Does Exercise Make Me More Attractive? Exploring the Relations Between Exercise and Mate Value. Evol. Psychol. Sci. 2021, 7, 124–133. [Google Scholar] [CrossRef]
- Darwin, C. The Descent of Man, and Selection in Relation to Sex; John Murray: London, UK, 1871. [Google Scholar]
- Darwin, C. On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life, 6th ed.; John Murray: London, UK, 1859. [Google Scholar]
- Archer, E. The Childhood Obesity Epidemic as a Result of Nongenetic Evolution: The Maternal Resources Hypothesis. Mayo Clin. Proc. 2015, 90, 77–92. [Google Scholar] [CrossRef]
- Maestripieri, D.; Mateo, J.M. (Eds.) Maternal Effects in Mammals; University of Chicago Press: Chicago, IL, USA, 2009. [Google Scholar]
- Bateson, P.; Barker, D.; Clutton-Brock, T.; Deb, D.; D’Udine, B.; Foley, R.A.; Gluckman, P.; Godfrey, K.; Kirkwood, T.; Lahr, M.M.; et al. Developmental plasticity and human health. Nature 2004, 430, 419–421. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Cooper, C.; Thornburg, K.L. Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 2008, 359, 61–73. [Google Scholar] [CrossRef]
- Archer, E.; Lavie, C.J.; Hill, J.O. The Contributions of ‘Diet’, ‘Genes’, and Physical Activity to the Etiology of Obesity: Contrary Evidence and Consilience. Prog. Cardiovasc. Dis. 2018, 61, 89–102. [Google Scholar] [CrossRef]
- Archer, E. The Failure of Gene-centrism. Behav. Brain Sci. 2023; Forthcoming. [Google Scholar]
- Sokolov, E.N. Neuronal models and the orienting reflex. In Proceedings of the Central Nervous System and Behavior, 3rd Conference, Princeton, NJ, USA, 22–25 February 1960; pp. 187–276. [Google Scholar]
- Archer, E.; Lavie, C.J. Obesity Subtyping: The Etiology, Prevention, and Management of Acquired versus Inherited Obese Phenotypes. Nutrients 2022, 14, 2286. [Google Scholar] [CrossRef]
- Sonagra, A.D.; Biradar, S.M.; Dattatreya, K.; Ds, J.M. Normal pregnancy-a state of insulin resistance. J. Clin. Diagn. Res. JCDR 2014, 8, CC01. [Google Scholar] [CrossRef]
- Barbour, L.A.; McCurdy, C.E.; Hernandez, T.L.; Kirwan, J.P.; Catalano, P.M.; Friedman, J.E. Cellular Mechanisms for Insulin Resistance in Normal Pregnancy and Gestational Diabetes. Diabetes Care 2007, 30, S112–S119. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, M.M.; Zeitler, P.S. Insulin Resistance of Puberty. Curr. Diab. Rep. 2016, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Amiel, S.A.; Sherwin, R.S.; Simonson, D.C.; Lauritano, A.A.; Tamborlane, W.V. Impaired Insulin Action in Puberty. N. Engl. J. Med. 1986, 315, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A.; Yakar, S.; Leroith, D. The intricate role of growth hormone in metabolism. Front. Endocrinol. 2011, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Archer, E.; Blair, S.N. Physical activity and the prevention of cardiovascular disease: From evolution to epidemiology. Prog. Cardiovasc. Dis. 2011, 53, 387–396. [Google Scholar] [CrossRef]
- Raichlen, D.A.; Polk, J.D. Linking brains and brawn: Exercise and the evolution of human neurobiology. Proc. R. Soc. B Biol. Sci. 2013, 280, 20122250. [Google Scholar] [CrossRef]
- Raichlen, D.A.; Pontzer, H.; Harris, J.A.; Mabulla, A.Z.P.; Marlowe, F.W.; Josh Snodgrass, J.; Eick, G.; Colette Berbesque, J.; Sancilio, A.; Wood, B.M. Physical activity patterns and biomarkers of cardiovascular disease risk in hunter-gatherers. Am. J. Hum. Biol. 2016, 29, 1–13. [Google Scholar] [CrossRef]
- Booth, F.W.; Roberts, C.K.; Laye, M.J. Lack of exercise is a major cause of chronic diseases. Compr. Physiol. 2012, 2, 1143–1211. [Google Scholar] [CrossRef]
- Pontzer, H.; Wood, B.M.; Raichlen, D.A. Hunter-gatherers as models in public health. Obes. Rev. 2018, 19, 24–35. [Google Scholar] [CrossRef]
- Archer, E.; Artero, E.G.; Blair, S.N. Sedentary Behavior and Cardiovascular Disease. In Sedentary Behavior and Health: Concepts, Assessment & Intervention–Human Kinetics; Zhu, W., Owen, N., Eds.; Human Kinetics: Champaign, IL, USA, 2017; pp. 203–225. [Google Scholar]
- Archer, E. Design and Validation of an Agent-Based Model of Human Energy Expenditure and Nutrient-Flux using the ‘Competition Model of Metabolism’. 2023; Forthcoming. [Google Scholar]
- Elia, M. Organ and tissue contribution to metabolic rate. In Energy Metabolism: Tissue Determinants and Cellular Corollaries; Kinney, J., Tucker, H., Eds.; Raven Press: New York, NY, USA, 1992; pp. 61–80. [Google Scholar]
- Bosy-Westphal, A.; Reinecke, U.; Schlorke, T.; Illner, K.; Kutzner, D.; Heller, M.; Muller, M.J. Effect of organ and tissue masses on resting energy expenditure in underweight, normal weight and obese adults. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Muller, M.J. Specific metabolic rates of major organs and tissues across adulthood: Evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 2010, 92, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.J.; Langemann, D.; Gehrke, I.; Later, W.; Heller, M.; Glüer, C.C.; Heymsfield, S.B.; Bosy-Westphal, A. Effect of constitution on mass of individual organs and their association with metabolic rate in humans—A detailed view on allometric scaling. PLoS ONE 2011, 6, e22732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Heller, M.; Later, W.; Heymsfield, S.B.; Muller, M.J. Evaluation of specific metabolic rates of major organs and tissues: Comparison between men and women. Am. J. Hum. Biol. 2011, 23, 333–338. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; Thomas, D.; Bosy-Westphal, A.; Shen, W.; Peterson, C.M.; Muller, M.J. Evolving concepts on adjusting human resting energy expenditure measurements for body size. Obes. Rev. 2012, 13, 1001–1014. [Google Scholar] [CrossRef]
- Bergouignan, A.; Kealey, E.H.; Schmidt, S.L.; Jackman, M.R.; Bessesen, D.H. Twenty-Four Hour Total and Dietary Fat Oxidation in Lean, Obese and Reduced-Obese Adults with and without a Bout of Exercise. PLoS ONE 2014, 9, e94181. [Google Scholar] [CrossRef]
- Bergouignan, A.; Trudel, G.; Simon, C.; Chopard, A.; Schoeller, D.A.; Momken, I.; Votruba, S.B.; Desage, M.; Burdge, G.C.; Gauquelin-Koch, G.; et al. Physical Inactivity Differentially Alters Dietary Oleate and Palmitate Trafficking. Diabetes 2009, 58, 367–376. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Soeters, M.R.; Soeters, P.B.; Schooneman, M.G.; Houten, S.M.; Romijn, J.A. Adaptive reciprocity of lipid and glucose metabolism in human short-term starvation. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1397–E1407. [Google Scholar] [CrossRef]
- Bergouignan, A.; Momken, I.; Lefai, E.; Antoun, E.; Schoeller, D.A.; Platat, C.; Chery, I.; Zahariev, A.; Vidal, H.; Gabert, L.; et al. Activity energy expenditure is a major determinant of dietary fat oxidation and trafficking, but the deleterious effect of detraining is more marked than the beneficial effect of training at current recommendations. Am. J. Clin. Nutr. 2013, 98, 648–658. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Katsiaras, A.; Kelley, D.E. Enhanced Fat Oxidation Through Physical Activity Is Associated With Improvements in Insulin Sensitivity in Obesity. Diabetes 2003, 52, 2191–2197. [Google Scholar] [CrossRef]
- Jackman, M.R.; Steig, A.; Higgins, J.A.; Johnson, G.C.; Fleming-Elder, B.K.; Bessesen, D.H.; MacLean, P.S. Weight regain after sustained weight reduction is accompanied by suppressed oxidation of dietary fat and adipocyte hyperplasia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1117–R1129. [Google Scholar] [CrossRef] [PubMed]
- Häger, A.; Sjörström, L.; Arvidsson, B.; Björntorp, P.; Smith, U. Adipose tissue cellularity in obese school girls before and after dietary treatment. Am. J. Clin. Nutr. 1978, 31, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Hager, A. Adipose tissue cellularity in childhood in relation to the development of obesity. Br. Med. Bull. 1981, 37, 287–290. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Lilly lecture 1987. The triumvirate: Beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988, 37, 667–687. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef]
- Barker, D.J. The fetal origins of hypertension. J. Hypertens. Suppl. Off. J. Int. Soc. Hypertens. 1996, 14, S117–S120. [Google Scholar] [CrossRef]
- Barker, D.J.; Lampl, M.; Roseboom, T.; Winder, N. Resource allocation in utero and health in later life. Placenta 2012, 33 (Suppl. S2), e30–e34. [Google Scholar] [CrossRef]
- Barker, D.J.; Martyn, C.N.; Osmond, C.; Hales, C.N.; Fall, C.H. Growth in utero and serum cholesterol concentrations in adult life. BMJ 1993, 307, 1524–1527. [Google Scholar] [CrossRef]
- Barker, D.J.P. The malnourished baby and infant: Relationship with Type 2 diabetes. Br. Med. Bull. 2001, 60, 69–88. [Google Scholar] [CrossRef]
- Archer, E.; McDonald, S.M. The Maternal Resources Hypothesis and Childhood Obesity. In Fetal and Early Postnatal Programming and Its Influence on Adult Health; Patel, M.S., Nielsen, J.S., Eds.; CRC Press; Taylor and Francis Group: New York, NY, USA, 2017; pp. 17–32. [Google Scholar]
- Archer, E. In reply-Maternal, paternal, and societal efforts are needed to “cure” childhood obesity. Mayo Clin. Proc. 2015, 90, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Archer, E. In reply—Epigenetics and childhood obesity. Mayo Clin. Proc. 2015, 90, 693–695. [Google Scholar] [CrossRef] [PubMed]
- Archer, E. The Mother of All Problems. New Sci. 2015, 225, 32–33. [Google Scholar] [CrossRef]
- McDonald, S.M.; Liu, J.; Wilcox, S.; Lau, E.Y.; Archer, E. Does dose matter in reducing gestational weight gain in exercise interventions? A systematic review of literature. J. Sci. Med. Sport 2016, 19, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Farahnik, B.; Park, K.; Kroumpouzos, G.; Murase, J. Striae gravidarum: Risk factors, prevention, and management. Int. J. Womens Dermatol. 2017, 3, 77–85. [Google Scholar] [CrossRef]
- Wang, J.X.; Davies, M.J.; Norman, R.J. Obesity increases the risk of spontaneous abortion during infertility treatment. Obes. Res. 2002, 10, 551–554. [Google Scholar] [CrossRef]
- Hahn, K.A.; Hatch, E.E.; Rothman, K.J.; Mikkelsen, E.M.; Brogly, S.B.; Sørensen, H.T.; Riis, A.H.; Wise, L.A. Body size and risk of spontaneous abortion among danish pregnancy planners. Paediatr. Perinat. Epidemiol. 2014, 28, 412–423. [Google Scholar] [CrossRef]
- Bellver, J.; Pellicer, A. Impact of obesity on spontaneous abortion. Am. J. Obstet. Gynecol. 2004, 190, 293–294. [Google Scholar] [CrossRef]
- Tian, L.; Shen, H.; Lu, Q.; Norman, R.J.; Wang, J. Insulin resistance increases the risk of spontaneous abortion after assisted reproduction technology treatment. J. Clin. Endocrinol. Metab. 2007, 92, 1430–1433. [Google Scholar] [CrossRef]
- Boots, C.; Stephenson, M.D. Does obesity increase the risk of miscarriage in spontaneous conception: A systematic review. In Proceedings of the Seminars in Reproductive Medicine; Thieme Medical Publishers: New York, NY, USA, 2011; pp. 507–513. [Google Scholar]
- Stothard, K.J.; Tennant, P.W.; Bell, R.; Rankin, J. Maternal overweight and obesity and the risk of congenital anomalies: A systematic review and meta-analysis. JAMA 2009, 301, 636–650. [Google Scholar] [CrossRef]
- Davies, G.A.; Maxwell, C.; McLeod, L.; Gagnon, R.; Basso, M.; Bos, H.; Delisle, M.-F.; Farine, D.; Hudon, L.; Menticoglou, S. Obesity in pregnancy. J. Obstet. Gynaecol. Can. 2010, 32, 165–173. [Google Scholar] [CrossRef]
- Rankin, J.; Tennant, P.; Stothard, K.; Bythell, M.; Summerbell, C.; Bell, R. Maternal body mass index and congenital anomaly risk: A cohort study. Int. J. Obes. 2010, 34, 1371–1380. [Google Scholar] [CrossRef]
- Chen, W.; Li, B.; Gan, K.; Liu, J.; Yang, Y.; Lv, X.; Ma, H. Gestational Weight Gain and Small for Gestational Age in Obese Women: A Systematic Review and Meta-Analysis. Int. J. Endocrinol. 2023, 2023, 3048171. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, R.; Chi, J.; Li, Y.; Bai, W. Pre-pregnancy body mass index has greater influence on newborn weight and perinatal outcome than weight control during pregnancy in obese women. Arch. Public Health 2023, 81, 5. [Google Scholar] [CrossRef]
- Vuguin, P.M. Animal models for small for gestational age and fetal programing of adult disease. Horm. Res. Paediatr. 2007, 68, 113–123. [Google Scholar] [CrossRef]
- Cobb, W.S.; Burns, J.M.; Kercher, K.W.; Matthews, B.D.; James Norton, H.; Todd Heniford, B. Normal intraabdominal pressure in healthy adults. J. Surg. Res. 2005, 129, 231–235. [Google Scholar] [CrossRef]
- Sugerman, H.; Windsor, A.; Bessos, M.; Wolfe, L. Intra-abdominal pressure, sagittal abdominal diameter and obesity comorbidity. J. Intern. Med. 1997, 241, 71–79. [Google Scholar] [CrossRef]
- Ueland, K.; Novy, M.J.; Metcalfe, J. Hemodynamic responses of patients with heart disease to pregnancy and exercise. Am. J. Obstet. Gynecol. 1972, 113, 47–59. [Google Scholar] [CrossRef]
- Fazzi, C.; Saunders, D.H.; Linton, K.; Norman, J.E.; Reynolds, R.M. Sedentary behaviours during pregnancy: A systematic review. Int. J. Behav. Nutr. Phys. Act. 2017, 14, 32. [Google Scholar] [CrossRef]
- Credeur, D.P.; Miller, S.M.; Jones, R.; Stoner, L.; Dolbow, D.R.; Fryer, S.M.; Stone, K.; McCoy, S.M. Impact of Prolonged Sitting on Peripheral and Central Vascular Health. Am. J. Cardiol. 2019, 123, 260–266. [Google Scholar] [CrossRef]
- Taylor, F.C.; Dunstan, D.W.; Homer, A.R.; Dempsey, P.C.; Kingwell, B.A.; Climie, R.E.; Owen, N.; Cohen, N.D.; Larsen, R.N.; Grace, M.; et al. Acute effects of interrupting prolonged sitting on vascular function in type 2 diabetes. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H393–H403. [Google Scholar] [CrossRef] [PubMed]
- Benabid, A.; Deslauriers, L.; Sinclair, I.; St-Pierre, M.; Vaillancourt, C.; Gagnon, S.; Dancause, K.N. Relationships between objectively measured sedentary behavior during pregnancy and infant birthweight. Int. J. Environ. Res. Public Health 2021, 18, 10000. [Google Scholar] [CrossRef] [PubMed]
- Archer, E.; Lavie, C.J.; McDonald, S.M.; Thomas, D.M.; Hebert, J.R.; Taverno Ross, S.E.; McIver, K.L.; Malina, R.M.; Blair, S.N. Maternal inactivity: 45-year trends in mothers’ use of time. Mayo Clin. Proc. 2013, 88, 1368–1377. [Google Scholar] [CrossRef]
- Archer, E.; Shook, R.P.; Thomas, D.M.; Church, T.S.; Katzmarzyk, P.T.; Hebert, J.R.; McIver, K.L.; Hand, G.A.; Lavie, C.J.; Blair, S.N. 45-Year trends in women’s use of time and household management energy expenditure. PLoS ONE 2013, 8, e56620. [Google Scholar] [CrossRef]
- Bonduriansky, R.; Day, T. Nongenetic Inheritance and Its Evolutionary Implications. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 103–125. [Google Scholar] [CrossRef]
- Day, T.; Bonduriansky, R. A unified approach to the evolutionary consequences of genetic and nongenetic inheritance. Am. Nat. 2011, 178, E18–E36. [Google Scholar] [CrossRef]
- Bonduriansky, R. Rethinking heredity, again. Trends Ecol. Evol. 2012, 27, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Bonduriansky, R.; Day, T. Extended Heredity: A New Understanding of Inheritance and Evolution; Princeton University Press: Princeton, NJ, USA, 2018. [Google Scholar]
- Santos, T.A.; El Shourbagy, S.; St John, J.C. Mitochondrial content reflects oocyte variability and fertilization outcome. Fertil. Steril. 2006, 85, 584–591. [Google Scholar] [CrossRef]
- Wang, Q.; Moley, K.H. Maternal diabetes and oocyte quality. Mitochondrion 2010, 10, 403–410. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Liu, X.; Chen, L.; Zhang, S.; Zhang, X.; Hao, C.; Miao, Y.-L. Advanced maternal age alters expression of maternal effect genes that are essential for human oocyte quality. Aging 2020, 12, 3950–3961. [Google Scholar] [CrossRef]
- Zhou, X.; McQueen, D.B.; Schufreider, A.; Lee, S.M.; Uhler, M.L.; Feinberg, E.C. Black recipients of oocyte donation experience lower live birth rates compared with White recipients. Reprod. BioMed. Online 2020, 40, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, D.; Lee, I.-W.; Yuen, W.S.; Carroll, J. Oocyte mitochondria—Key regulators of oocyte function and potential therapeutic targets for improving fertility. Biol. Reprod. 2022, 106, 366–377. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The significance of responses of the genome to challenge. Science 1984, 226, 792–801. [Google Scholar] [CrossRef]
- Church, T.S.; Thomas, D.M.; Tudor-Locke, C.; Katzmarzyk, P.T.; Earnest, C.P.; Rodarte, R.Q.; Martin, C.K.; Blair, S.N.; Bouchard, C. Trends over 5 decades in U.S. occupation-related physical activity and their associations with obesity. PLoS ONE 2011, 6, e19657. [Google Scholar] [CrossRef]
- McDonald, N.C. Active transportation to school: Trends among U.S. schoolchildren, 1969–2001. Am. J. Prev. Med. 2007, 32, 509–516. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Archer, E.; Lavie, C.J.; Dobersek, U.; Hill, J.O. Metabolic Inheritance and the Competition for Calories between Mother and Fetus. Metabolites 2023, 13, 545. https://doi.org/10.3390/metabo13040545
Archer E, Lavie CJ, Dobersek U, Hill JO. Metabolic Inheritance and the Competition for Calories between Mother and Fetus. Metabolites. 2023; 13(4):545. https://doi.org/10.3390/metabo13040545
Chicago/Turabian StyleArcher, Edward, Carl J. Lavie, Urska Dobersek, and James O. Hill. 2023. "Metabolic Inheritance and the Competition for Calories between Mother and Fetus" Metabolites 13, no. 4: 545. https://doi.org/10.3390/metabo13040545
APA StyleArcher, E., Lavie, C. J., Dobersek, U., & Hill, J. O. (2023). Metabolic Inheritance and the Competition for Calories between Mother and Fetus. Metabolites, 13(4), 545. https://doi.org/10.3390/metabo13040545