
A Strategy for Uncovering the Serum Metabolome by Direct-Infusion High-Resolution Mass Spectrometry



Abstract
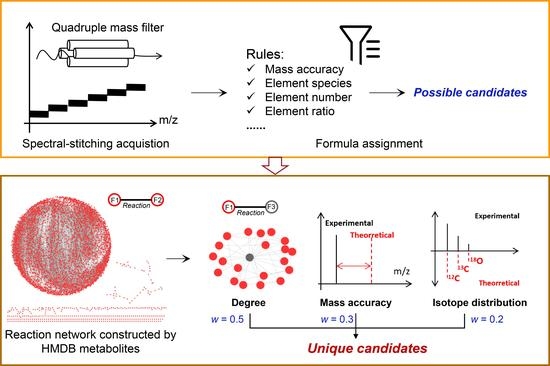
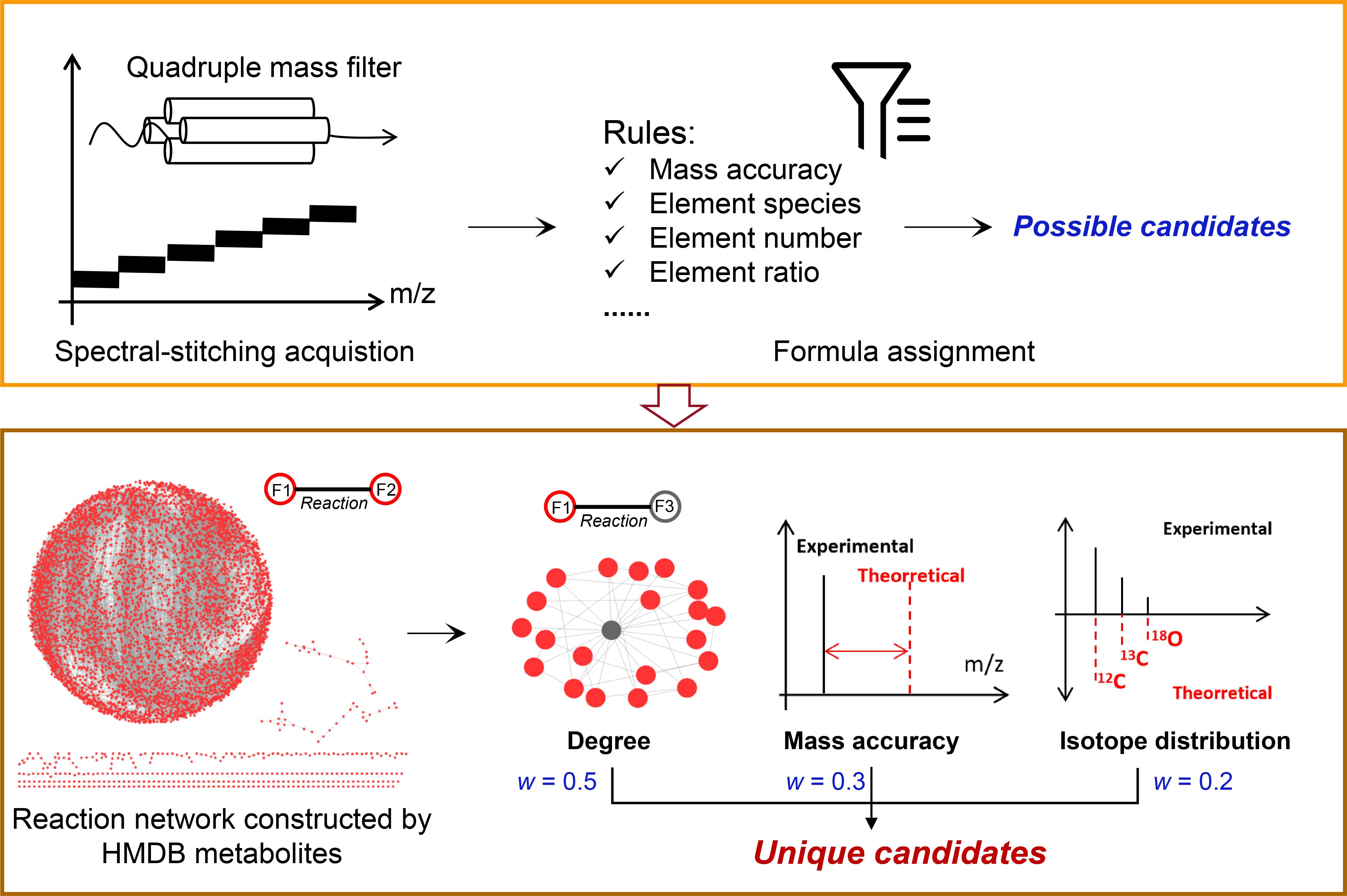{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Sample Information and Preparation
2.3. DI-nESI HRMS Analysis
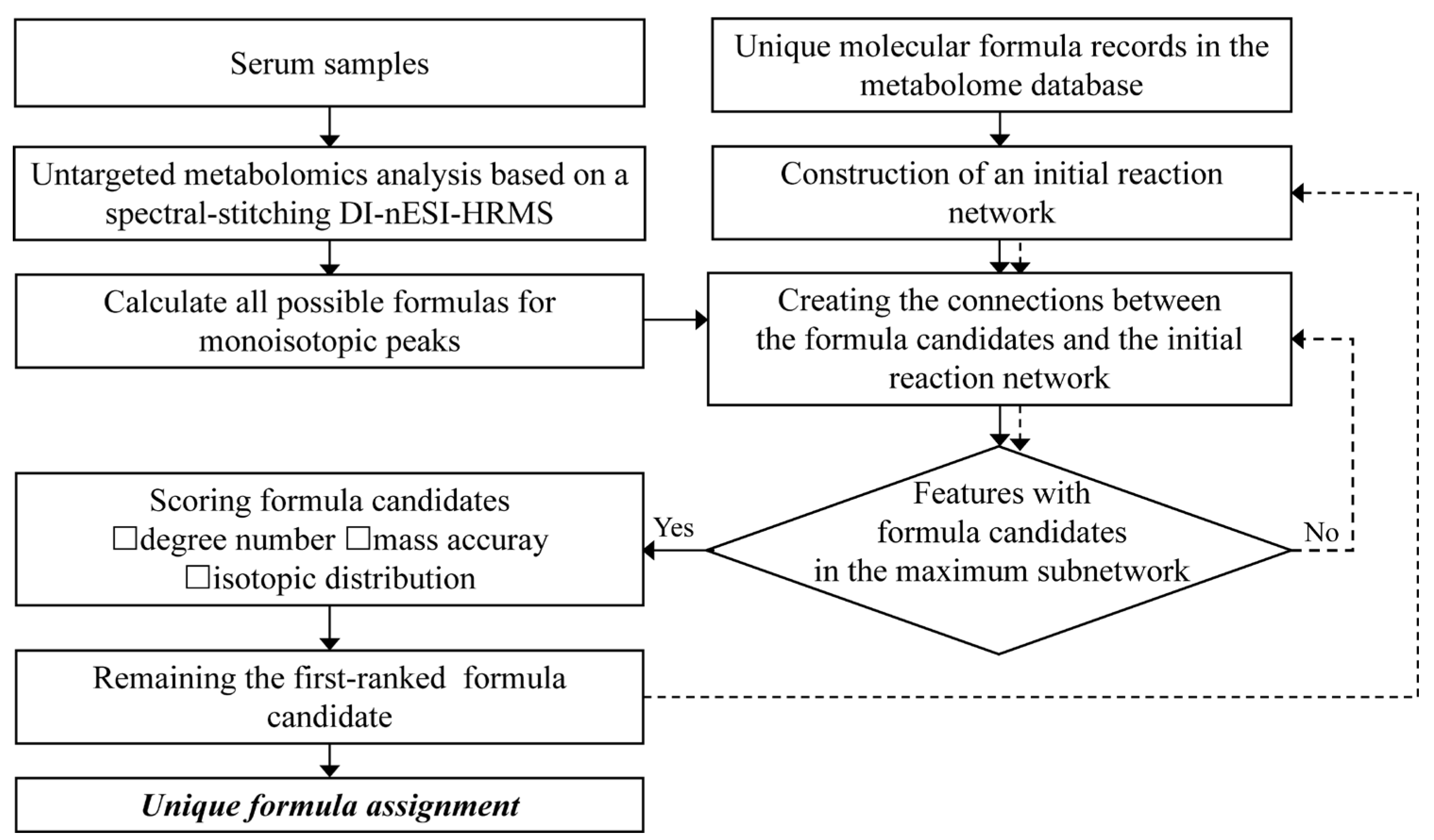
2.4. Data Processing
3. Results
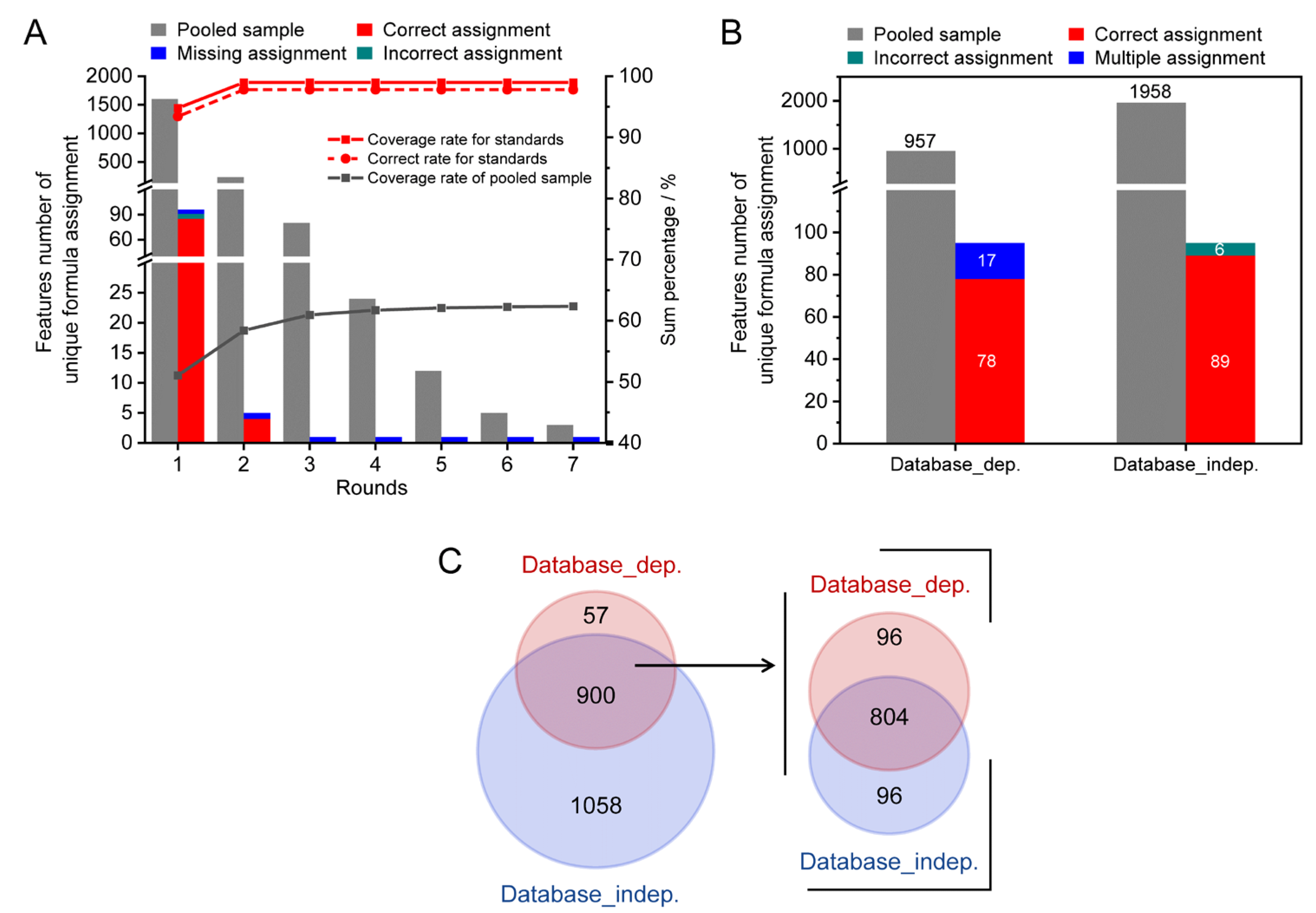
3.1. Workflow of the Developed Method
3.2. The Method Establishment
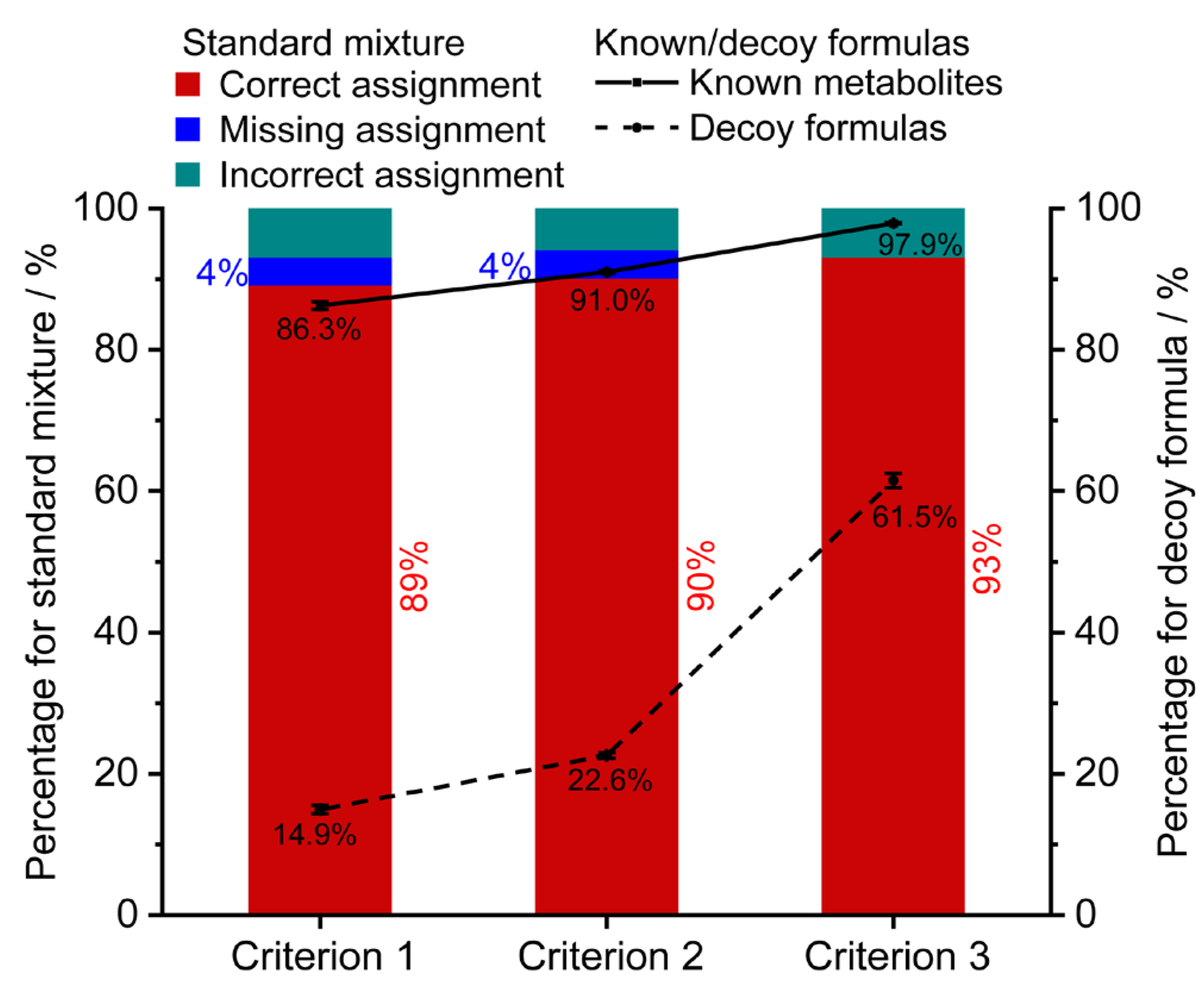
3.3. Method Validation
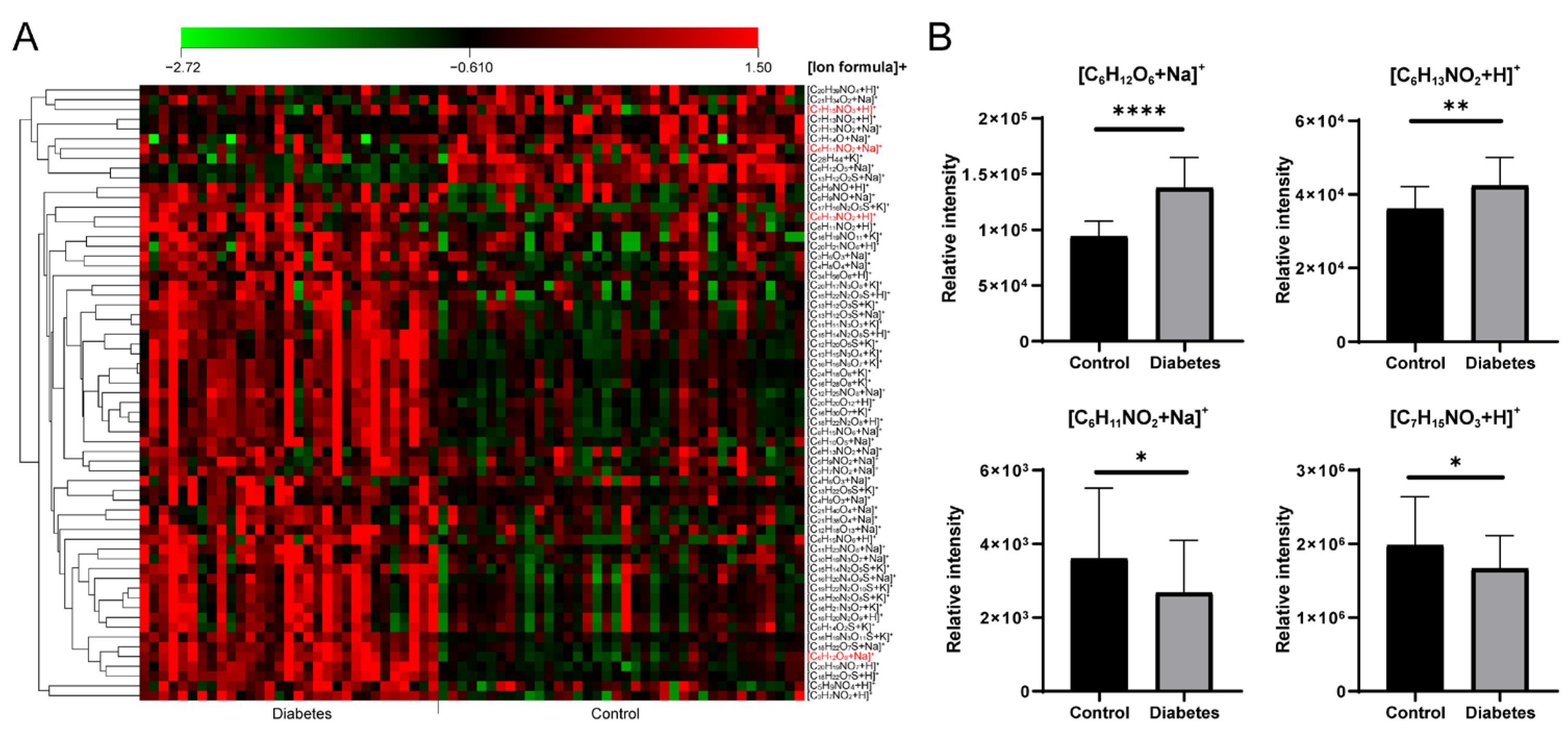
3.4. Application for Serum Metabolomic Analysis in Diabetes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Southam, A.D.; Weber, R.J.M.; Engel, J.; Jones, M.R.; Viant, M.R. A complete workflow for high-resolution spectral-stitching nanoelectrospray direct-infusion mass-spectrometry-based metabolomics and lipidomics. Nat. Protoc. 2016, 12, 310–328. [Google Scholar] [CrossRef] [PubMed]
- Habchi, B.; Alves, S.; Jouan-Rimbaud Bouveresse, D.; Moslah, B.; Paris, A.; Lécluse, Y.; Gauduchon, P.; Lebailly, P.; Rutledge, D.N.; Rathahao-Paris, E. An innovative chemometric method for processing direct introduction high resolution mass spectrometry metabolomic data: Independent component–discriminant analysis (IC–DA). Metabolomics 2017, 13, 45. [Google Scholar] [CrossRef]
- Chekmeneva, E.; Dos Santos Correia, G.; Gómez-Romero, M.; Stamler, J.; Chan, Q.; Elliott, P.; Nicholson, J.K.; Holmes, E. Ultra-Performance Liquid Chromatography–High-Resolution Mass Spectrometry and Direct Infusion–High-Resolution Mass Spectrometry for Combined Exploratory and Targeted Metabolic Profiling of Human Urine. J. Proteome Res. 2018, 17, 3492–3502. [Google Scholar] [CrossRef]
- Pöhö, P.; Lipponen, K.; Bespalov, M.M.; Sikanen, T.; Kotiaho, T.; Kostiainen, R. Comparison of liquid chromatography-mass spectrometry and direct infusion microchip electrospray ionization mass spectrometry in global metabolomics of cell samples. Eur. J. Pharm. Sci. 2019, 138, 104991. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, J.M.; Palosaari, T.; Sund, J.; Carpi, D.; Bouhifd, M.; Weber, R.J.M.; Whelan, M.; Viant, M.R. Integrating in vitro metabolomics with a 96-well high-throughput screening platform. Metabolomics 2022, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.J.; Hall, A.R.; Lloyd, G.R.; Weber, R.J.M.; Wilson, A.; Pointon, A.; Viant, M.R. An Extensive Metabolomics Workflow to Discover Cardiotoxin-Induced Molecular Perturbations in Microtissues. Metabolites 2021, 11, 644. [Google Scholar] [CrossRef]
- González-Domínguez, R.; Sayago, A.; Fernández-Recamales, Á. Direct infusion mass spectrometry for metabolomic phenotyping of diseases. Bioanalysis 2017, 9, 131–148. [Google Scholar] [CrossRef]
- Chekmeneva, E.; Dos Santos Correia, G.; Chan, Q.; Wijeyesekera, A.; Tin, A.; Young, J.H.; Elliott, P.; Nicholson, J.K.; Holmes, E. Optimization and Application of Direct Infusion Nanoelectrospray HRMS Method for Large-Scale Urinary Metabolic Phenotyping in Molecular Epidemiology. J. Proteome Res. 2017, 16, 1646–1658. [Google Scholar] [CrossRef]
- de Sain-van der Velden, M.G.M.; van der Ham, M.; Gerrits, J.; Prinsen, H.C.M.T.; Willemsen, M.; Pras-Raves, M.L.; Jans, J.J.; Verhoeven-Duif, N.M. Quantification of metabolites in dried blood spots by direct infusion high resolution mass spectrometry. Anal. Chim. Acta 2017, 979, 45–50. [Google Scholar] [CrossRef]
- Haijes, H.A.; van der Ham, M.; Gerrits, J.; van Hasselt, P.M.; Prinsen, H.C.M.T.; de Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.M. Direct-infusion based metabolomics unveils biochemical profiles of inborn errors of metabolism in cerebrospinal fluid. Mol. Genet. Metab. 2019, 127, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; Willemsen, M.; Van der Ham, M.; Gerrits, J.; Pras-Raves, M.L.; Prinsen, H.C.M.T.; Van Hasselt, P.M.; De Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.M. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lv, W.; Sun, X.; Zheng, F.; Xu, T.; Liu, X.; Li, H.; Lu, X.; Peng, X.; Hu, C.; et al. Strategy for Nontargeted Metabolomic Annotation and Quantitation Using a High-Resolution Spectral-Stitching Nanoelectrospray Direct-Infusion Mass Spectrometry with Data-Independent Acquisition. Anal. Chem. 2021, 93, 10528–10537. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.; Nothias, L.-F.; Dührkop, K.; Koester, I.; Fleischauer, M.; Hoffmann, M.A.; Petras, D.; Vargas, F.; Morsy, M.; Aluwihare, L.; et al. Database-independent molecular formula annotation using Gibbs sampling through ZODIAC. Nat. Mach. Intell. 2020, 2, 629–641. [Google Scholar] [CrossRef]
- Wolthuis, J.C.; Magnusdottir, S.; Pras-Raves, M.; Moshiri, M.; Jans, J.J.M.; Burgering, B.; van Mil, S.; de Ridder, J. MetaboShiny: Interactive analysis and metabolite annotation of mass spectrometry-based metabolomics data. Metabolomics 2020, 16, 99. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Flight, R.M.; Moseley, H.N.B. Small Molecule Isotope Resolved Formula Enumeration: A Methodology for Assigning Isotopologues and Metabolite Formulas in Fourier Transform Mass Spectra. Anal. Chem. 2019, 91, 8933–8940. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; Willemse, E.A.; Gerrits, J.; van der Flier, W.M.; Teunissen, C.E.; Verhoeven-Duif, N.M.; Jans, J.J.M. Assessing the Pre-Analytical Stability of Small-Molecule Metabolites in Cerebrospinal Fluid Using Direct-Infusion Metabolomics. Metabolites 2019, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Sarvin, B.; Lagziel, S.; Sarvin, N.; Mukha, D.; Kumar, P.; Aizenshtein, E.; Shlomi, T. Fast and sensitive flow-injection mass spectrometry metabolomics by analyzing sample-specific ion distributions. Nat. Commun. 2020, 11, 3186. [Google Scholar] [CrossRef]
- Southam, A.D.; Payne, T.G.; Cooper, H.J.; Arvanitis, T.N.; Viant, M.R. Dynamic Range and Mass Accuracy of Wide-Scan Direct Infusion Nanoelectrospray Fourier Transform Ion Cyclotron Resonance Mass Spectrometry-Based Metabolomics Increased by the Spectral Stitching Method. Anal. Chem. 2007, 79, 4595–4602. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Bouza, M.; Gaul, D.A.; Leach, F.E.; Amster, I.J.; Schroeder, F.C.; Edison, A.S.; Fernández, F.M. Comparison of High-Resolution Fourier Transform Mass Spectrometry Platforms for Putative Metabolite Annotation. Anal. Chem. 2021, 93, 12374–12382. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, A.T.; Kourtchev, I.; Bortolini, C.; Fuller, S.J.; Giorio, C.; Popoola, O.A.M.; Bogialli, S.; Tapparo, A.; Jones, R.L.; Kalberer, M. A new processing scheme for ultra-high resolution direct infusion mass spectrometry data. Atmospheric Environ. 2018, 178, 129–139. [Google Scholar] [CrossRef]
- Cao, D.; Hao, Z.; Hu, M.; Geng, F.; Rao, Z.; Niu, H.; Shi, Y.; Cai, Y.; Zhou, Y.; Liu, J.; et al. A feasible strategy to improve confident elemental composition determination of compounds in complex organic mixture such as natural organic matter by FTICR-MS without internal calibration. Sci. Total. Environ. 2021, 751, 142255. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinform. 2006, 7, 234. [Google Scholar] [CrossRef] [PubMed]
- Alden, N.; Krishnan, S.; Porokhin, V.; Raju, R.; McElearney, K.; Gilbert, A.; Lee, K. Biologically Consistent Annotation of Metabolomics Data. Anal. Chem. 2017, 89, 13097–13104. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J. Systems biology of metabolism. Annu. Rev. Biochem. 2017, 86, 245–275. [Google Scholar] [CrossRef]
- Moritz, F.; Kaling, M.; Schnitzler, J.-P.; Schmitt-Kopplin, P. Characterization of poplar metabotypes via mass difference enrichment analysis. Plant, Cell Environ. 2017, 40, 1057–1073. [Google Scholar] [CrossRef]
- Tziotis, D.; Hertkorn, N.; Schmitt-Kopplin, P. Kendrick-Analogous Network Visualisation of Ion Cyclotron Resonance Fourier Transform Mass Spectra: Improved Options for the Assignment of Elemental Compositions and the Classification of Organic Molecular Complexity. Eur. J. Mass Spectrom. 2011, 17, 415–421. [Google Scholar] [CrossRef]
- Witting, M.; Lucio, M.; Tziotis, D.; Wägele, B.; Suhre, K.; Voulhoux, R.; Garvis, S.; Schmitt-Kopplin, P. DI-ICR-FT-MS-based high-throughput deep metabotyping: A case study of the Caenorhabditis elegans–Pseudomonas aeruginosa infection model. Anal. Bioanal. Chem. 2015, 407, 1059–1073. [Google Scholar] [CrossRef]
- Amara, A.; Frainay, C.; Jourdan, F.; Naake, T.; Neumann, S.; Novoa-Del-Toro, E.M.; Salek, R.M.; Salzer, L.; Scharfenberg, S.; Witting, M. Networks and Graphs Discovery in Metabolomics Data Analysis and Interpretation. Front. Mol. Biosci. 2022, 9, 841373. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Kopplin, P.; Hemmler, D.; Moritz, F.; Gougeon, R.D.; Lucio, M.; Meringer, M.; Müller, C.; Harir, M.; Hertkorn, N. Systems chemical analytics: Introduction to the challenges of chemical complexity analysis. Faraday Discuss. 2019, 218, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Traquete, F.; Luz, J.; Cordeiro, C.; Silva, M.S.; Ferreira, A.E.N. Graph Properties of Mass-Difference Networks for Profiling and Discrimination in Untargeted Metabolomics. Front. Mol. Biosci. 2022, 9, 917911. [Google Scholar] [CrossRef]
- Chen, L.; Lu, W.; Wang, L.; Xing, X.; Chen, Z.; Teng, X.; Zeng, X.; Muscarella, A.D.; Shen, Y.; Cowan, A.; et al. Metabolite discovery through global annotation of untargeted metabolomics data. Nat. Methods 2021, 18, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Forcisi, S.; Moritz, F.; Lucio, M.; Lehmann, R.; Stefan, N.; Schmitt-Kopplin, P. Solutions for Low and High Accuracy Mass Spectrometric Data Matching: A Data-Driven Annotation Strategy in Nontargeted Metabolomics. Anal. Chem. 2015, 87, 8917–8924. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, R.; Xiong, X.; Yin, Y.; Cai, Y.; Ma, Z.; Liu, N.; Zhu, Z.-J. Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat. Commun. 2019, 10, 1516. [Google Scholar] [CrossRef]
- Sun, L.; Liang, L.; Gao, X.; Zhang, H.; Yao, P.; Hu, Y.; Ma, Y.; Wang, F.; Jin, Q.; Li, H.; et al. Early Prediction of Developing Type 2 Diabetes by Plasma Acyl-carnitines: A Population-Based Study. Diabetes Care 2016, 39, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Guasch-Ferré, M.; Ruiz-Canela, M.; Li, J.; Zheng, Y.; Bullo, M.; Wang, D.D.; Toledo, E.; Clish, C.; Corella, D.; Estruch, R.; et al. Plasma Acylcarnitines and Risk of Type 2 Diabetes in a Mediterranean Population at High Cardiovascular Risk. J. Clin. Endocrinol. Metab. 2019, 104, 1508–1519. [Google Scholar] [CrossRef]
- Guasch-Ferré, M.; Hruby, A.; Toledo, E.; Clish, C.B.; Martínez-González, M.A.; Salas-Salvadó, J.; Hu, F.B. Metabolomics in Prediabetes and Diabetes: A Systematic Review and Meta-analysis. Diabetes Care 2016, 39, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Vanweert, F.; Schrauwen, P.; Phielix, E. Role of branched-chain amino acid metabolism in the pathogenesis of obesity and type 2 diabetes-related metabolic disturbances BCAA metabolism in type 2 diabetes. Nutr. Diabetes 2022, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Qiu, G.; Zhao, X.; Su, B.; Feng, D.; Lv, W.; Xuan, Q.; Wang, L.; Yu, D.; Wang, Q.; et al. Metabolome-Genome-Wide Association Study (mGWAS) Reveals Novel Metabolites Associated with Future Type 2 Diabetes Risk and Susceptibility Loci in a Case-Control Study in a Chinese Prospective Cohort. Glob. Chall. 2021, 5, 2000088. [Google Scholar] [CrossRef]
- Fuhrer, T.; Zamboni, N. High-throughput discovery metabolomics. Curr. Opin. Biotechnol. 2015, 31, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Kempa, E.E.; Hollywood, K.A.; Smith, C.A.; Barran, P.E. High throughput screening of complex biological samples with mass spectrometry–from bulk measurements to single cell analysis. Analyst 2019, 144, 872–891. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, A.; Shkrigunov, T.; Gusev, S.; Guseva, M.; Ponomarenko, E.; Lisitsa, A. An Open-Source Pipeline for Processing Direct Infusion Mass Spectrometry Data of the Human Plasma Metabolome. Metabolites 2022, 12, 768. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, E.M.; Huan, T.; Rinehart, D.; Benton, H.P.; Warth, B.; Hilmers, B.; Siuzdak, G. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nat. Protoc. 2018, 13, 633–651. [Google Scholar] [CrossRef]
- Thompson, C.J.; Witt, M.; Forcisi, S.; Moritz, F.; Kessler, N.; Laukien, F.H.; Schmitt-Kopplin, P. An Enhanced Isotopic Fine Structure Method for Exact Mass Analysis in Discovery Metabolomics: FIA-CASI-FTMS. J. Am. Soc. Mass Spectrom. 2020, 31, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef]
- Baygi, S.F.; Banerjee, S.K.; Chakraborty, P.; Kumar, Y.; Barupal, D.K. IDSL.UFA Assigns High-Confidence Molecular Formula Annotations for Untargeted LC/HRMS Data Sets in Metabolomics and Exposomics. Anal. Chem. 2022, 94, 13315–13322. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Jia, Z.; Zhang, Y.; Zhao, X.; Zhao, C.; Lu, X.; Xu, G. A Strategy for Uncovering the Serum Metabolome by Direct-Infusion High-Resolution Mass Spectrometry. Metabolites 2023, 13, 460. https://doi.org/10.3390/metabo13030460
Sun X, Jia Z, Zhang Y, Zhao X, Zhao C, Lu X, Xu G. A Strategy for Uncovering the Serum Metabolome by Direct-Infusion High-Resolution Mass Spectrometry. Metabolites. 2023; 13(3):460. https://doi.org/10.3390/metabo13030460
Chicago/Turabian StyleSun, Xiaoshan, Zhen Jia, Yuqing Zhang, Xinjie Zhao, Chunxia Zhao, Xin Lu, and Guowang Xu. 2023. "A Strategy for Uncovering the Serum Metabolome by Direct-Infusion High-Resolution Mass Spectrometry" Metabolites 13, no. 3: 460. https://doi.org/10.3390/metabo13030460
APA StyleSun, X., Jia, Z., Zhang, Y., Zhao, X., Zhao, C., Lu, X., & Xu, G. (2023). A Strategy for Uncovering the Serum Metabolome by Direct-Infusion High-Resolution Mass Spectrometry. Metabolites, 13(3), 460. https://doi.org/10.3390/metabo13030460