



Muscle Pathology in Dystrophic Rats and Zebrafish Is Unresponsive to Taurine Treatment, Compared to the mdx Mouse Model for Duchenne Muscular Dystrophy

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals—Rats
2.2. Animals—Zebrafish
2.3. Human Plasma Samples
2.4. HPLC Quantification of Taurine in Rat and Human Plasma, Rat Muscles, and Zebrafish Tails
2.5. Plasma Creatine Kinase (CK)
2.6. Quantification of Myeloperoxidase (MPO) in the Muscle as a Measure of Neutrophil Activity
2.7. Quantification of Muscle Oxidative Stress (Protein Thiol Oxidation)
2.8. Statistics
3. Results
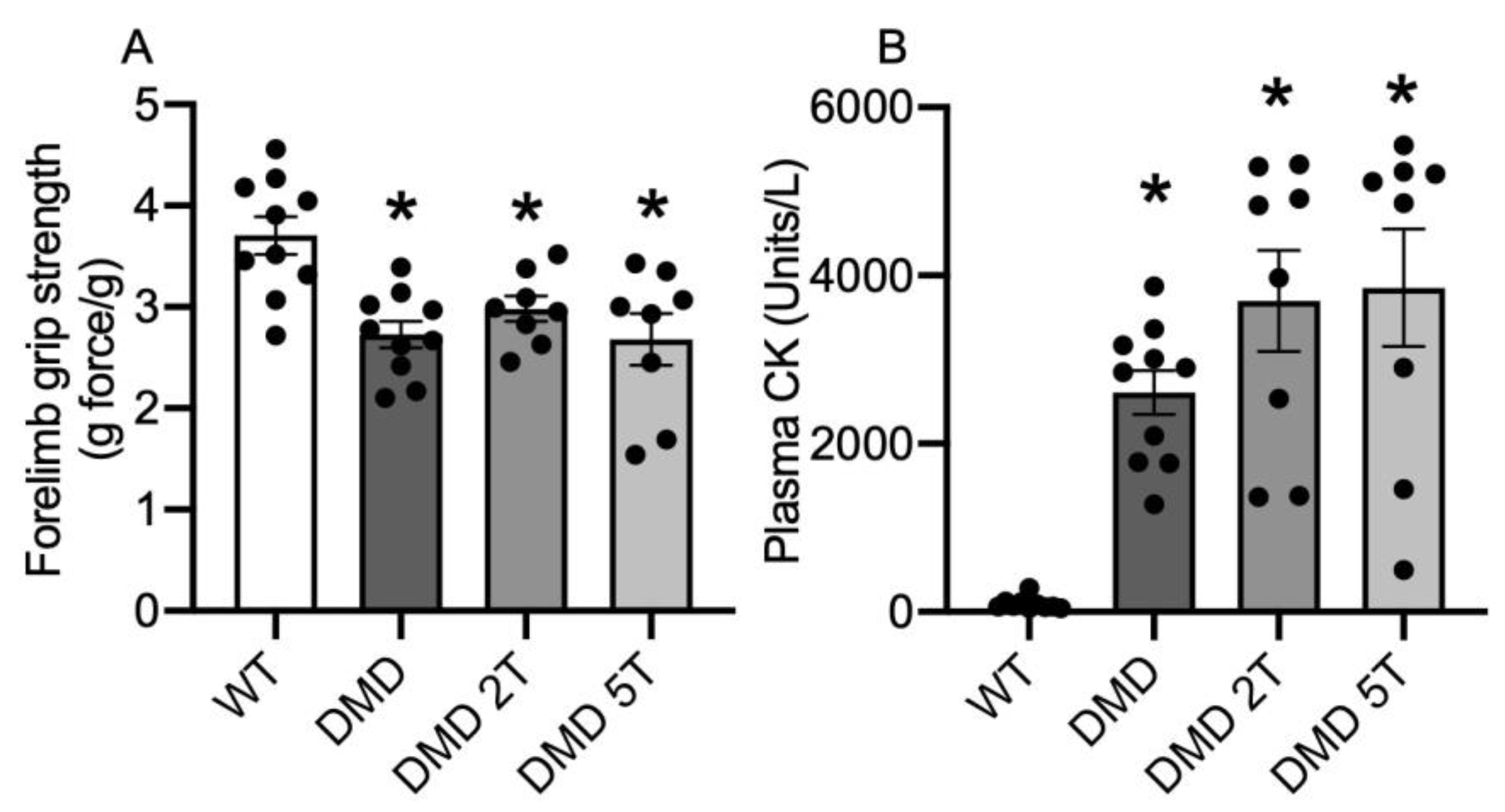
3.1. Dystropathology (Grip Strength, Plasma CK, and Other Parameters) in WT Compared with Untreated and Taurine-Treated DMDmdx Rats
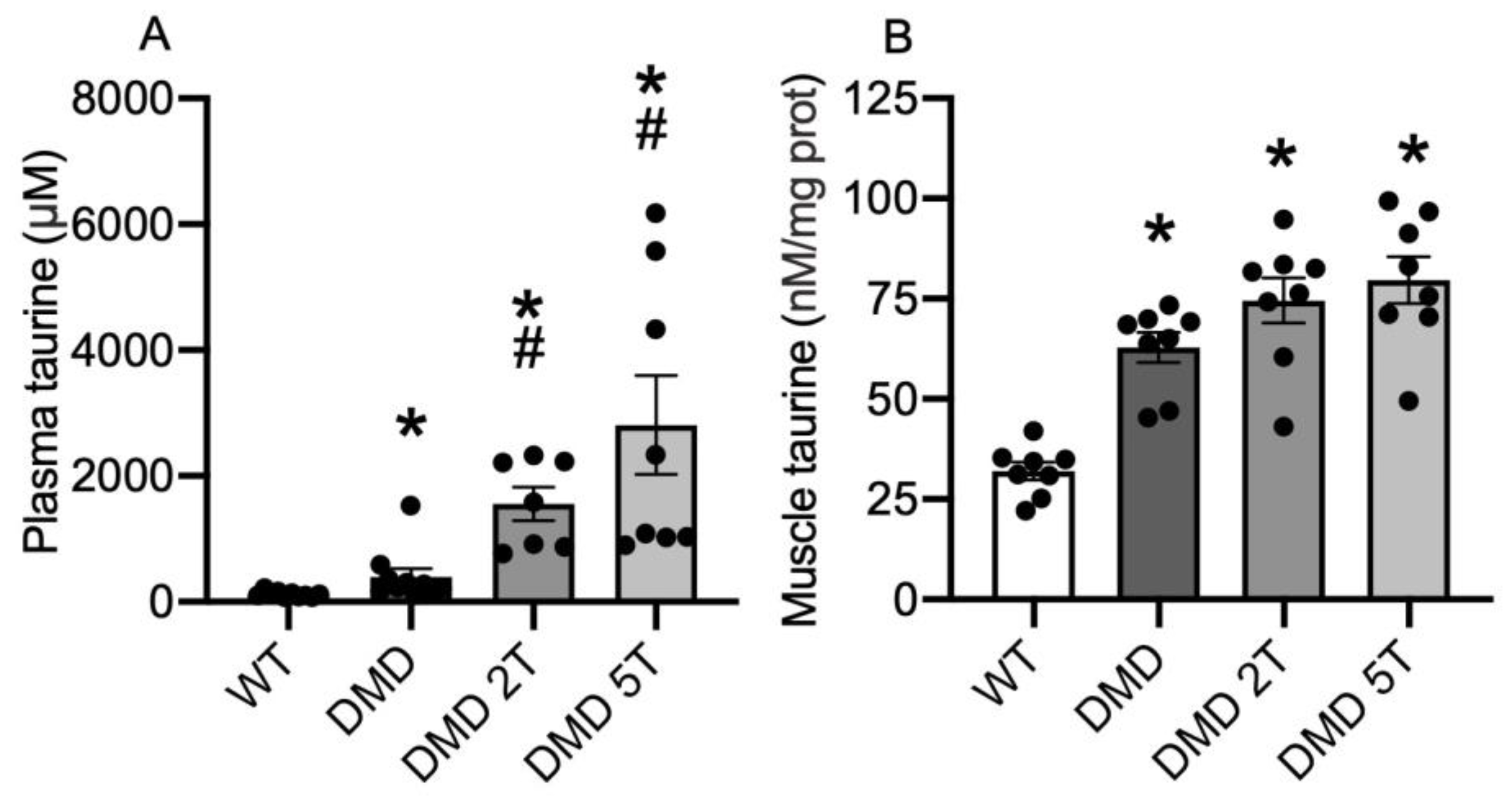
3.2. Plasma and Muscle Taurine Content in WT Compared with Untreated and Taurine-Treated DMDmdx Rats
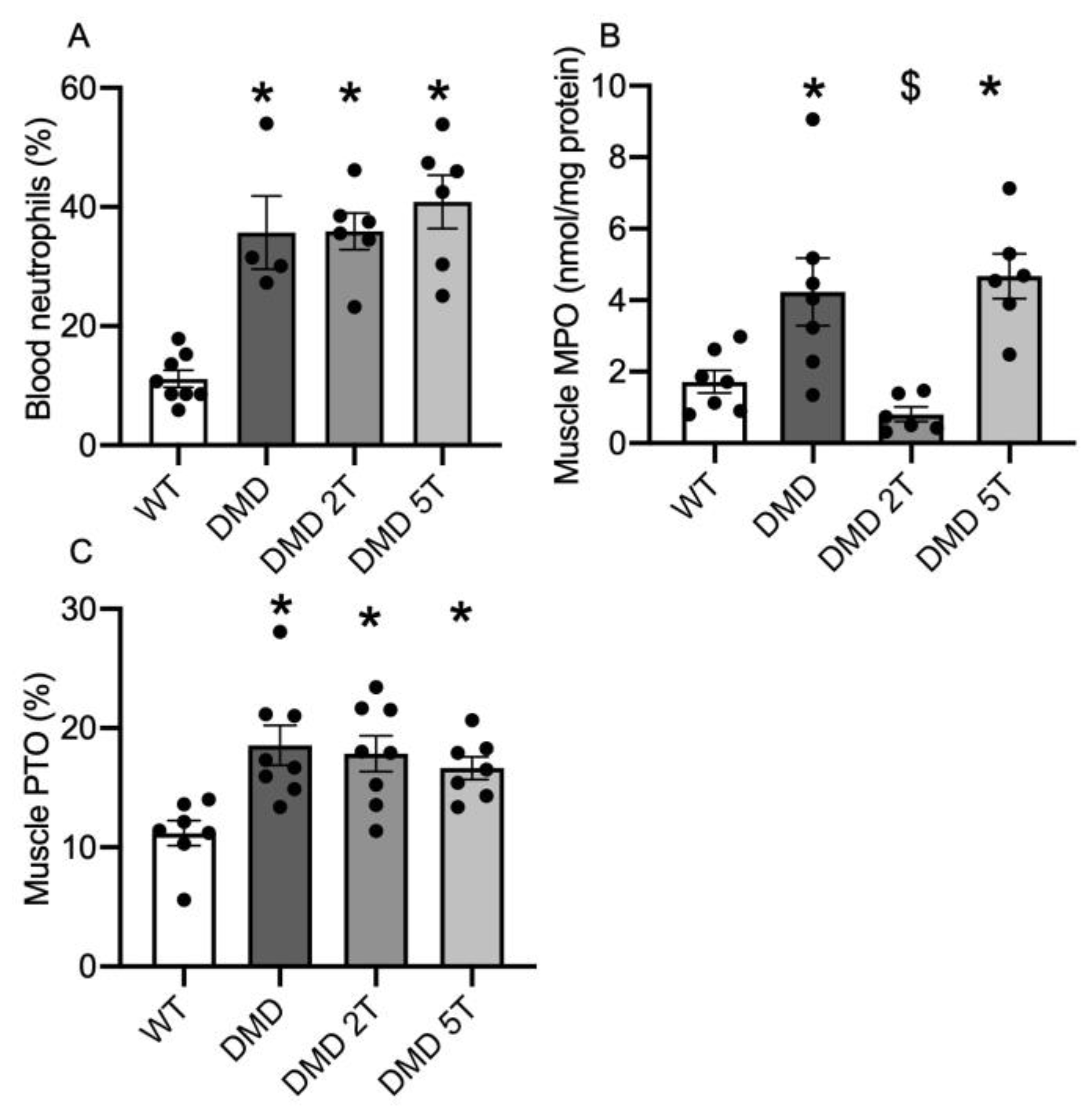
3.3. Inflammation and Oxidative Stress in the Plasma and Muscles of Taurine-Treated DMDmdx Rats
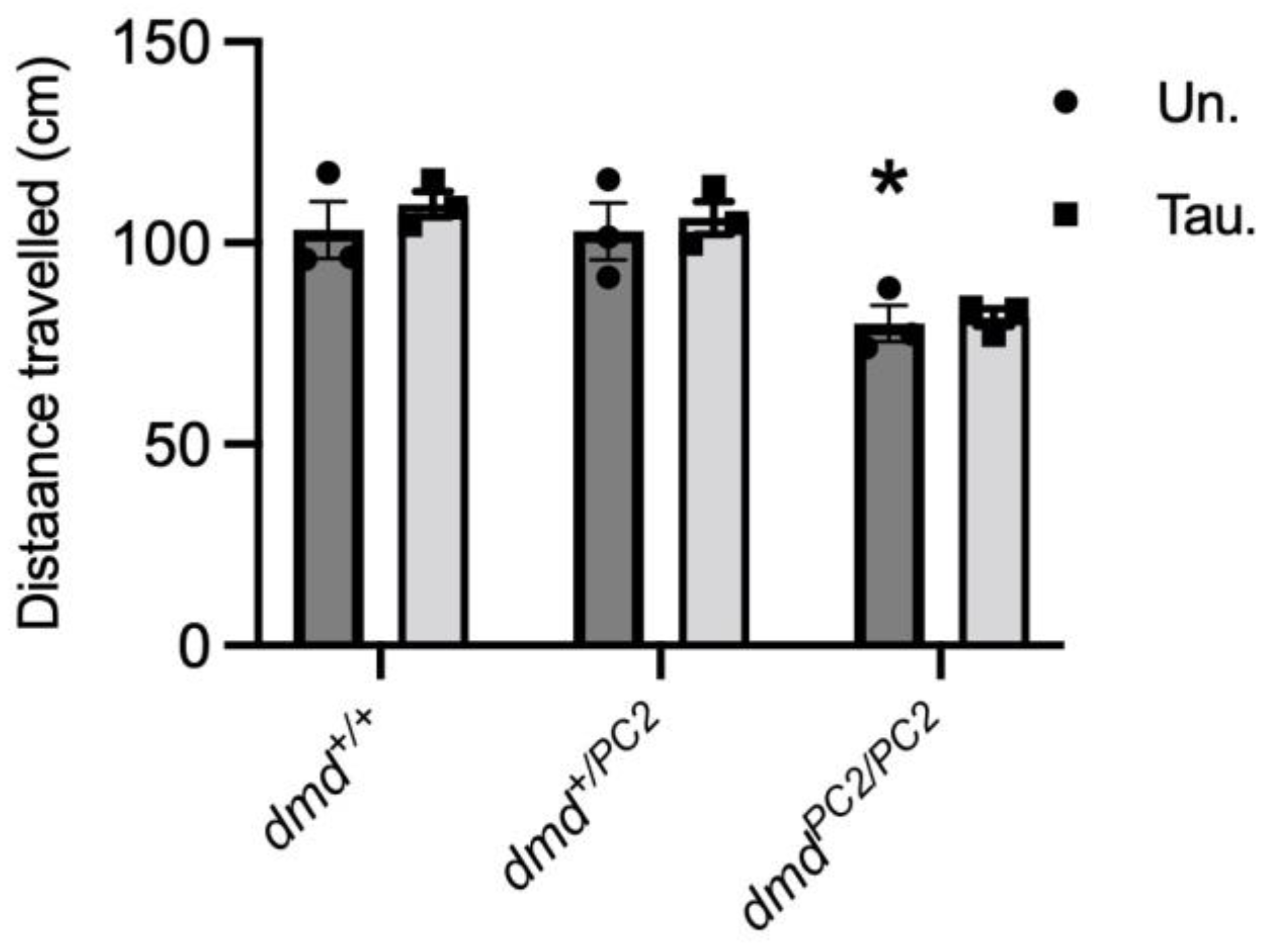
3.4. Muscle Function in Untreated and Taurine-Treated WT (dmd+/+), Heterozygous (dmd+/pc2) and Dystrophic Homozygous (dmdpc2/pc2) Zebrafish
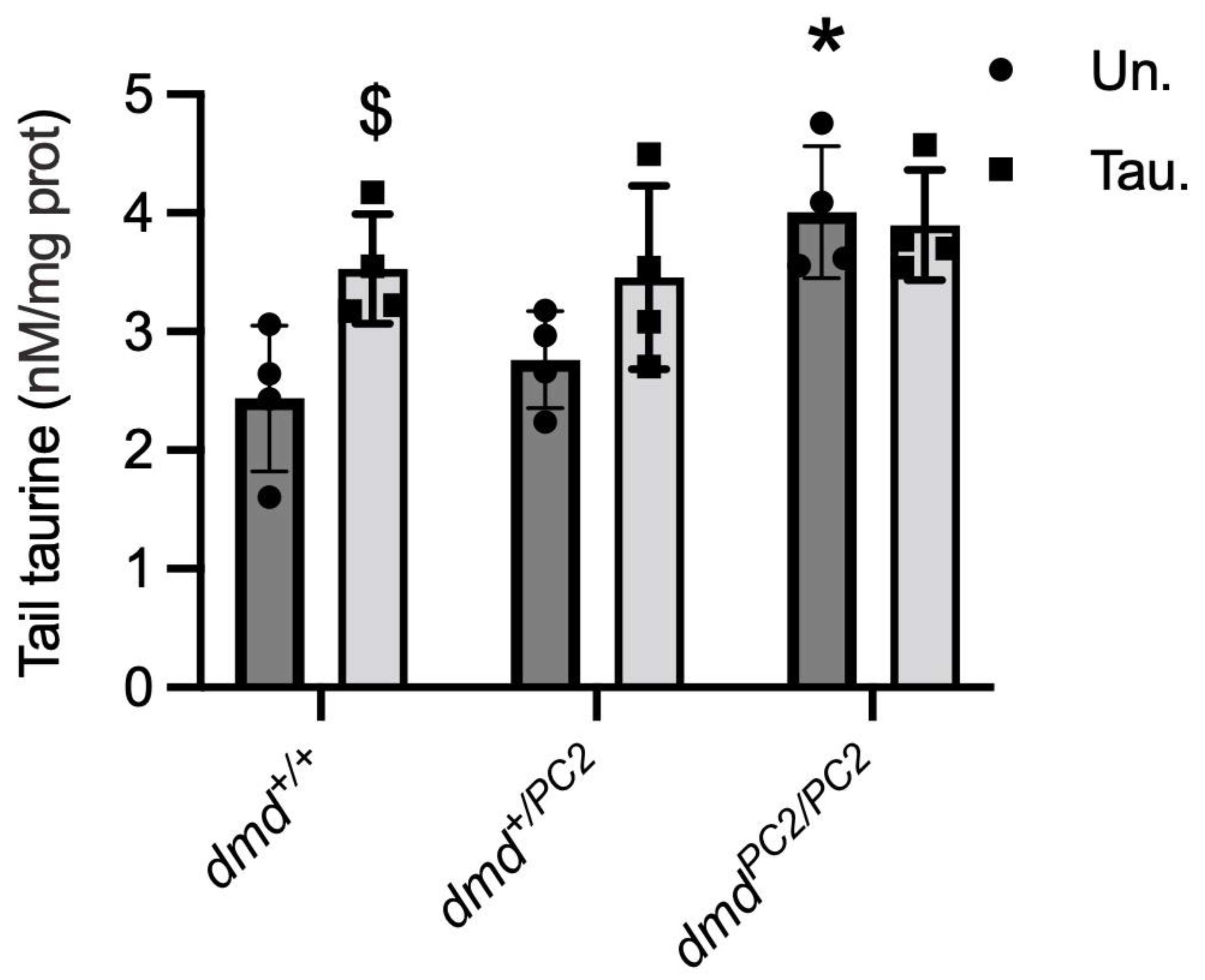
3.5. Taurine Content in Untreated and Taurine-Treated WT (dmd+/+), Heterozygous (dmd+/pc2) and Dystrophic Homozygous (dmdpc2/pc2) Zebrafish
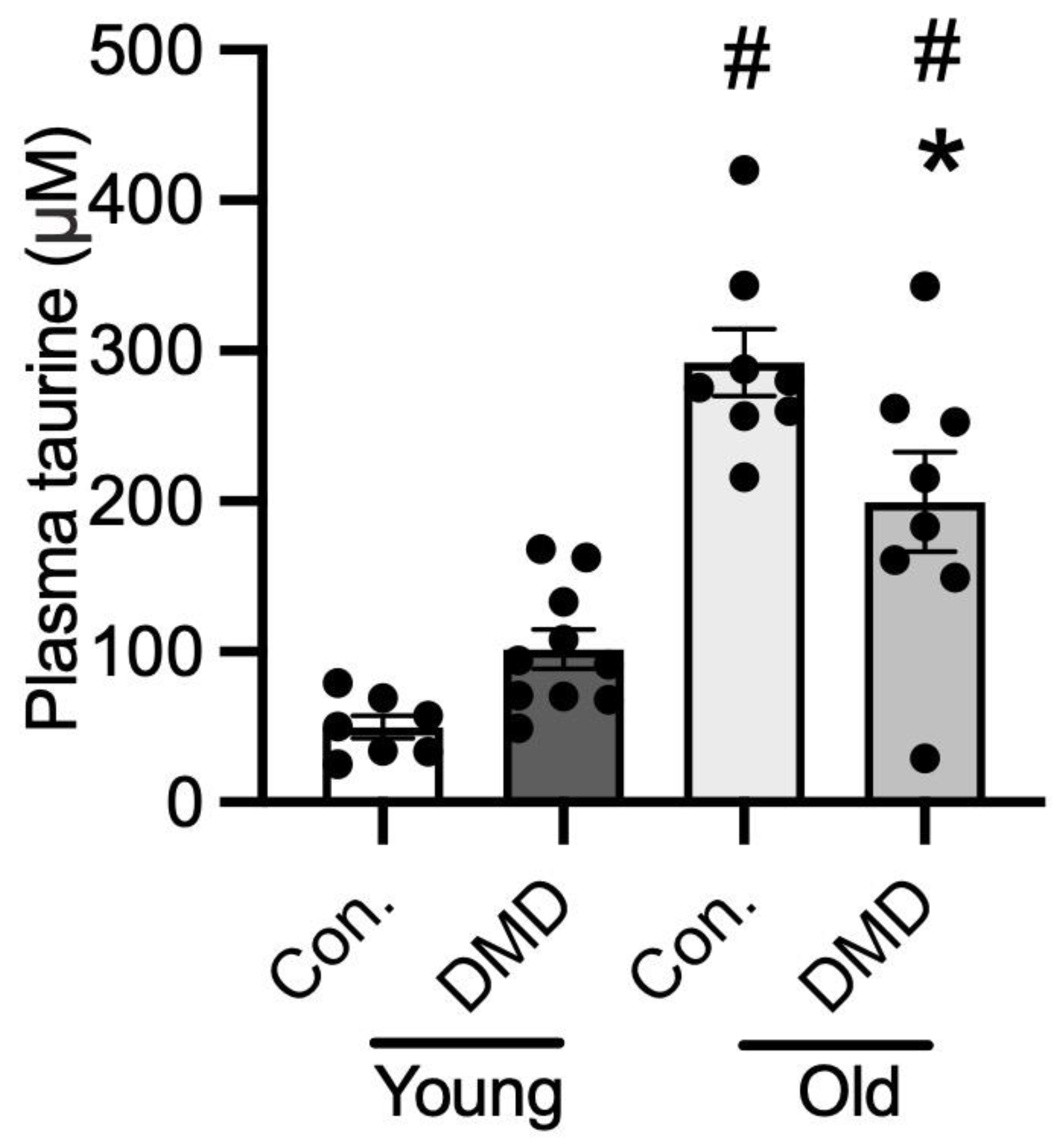
3.6. Taurine Content in Human Plasma of Normal Controls and Patients with DMD
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.I.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D. Two-tiered hypotheses for Duchenne muscular dystrophy. Cell. Mol. Life Sci. 2008, 65, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Munoz-Canoves, P. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed. Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef] [PubMed]
- Lapidos, K.A.; Kakkar, R.; McNally, E.M. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 2004, 94, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model. Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef]
- Biggar, W.D. Duchenne muscular dystrophy. Pediatr. Rev. 2006, 27, 83–88. [Google Scholar] [CrossRef]
- Verhaart, I.E.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef]
- Bulfield, G.; Siller, W.; Wight, P.; Moore, K. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef]
- McGeachie, J.K.; Grounds, M.D.; Partridge, T.A.; Morgan, J.E. Age-related changes in replication of myogenic cells in mdx mice: Quantitative autoradiographic studies. J. Neurol. Sci. 1993, 119, 169–179. [Google Scholar] [CrossRef]
- Radley, H.G.; Grounds, M.D. Cromolyn administration (to block mast cell degranulation) reduces necrosis of dystrophic muscle in mdx mice. Neurobiol. Dis. 2006, 23, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Radley, H.G.; Davies, M.J.; Grounds, M.D. Reduced muscle necrosis and long-term benefits in dystrophic mdx mice after cV1q (blockade of TNF) treatment. Neuromuscul. Disord. 2008, 18, 227–238. [Google Scholar] [CrossRef]
- Grounds, M.D.; Torrisi, J. Anti-TNF alpha (Remicade (R)) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. 2004, 18, 676–682. [Google Scholar] [CrossRef]
- Lefaucheur, J.P.; Pastoret, C.; Sebille, A. Phenotype of dystrophinopathy in old mdx mice. Anat. Rec. 1995, 242, 70–76. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Pierno, S.; Camerino, D.C. Taurine: The appeal of a safe amino acid for skeletal muscle disorders. J. Transl. Med. 2015, 13, 243. [Google Scholar] [CrossRef]
- Terrill, J.R.; Webb, S.M.; Arthur, P.G.; Hackett, M.J. Investigation of the effect of taurine supplementation on muscle taurine content in the mdx mouse model of Duchenne muscular dystrophy using chemically specific synchrotron imaging. Analyst 2020, 145, 7242–7251. [Google Scholar] [CrossRef]
- Terrill, J.R.; Pinniger, G.J.; Graves, J.A.; Grounds, M.D.; Arthur, P.G. Increasing taurine intake and taurine synthesis improves skeletal muscle function in the mdx mouse model for Duchenne muscular dystrophy. J. Physiol. 2016, 594, 3095–3110. [Google Scholar] [CrossRef]
- Terrill, J.R.; Grounds, M.D.; Arthur, P.G. Increased taurine in pre-weaned juvenile mdx mice greatly reduces the acute onset of myofibre necrosis and dystropathology and prevents inflammation. PLoS Curr. 2016, 8, ecurrents. [Google Scholar] [CrossRef] [PubMed]
- Cozzoli, A.; Rolland, J.F.; Capogrosso, R.F.; Sblendorio, V.T.; Longo, V.; Simonetti, S.; Nico, B.; De Luca, A. Evaluation of potential synergistic action of a combined treatment with alpha-methyl-prednisolone and taurine on the mdx mouse model of Duchenne muscular dystrophy. Neuropathol. Appl. Neurobiol. 2011, 37, 243–256. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Pierno, S.; Liantonio, A.; Cetrone, M.; Camerino, C.; Fraysse, B.; Mirabella, M.; Servidei, S.; Ruegg, U.T.; Conte Camerino, D. Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J. Pharmacol. Exp. Ther. 2003, 304, 453–463. [Google Scholar] [CrossRef]
- De Luca, A.; Pierno, S.; Liantonio, A.; Cetrone, M.; Camerino, C.; Simonetti, S.; Papadia, F.; Camerino, D.C. Alteration of excitation-contraction coupling mechanism in extensor digitorum longus muscle fibres of dystrophic mdx mouse and potential efficacy of taurine. Br. J. Pharmacol. 2001, 132, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Capogrosso, R.F.; Cozzoli, A.; Mantuano, P.; Camerino, G.M.; Massari, A.M.; Sblendorio, V.T.; De Bellis, M.; Tamma, R.; Giustino, A.; Nico, B.; et al. Assessment of resveratrol, apocynin and taurine on mechanical-metabolic uncoupling and oxidative stress in a mouse model of duchenne muscular dystrophy: A comparison with the gold standard, alpha-methyl prednisolone. Pharmacol. Res. 2016, 106, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Mele, A.; Mantuano, P.; De Bellis, M.; Rana, F.; Sanarica, F.; Conte, E.; Morgese, M.G.; Bove, M.; Rolland, J.-F.; Capogrosso, R.F. A long-term treatment with taurine prevents cardiac dysfunction in mdx mice. Transl. Res. 2019, 204, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D.M.; Murphy, R.M.; Mollica, J.P.; Hayes, A.; Goodman, C.A. The effect of taurine and β-alanine supplementation on taurine transporter protein and fatigue resistance in skeletal muscle from mdx mice. Amino Acids 2016, 48, 2635–2645. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.G.; Horvath, D.; van der Poel, C.; Murphy, R.M. Benefits of prenatal taurine supplementation in preventing the onset of acute damage in the Mdx mouse. PLoS Curr. 2017, 9, ecurrents.md.9a3e357a0154d01050b591601cbd4fdb. [Google Scholar]
- Terrill, J.R.; Boyatzis, A.; Grounds, M.D.; Arthur, P.G. Treatment with the cysteine precursor l-2-oxothiazolidine-4-carboxylate (OTC) implicates taurine deficiency in severity of dystropathology in mdx mice. Int. J. Biochem. Cell Biol. 2013, 45, 2097–2108. [Google Scholar] [CrossRef]
- Terrill, J.R.; Grounds, M.D.; Arthur, P.G. Taurine deficiency, synthesis and transport in the mdx mouse model for Duchenne Muscular Dystrophy. Int. J. Biochem. Cell Biol. 2015, 66, 141–148. [Google Scholar] [CrossRef]
- Griffin, J.; Williams, H.; Sang, E.; Clarke, K.; Rae, C.; Nicholson, J. Metabolic Profiling of Genetic Disorders: A Multitissue1 H Nuclear Magnetic Resonance Spectroscopic and Pattern Recognition Study into Dystrophic Tissue. Anal. Biochem. 2001, 293, 16–21. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, L.; Granberg, K.E.; Brière, K.M.; Anderson, J.E. Nuclear magnetic resonance spectroscopy study of muscle growth, mdx dystrophy and glucocorticoid treatments: Correlation with repair. NMR Biomed. 1998, 11, 1–10. [Google Scholar] [CrossRef]
- Terrill, J.R.; Duong, M.N.; Turner, R.; Le Guiner, C.; Boyatzis, A.; Kettle, A.J.; Grounds, M.D.; Arthur, P.G. Levels of inflammation and oxidative stress, and a role for taurine in dystropathology of the Golden Retriever Muscular Dystrophy dog model for Duchenne Muscular Dystrophy. Redox Biol. 2016, 9, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Wingate, C.D.; Abbas, Z.; Baustista, A.P.; Bettis, A.K.; Balog-Alvarez, C.J.; Kornegay, J.N.; Nghiem, P.P.; et al. Oxidative damage to urinary proteins from the GRMD dog and mdx mouse as biomarkers of dystropathology in Duchenne muscular dystrophy. PLoS ONE 2020, 15, e0240317. [Google Scholar] [CrossRef]
- Berger, J.; Berger, S.; Jacoby, A.S.; Wilton, S.D.; Currie, P.D. Evaluation of exon-skipping strategies for Duchenne muscular dystrophy utilizing dystrophin-deficient zebrafish. J. Cell. Mol. Med. 2011, 15, 2643–2651. [Google Scholar] [CrossRef]
- Gibbs, E.M.; Horstick, E.J.; Dowling, J.J. Swimming into prominence: The zebrafish as a valuable tool for studying human myopathies and muscular dystrophies. FEBS J. 2013, 280, 4187–4197. [Google Scholar] [CrossRef]
- Kawahara, G.; Karpf, J.A.; Myers, J.A.; Alexander, M.S.; Guyon, J.R.; Kunkel, L.M. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2011, 108, 5331–5336. [Google Scholar] [CrossRef]
- Larcher, T.; Lafoux, A.; Tesson, L.; Remy, S.; Thepenier, V.; François, V.; Le Guiner, C.; Goubin, H.; Dutilleul, M.; Guigand, L. Characterization of dystrophin deficient rats: A new model for Duchenne muscular dystrophy. PLoS ONE 2014, 9, e110371. [Google Scholar] [CrossRef]
- Szabó, P.L.; Ebner, J.; Koenig, X.; Hamza, O.; Watzinger, S.; Trojanek, S.; Abraham, D.; Todt, H.; Kubista, H.; Schicker, K. Cardiovascular phenotype of the Dmdmdx rat–a suitable animal model for Duchenne muscular dystrophy. Dis. Model. Mech. 2021, 14, dmm047704. [Google Scholar] [CrossRef]
- Caudal, D.; François, V.; Lafoux, A.; Ledevin, M.; Anegon, I.; Le Guiner, C.; Larcher, T.; Huchet, C. Characterization of brain dystrophins absence and impact in dystrophin-deficient Dmdmdx rat model. PLoS ONE 2020, 15, e0230083. [Google Scholar] [CrossRef]
- Krishnan, V.S.; Thanigaiarasu, L.P.; White, R.; Crew, R.; Larcher, T.; Le Guiner, C.; Grounds, M.D. Dystrophic Dmdmdx rats show early neuronal changes (increased S100β and Tau5) at 8 months, supporting severe dystropathology in this rodent model of Duchenne muscular dystrophy. Mol. Cell. Neurosci. 2020, 108, 103549. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, A.; François, V.; Zhang, L.; Lafoux, A.; Fraysse, B.; Toumaniantz, G.; Larcher, T.; Girard, T.; Ledevin, M.; Lebreton, C. Evaluation of the dystrophin carboxy-terminal domain for micro-dystrophin gene therapy in cardiac and skeletal muscles in the DMDmdx rat model. Gene Ther. 2022, 29, 520–535. [Google Scholar] [CrossRef]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish. 2000. Available online: http://zfin.org/zf_info/zfbook/zfbk.html (accessed on 11 May 2021).
- Messineo, A.M.; Gineste, C.; Sztal, T.E.; McNamara, E.L.; Vilmen, C.; Ogier, A.C.; Hahne, D.; Bendahan, D.; Laing, N.G.; Bryson-Richardson, R.J. L-tyrosine supplementation does not ameliorate skeletal muscle dysfunction in zebrafish and mouse models of dominant skeletal muscle α-actin nemaline myopathy. Sci. Rep. 2018, 8, 11490. [Google Scholar] [CrossRef] [PubMed]
- Sztal, T.E.; Ruparelia, A.A.; Williams, C.; Bryson-Richardson, R.J. Using touch-evoked response and locomotion assays to assess muscle performance and function in zebrafish. JoVE (J. Vis. Exp.) 2016, e54431. [Google Scholar] [CrossRef]
- Burch, P.M.; Pogoryelova, O.; Goldstein, R.; Bennett, D.; Guglieri, M.; Straub, V.; Bushby, K.; Lochmüller, H.; Morris, C. Muscle-derived proteins as serum biomarkers for monitoring disease progression in three forms of muscular dystrophy. J. Neuromuscul. Dis. 2015, 2, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Setsukinai, K.; Urano, Y.; Kakinuma, K.; Majima, H.J.; Nagano, T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem. 2003, 278, 3170–3175. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Vissers, M.C.M.; Kettle, A.J. Myeloperoxidase. Curr. Opin. Hematol. 2000, 7, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J. Biomarkers of myeloperoxidase-derived hypochlorous acid. Free Radic. Biol. Med. 2000, 29, 403–409. [Google Scholar] [CrossRef]
- Armstrong, A.E.; Zerbes, R.; Fournier, P.A.; Arthur, P.G. A fluorescent dual labeling technique for the quantitative measurement of reduced and oxidized protein thiols in tissue samples. Free Radic. Biol. Med. 2011, 50, 510–517. [Google Scholar] [CrossRef]
- Bakker, A.J.; Berg, H.M. Effect of taurine on sarcoplasmic reticulum function and force in skinned fast-twitch skeletal muscle fibres of the rat. J. Physiol. 2004, 538, 185–194. [Google Scholar] [CrossRef]
- Hamilton, E.J.; Berg, H.M.; Easton, C.J.; Bakker, A.J. The effect of taurine depletion on the contractile properties and fatigue in fast-twitch skeletal muscle of the mouse. Amino Acids 2006, 31, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Huxtable, R.J. Physiological actions of taurine. Physiol. Rev. 1992, 72, 101–163. [Google Scholar] [CrossRef] [PubMed]
- Warskulat, U.; Flogel, U.; Jacoby, C.; Hartwig, H.G.; Thewissen, M.; Merx, M.W.; Molojavyi, A.; Heller-Stilb, B.; Schrader, J.; Haussinger, D. Taurine transporter knockout depletes muscle taurine levels and results in severe skeletal muscle impairment but leaves cardiac function uncompromised. FASEB J. 2004, 18, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Warskulat, U.; Heller-Stilb, B.; Oermann, E.; Zilles, K.; Haas, H.; Lang, F.; Haussinger, D. Phenotype of the taurine transporter knockout mouse. Methods Enzymol. 2007, 428, 439–458. [Google Scholar] [CrossRef]
- Camerino, D.C.; Tricarico, D.; Pierno, S.; Desaphy, J.F.; Liantonio, A.; Pusch, M.; Burdi, R.; Camerino, C.; Fraysse, B.; De Luca, A. Taurine and skeletal muscle disorders. Neurochem. Res. 2004, 29, 135–142. [Google Scholar] [CrossRef]
- Terrill, J.R.; Pinniger, G.J.; Nair, K.V.; Grounds, M.D.; Arthur, P.G. Beneficial effects of high dose taurine treatment in juvenile dystrophic mdx mice are offset by growth restriction. PLoS ONE 2017, 12, e0187317. [Google Scholar] [CrossRef]
- Gregory, D.M.; Sovetts, D.; Clow, C.L.; Scriver, C.R. Plasma free amino acid values in normal children and adolescents. Metabolism 1986, 35, 967–969. [Google Scholar] [CrossRef]
- Radley-Crabb, H.G.; Marini, J.C.; Sosa, H.A.; Castillo, L.I.; Grounds, M.D.; Fiorotto, M.L. Dystropathology increases energy expenditure and protein turnover in the mdx mouse model of duchenne muscular dystrophy. PLoS ONE 2014, 9, e89277. [Google Scholar] [CrossRef]
- Nakamura, K.; Fujii, W.; Tsuboi, M.; Tanihata, J.; Teramoto, N.; Takeuchi, S.; Naito, K.; Yamanouchi, K.; Nishihara, M. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci. Rep. 2014, 4, 5635. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Wildtype | DMDmdx | DMDmdx Taurine Treated- 2 g/kg/day | DMDmdx Taurine Treated- 5 g/kg/day |
|---|---|---|---|---|
| Body weight (g) | 497 ± 42 | 433 ± 35 | 438 ± 27 | 433 ± 33 |
| TA weight (mg) | 831 ± 12 | 952 ± 38 * | 987 ± 29 | 1019 ± 71 |
| EDL weight (mg) | 221 ± 5 | 237 ± 9 | 241 ± 9 | 218 ± 13 |
| Sol weight (mg) | 207 ± 4 | 213 ± 7 | 214 ± 8 | 192 ± 12 |
| Distance travelled (cm) | 2305 ± 209 | 1937 ± 197 | 1885 ± 162 | 2012 ± 113 |
| Activity time (s) | 196 ± 9 | 181 ± 13 | 179 ± 10 | 191 ± 6 |
| Number rearings | 18 ± 2 | 11 ± 2 | 7 ± 2 * | 12 ± 2 |
| Platelets (Giga/L) | 963 ± 35 | 1645 ± 52 * | 1527 ± 37 * | 1543 ± 50 * |
| Neutrophils (%) | 11 ± 1 | 36 ± 6 * | 36 ± 3 * | 41 ± 5 * |
| Lymphocytes (%) | 87 ± 2 | 60 ± 7 * | 58 ± 3 * | 54 ± 5 * |
| Hematocrit (%) | 45 ± 1 | 48 ± 1 * | 44 ± 0.4 $ | 45 ± 1$ |
| Leucocytes (Giga/L) | 8 ± 1 | 10 ± 1 | 10 ± 1 | 11 ± 1 |
| Monocytes (%) | 1.2 ± 1 | 3.7 ± 1 | 4.7 ± 0.5 | 3.5 ± 1 |
| Model | Age | Taurine in Muscle | Taurine in Plasma | Taurine Efficacy | Reference |
|---|---|---|---|---|---|
| mdx mouse | 18 days | — | ↓ | ✓ | [30] |
| <3 weeks | ↓ | N | N | [32] | |
| 23 days | — | N | ✓ | [21] | |
| 4 weeks | ↓/— | — | ✓ | [30]/[28] | |
| 6 weeks | — | — | ✓ | [19,20,30,57] | |
| 10 weeks | ↑ | — | ✘ | [28] | |
| 12 weeks | ↓ | ↑ | N | [29] | |
| 6 months | ↓ | N | ✓ | [27] | |
| 6–8 months | — | ↑ | N | [24] | |
| 12 months | ↓ | ↑ | Cardiac muscle only | [26] | |
| DMDmdx rats | 12 weeks | ↑ | ↑ | ✘ | |
| dmdzebrafish | 6 dpf | ↑ (tails) | N | ✘ | |
| GRMD dogs | 8 months | ↑ | ↓ | N | [33] |
| DMD patients | 2–6 years | N | — | N | |
| 16–20 years | N | ↓ | N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrill, J.R.; Huchet, C.; Le Guiner, C.; Lafoux, A.; Caudal, D.; Tulangekar, A.; Bryson-Richardson, R.J.; Sztal, T.E.; Grounds, M.D.; Arthur, P.G. Muscle Pathology in Dystrophic Rats and Zebrafish Is Unresponsive to Taurine Treatment, Compared to the mdx Mouse Model for Duchenne Muscular Dystrophy. Metabolites 2023, 13, 232. https://doi.org/10.3390/metabo13020232
Terrill JR, Huchet C, Le Guiner C, Lafoux A, Caudal D, Tulangekar A, Bryson-Richardson RJ, Sztal TE, Grounds MD, Arthur PG. Muscle Pathology in Dystrophic Rats and Zebrafish Is Unresponsive to Taurine Treatment, Compared to the mdx Mouse Model for Duchenne Muscular Dystrophy. Metabolites. 2023; 13(2):232. https://doi.org/10.3390/metabo13020232
Chicago/Turabian StyleTerrill, Jessica R., Corinne Huchet, Caroline Le Guiner, Aude Lafoux, Dorian Caudal, Ankita Tulangekar, Robert J. Bryson-Richardson, Tamar E. Sztal, Miranda D. Grounds, and Peter G. Arthur. 2023. "Muscle Pathology in Dystrophic Rats and Zebrafish Is Unresponsive to Taurine Treatment, Compared to the mdx Mouse Model for Duchenne Muscular Dystrophy" Metabolites 13, no. 2: 232. https://doi.org/10.3390/metabo13020232
APA StyleTerrill, J. R., Huchet, C., Le Guiner, C., Lafoux, A., Caudal, D., Tulangekar, A., Bryson-Richardson, R. J., Sztal, T. E., Grounds, M. D., & Arthur, P. G. (2023). Muscle Pathology in Dystrophic Rats and Zebrafish Is Unresponsive to Taurine Treatment, Compared to the mdx Mouse Model for Duchenne Muscular Dystrophy. Metabolites, 13(2), 232. https://doi.org/10.3390/metabo13020232