
Role of Oxidative Stress in Ocular Diseases: A Balancing Act

 ,
,  ,
,  , , ,
, , ,  and
and
Abstract
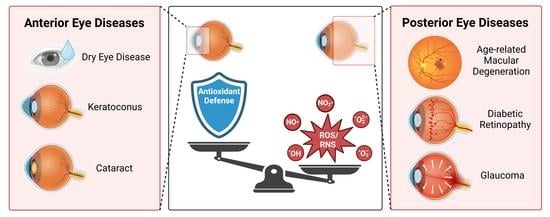
1. Introduction
2. Sources of Oxidative Stress
3. Cornea
3.1. Dry Eye Disease
3.2. Altered Metabolism and Bioenergetics in Keratoconus
4. Oxidative Stress in Cataract
5. Retina
5.1. Oxidative Stress and Retinal Vasculature
5.2. Oxidative Stress during Age-Related Macular Degeneration
5.3. Oxidative Stress and Proliferative Vitreoretinopathy
5.4. Oxidative Stress and Diabetic Retinopathy
5.5. Oxidative Stress Mechanisms in Glaucoma
6. Targeting Oxidative Stress in Aging
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, health, and hallmarks of aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Samuels, D.C.; Elson, J.; Turnbull, D.M. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: Is there a common mechanism? Lancet 2002, 360, 1323–1325. [Google Scholar] [CrossRef]
- Marcelino, L.A.; Thilly, W.G. Mitochondrial mutagenesis in human cells and tissues. Mutat. Res. DNA Repair 1999, 434, 177–203. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Shibata, D.; Soong, N.W.; Arnheim, N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 7370–7374. [Google Scholar] [CrossRef]
- Song, S.; Pursell, Z.F.; Copeland, W.C.; Longley, M.J.; Kunkel, T.A.; Mathews, C.K. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc. Natl. Acad. Sci. USA 2005, 102, 4990–4995. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Kaiser, C.A.; Amom, A.; Ploegh, H.; Bretscher, A.; Kriefer, M.; Martin, K.C. Molecular Cell Biology, 8th ed.; Macmillan: New York, NY, USA, 2016. [Google Scholar]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef]
- Sakurai, T.; Tsuchiya, S. Superoxide production from nonenzymatically glycated protein. FEBS Lett. 1988, 236, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Badwey, J.A.; Curnutte, J.T.; Karnovsky, M.L. Cis-Polyunsaturated fatty acids induce high levels of superoxide production by human neutrophils. J. Biol. Chem. 1981, 256, 12640–12643. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Kawanishi, S. Hydroxyl radical production and human DNA damage induced by ferric nitrilotriacetate and hydrogen peroxide. Cancer Res. 1987, 47, 6522–6527. [Google Scholar]
- Wagner, J.R.; Hu, C.-C.; Ames, B.N. Endogenous oxidative damage of deoxycytidine in DNA. Proc. Natl. Acad. Sci. USA 1992, 89, 3380–3384. [Google Scholar] [CrossRef]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.-X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef]
- Traverso, N.; Menini, S.; Maineri, E.P.; Patriarca, S.; Odetti, P.; Cottalasso, D.; Marinari, U.M.; Pronzato, M.A. Malondialdehyde, a lipoperoxidation-derived aldehyde, can bring about secondary oxidative damage to proteins. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, B890–B895. [Google Scholar] [CrossRef]
- Refsgaard, H.H.; Tsai, L.; Stadtman, E.R. Modifications of proteins by polyunsaturated fatty acid peroxidation products. Proc. Natl. Acad. Sci. USA 2000, 97, 611–616. [Google Scholar] [CrossRef]
- Iles, K.E.; Liu, R.-M. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free Radic. Biol. Med. 2005, 38, 547–556. [Google Scholar] [CrossRef]
- Uchida, K.; Shiraishi, M.; Naito, Y.; Torii, Y.; Nakamura, Y.; Osawa, T. Activation of stress signaling pathways by the end product of lipid peroxidation: 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J. Biol. Chem. 1999, 274, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Behndig, A.; Svensson, B.; Marklund, S.L.; Karlsson, K. Superoxide dismutase isoenzymes in the human eye. Investig. Ophthalmol. Vis. Sci. 1998, 39, 471–475. [Google Scholar]
- Deliyanti, D.; Alrashdi, S.F.; Tan, S.M.; Meyer, C.; Ward, K.W.; de Haan, J.B.; Wilkinson-Berka, J.L. Nrf2 Activation Is a Potential Therapeutic Approach to Attenuate Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 815–825. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2017, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Balboa, M.A.; Balsinde, J. Oxidative stress and arachidonic acid mobilization. Biochim. Biophys. Acta 2006, 1761, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef]
- Cummings, B.S.; McHowat, J.; Schnellmann, R.G. Phospholipase A(2)s in cell injury and death. J. Pharmacol. Exp. Ther. 2000, 294, 793–799. [Google Scholar] [PubMed]
- Colston, J.T.; de la Rosa, S.D.; Strader, J.R.; Anderson, M.A.; Freeman, G.L. H2O2 activates Nox4 through PLA2-dependent arachidonic acid production in adult cardiac fibroblasts. FEBS Lett. 2005, 579, 2533–2540. [Google Scholar] [CrossRef]
- Sporn, P.H.; Marshall, T.M.; Peters-Golden, M. Hydrogen peroxide increases the availability of arachidonic acid for oxidative metabolism by inhibiting acylation into phospholipids in the alveolar macrophage. Am. J. Respir. Cell Mol. Biol. 1992, 7, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Cane, A.; Breton, M.; Koumanov, K.; Béréziat, G.; Colard, O. Oxidant-induced arachidonic acid release and impairment of fatty acid acylation in vascular smooth muscle cells. Am. J. Physiol. 1998, 274, C1040–C1046. [Google Scholar] [CrossRef] [PubMed]
- Matono, R.; Miyano, K.; Kiyohara, T.; Sumimoto, H. Arachidonic acid induces direct interaction of the p67(phox)-Rac complex with the phagocyte oxidase Nox2, leading to superoxide production. J. Biol. Chem. 2014, 289, 24874–24884. [Google Scholar] [CrossRef]
- Wang, Z.J.; Liang, C.L.; Li, G.M.; Yu, C.Y.; Yin, M. Neuroprotective effects of arachidonic acid against oxidative stress on rat hippocampal slices. Chem. Biol. Interact. 2006, 163, 207–217. [Google Scholar] [CrossRef]
- DelMonte, D.W.; Kim, T. Anatomy and physiology of the cornea. J. Cataract. Refract. Surg. 2011, 37, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Hassell, J.R.; Birk, D.E. The molecular basis of corneal transparency. Exp. Eye Res. 2010, 91, 326–335. [Google Scholar] [CrossRef]
- Wu, M.; Hill, L.J.; Downie, L.E.; Chinnery, H.R. Neuroimmune crosstalk in the cornea: The role of immune cells in corneal nerve maintenance during homeostasis and inflammation. Prog. Retin. Eye Res. 2022, 91, 101105. [Google Scholar] [CrossRef]
- Alves, M.; Novaes, P.; Morraye Mde, A.; Reinach, P.S.; Rocha, E.M. Is dry eye an environmental disease? Arq. Bras. Oftalmol. 2014, 77, 193–200. [Google Scholar] [CrossRef]
- Seen, S.; Tong, L. Dry eye disease and oxidative stress. Acta Ophthalmol. 2018, 96, e412–e420. [Google Scholar] [CrossRef]
- Junqueira, V.B.; Barros, S.B.; Chan, S.S.; Rodrigues, L.; Giavarotti, L.; Abud, R.L.; Deucher, G.P. Aging and oxidative stress. Mol. Asp. Med. 2004, 25, 5–16. [Google Scholar] [CrossRef]
- Nakamura, S.; Shibuya, M.; Nakashima, H.; Hisamura, R.; Masuda, N.; Imagawa, T.; Uehara, M.; Tsubota, K. Involvement of oxidative stress on corneal epithelial alterations in a blink-suppressed dry eye. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, T.H.; Dogru, M.; Matsumoto, Y.; Kojima, T.; Kaido, M.; Ibrahim, O.M.; Sato, E.A.; Igarashi, A.; Ichihashi, Y.; Satake, Y.; et al. Evaluation of lipid oxidative stress status in Sjögren syndrome patients. Investig. Ophthalmol. Vis. Sci. 2013, 54, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Wakamatsu, T.H.; Dogru, M.; Ogawa, Y.; Igarashi, A.; Ibrahim, O.M.; Inaba, T.; Shimizu, T.; Noda, S.; Obata, H.; et al. Age-related dysfunction of the lacrimal gland and oxidative stress: Evidence from the Cu,Zn-superoxide dismutase-1 (Sod1) knockout mice. Am. J. Pathol. 2012, 180, 1879–1896. [Google Scholar] [CrossRef]
- Uchino, Y.; Kawakita, T.; Miyazawa, M.; Ishii, T.; Onouchi, H.; Yasuda, K.; Ogawa, Y.; Shimmura, S.; Ishii, N.; Tsubota, K. Oxidative stress induced inflammation initiates functional decline of tear production. PLoS ONE 2012, 7, e45805. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Alves, M.; Rios, J.D.; Dartt, D.A. The aging lacrimal gland: Changes in structure and function. Ocul. Surf. 2008, 6, 162–174. [Google Scholar] [CrossRef]
- Nichols, K.K.; Foulks, G.N.; Bron, A.J.; Glasgow, B.J.; Dogru, M.; Tsubota, K.; Lemp, M.A.; Sullivan, D.A. The international workshop on meibomian gland dysfunction: Executive summary. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1922–1929. [Google Scholar] [CrossRef]
- Ibrahim, O.M.; Dogru, M.; Matsumoto, Y.; Igarashi, A.; Kojima, T.; Wakamatsu, T.H.; Inaba, T.; Shimizu, T.; Shimazaki, J.; Tsubota, K. Oxidative stress induced age dependent meibomian gland dysfunction in Cu, Zn-superoxide dismutase-1 (Sod1) knockout mice. PLoS ONE 2014, 9, e993282014. [Google Scholar] [CrossRef]
- Macri, A.; Scanarotti, C.; Bassi, A.M.; Giuffrida, S.; Sangalli, G.; Traverso, C.E.; Iester, M. Evaluation of oxidative stress levels in the conjunctival epithelium of patients with or without dry eye, and dry eye patients treated with preservative-free hyaluronic acid 0.15 % and vitamin B12 eye drops. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 425–430. [Google Scholar] [CrossRef]
- Horwath-Winter, J.; Schmut, O.; Haller-Schober, E.M.; Gruber, A.; Rieger, G. Iodide iontophoresis as a treatment for dry eye syndrome. Br. J. Ophthalmol. 2005, 89, 40–44. [Google Scholar] [CrossRef]
- Downie, L.E.; Ng, S.M.; Lindsley, K.B.; Akpek, E.K. Omega-3 and omega-6 polyunsaturated fatty acids for dry eye disease. Cochrane Database Syst. Rev. 2019, 12, Cd011016. [Google Scholar] [CrossRef]
- Wang, B.; Zuo, X.; Peng, L.; Wang, X.; Zeng, H.; Zhong, J.; Li, S.; Xiao, Y.; Wang, L.; Ouyang, H.; et al. Melatonin ameliorates oxidative stress-mediated injuries through induction of HO-1 and restores autophagic flux in dry eye. Exp. Eye Res. 2021, 205, 108491. [Google Scholar] [CrossRef]
- Sullivan, D.A.; Rocha, E.M.; Aragona, P.; Clayton, J.A.; Ding, J.; Golebiowski, B.; Hampel, U.; McDermott, A.M.; Schaumberg, D.A.; Srinivasan, S.; et al. TFOS DEWS II Sex, Gender, and Hormones Report. Ocul. Surf. 2017, 15, 284–333. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Fjærvoll, H.K.; Fjærvoll, K.A.; Wang, N.H.; Utheim, T.P.; Serhan, C.N.; Dartt, D.A. Sex-based differences in conjunctival goblet cell responses to pro-inflammatory and pro-resolving mediators. Sci. Rep. 2022, 12, 16305. [Google Scholar] [CrossRef]
- Garriz, A.; Morokuma, J.; Bowman, M.; Pagni, S.; Zoukhri, D. Effects of proinflammatory cytokines on lacrimal gland myoepithelial cells contraction. Front. Ophthalmol. 2022, 2, 873486. [Google Scholar] [CrossRef] [PubMed]
- McKay, T.B.; Priyadarsini, S.; Karamichos, D. Sex Hormones, Growth Hormone, and the Cornea. Cells 2022, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, H.; Heydarian, S.; Hooshmand, E.; Saatchi, M.; Yekta, A.; Aghamirsalim, M.; Valadkhan, M.; Mortazavi, M.; Hashemi, A.; Khabazkhoob, M. The prevalence and risk factors for keratoconus: A systematic review and meta-analysis. Cornea 2020, 39, 263–270. [Google Scholar] [CrossRef]
- Tur, V.M.; MacGregor, C.; Jayaswal, R.; O’Brart, D.; Maycock, N. A review of keratoconus: Diagnosis, pathophysiology, and genetics. Surv. Ophthalmol. 2017, 62, 770–783. [Google Scholar]
- De Barros, M.R.M.; Chakravarti, S. Pathogenesis of keratoconus: NRF2-antioxidant, extracellular matrix and cellular dysfunctions. Exp. Eye Res. 2022, 219, 109062. [Google Scholar]
- Navel, V.; Malecaze, J.; Pereira, B.; Baker, J.S.; Malecaze, F.; Sapin, V.; Chiambaretta, F.; Dutheil, F. Oxidative and antioxidative stress markers in keratoconus: A systematic review and meta-analysis. Acta Ophthalmol. 2021, 99, e777–e794. [Google Scholar] [CrossRef]
- Arnal, E.; Peris-Martínez, C.; Menezo, J.L.; Johnsen-Soriano, S.; Romero, F.J. Oxidative stress in keratoconus? Invest. Ophthalmol. Vis. Sci. 2011, 52, 8592–8597. [Google Scholar] [CrossRef] [PubMed]
- Balmus, I.M.; Alexa, A.I.; Ciuntu, R.E.; Danielescu, C.; Stoica, B.; Cojocaru, S.I.; Ciobica, A.; Cantemir, A. Oxidative stress markers dynamics in keratoconus patients’ tears before and after corneal collagen crosslinking procedure. Exp. Eye Res. 2020, 190, 107897. [Google Scholar] [CrossRef] [PubMed]
- López-López, M.; Regueiro, U.; Bravo, S.B.; Chantada-Vázquez, M.D.P.; Varela-Fernández, R.; Ávila-Gómez, P.; Hervella, P.; Lema, I. Tear Proteomics in Keratoconus: A Quantitative SWATH-MS Analysis. Investig. Ophthalmol. Vis. Sci. 2021, 62, 30. [Google Scholar] [CrossRef] [PubMed]
- Toprak, I.; Kucukatay, V.; Yildirim, C.; Kilic-Toprak, E.; Kilic-Erkek, O. Increased systemic oxidative stress in patients with keratoconus. Eye 2014, 28, 285–289. [Google Scholar] [CrossRef]
- Karamichos, D.; Hutcheon, A.E.; Rich, C.B.; Trinkaus-Randall, V.; Asara, J.M.; Zieske, J.D. In vitro model suggests oxidative stress involved in keratoconus disease. Sci. Rep. 2014, 4, 4608. [Google Scholar] [CrossRef]
- Shinde, V.; Hu, N.; Mahale, A.; Maiti, G.; Daoud, Y.; Eberhart, C.G.; Maktabi, A.; Jun, A.S.; Al-Swailem, S.A.; Chakravarti, S. RNA sequencing of corneas from two keratoconus patient groups identifies potential biomarkers and decreased NRF2-antioxidant responses. Sci. Rep. 2020, 10, 9907. [Google Scholar] [CrossRef] [PubMed]
- Lupasco, T.; He, Z.; Cassagne, M.; Sagnial, T.; Brion, L.; Fournié, P.; Gain, P.; Thuret, G.; Allouche, M.; Malecaze, F.; et al. Corneal epithelium in keratoconus underexpresses active NRF2 and a subset of oxidative stress-related genes. PLoS ONE 2022, 17, e0273807. [Google Scholar] [CrossRef]
- Chaerkady, R.; Shao, H.; Scott, S.G.; Pandey, A.; Jun, A.S.; Chakravarti, S. The keratoconus corneal proteome: Loss of epithelial integrity and stromal degeneration. J. Proteom. 2013, 87, 122–131. [Google Scholar] [CrossRef]
- Rabinowitz, Y.S. Keratoconus. Surv. Ophthalmol. 1998, 42, 297–319. [Google Scholar] [CrossRef]
- Wisse, R.P.L.; Kuiper, J.J.W.; Radstake, T.R.D.; Broen, J.C.A. Quantification of Double Stranded DNA Breaks and Telomere Length as Proxies for Corneal Damage and Replicative Stress in Human Keratoconus Corneas. Transl. Vis. Sci. Technol. 2019, 8, 10. [Google Scholar] [CrossRef]
- Kenney, M.C.; Brown, D.J.; Rajeev, B. Everett Kinsey lecture. The elusive causes of keratoconus: A working hypothesis. Clao J. 2000, 26, 10–13. [Google Scholar] [PubMed]
- Behndig, A.; Karlsson, K.; Johansson, B.O.; Brännström, T.; Marklund, S.L. Superoxide dismutase isoenzymes in the normal and diseased human cornea. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2293–2296. [Google Scholar] [PubMed]
- Udar, N.; Atilano, S.R.; Small, K.; Nesburn, A.B.; Kenney, M.C. SOD1 haplotypes in familial keratoconus. Cornea 2009, 28, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Udar, N.; Atilano, S.R.; Brown, D.J.; Holguin, B.; Small, K.; Nesburn, A.B.; Kenney, M.C. SOD1: A candidate gene for keratoconus. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3345–3351. [Google Scholar] [CrossRef]
- Moschos, M.M.; Kokolakis, N.; Gazouli, M.; Chatziralli, I.P.; Droutsas, D.; Anagnou, N.P.; Ladas, I.D. Polymorphism Analysis of VSX1 and SOD1 Genes in Greek Patients with Keratoconus. Ophthalmic Genet. 2015, 36, 213–217. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Azad, T.A.; Kalantan, H.; Sultan, T.; Al-Muammar, A.M. Mitochondrial sequence changes in keratoconus patients. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1706–1710. [Google Scholar] [CrossRef]
- Atilano, S.R.; Coskun, P.; Chwa, M.; Jordan, N.; Reddy, V.; Le, K.; Wallace, D.C.; Kenney, M.C. Accumulation of mitochondrial DNA damage in keratoconus corneas. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1256–1263. [Google Scholar] [CrossRef]
- Gadelha, D.N.B.; Feitosa, A.F.B.; da Silva, R.G.; Antunes, L.T.; Muniz, M.C.; de Oliveira, M.A.; Andrade, D.O.; da Paz Silva, N.M.; Cronemberger, S.; Schamber-Reis, B.L.F. Screening for Novel LOX and SOD1 Variants in Keratoconus Patients from Brazil. J. Ophthalmic Vis. Res. 2020, 15, 138–148. [Google Scholar] [CrossRef]
- Nejabat, M.; Naghash, P.; Dastsooz, H.; Mohammadi, S.; Alipour, M.; Fardaei, M. VSX1 and SOD1 Mutation Screening in Patients with Keratoconus in the South of Iran. J. Ophthalmic Vis. Res. 2017, 12, 135–140. [Google Scholar] [CrossRef]
- Stabuc-Silih, M.; Strazisar, M.; Hawlina, M.; Glavac, D. Absence of pathogenic mutations in VSX1 and SOD1 genes in patients with keratoconus. Cornea 2010, 29, 172–176. [Google Scholar] [CrossRef]
- Al-Muammar, A.M.; Kalantan, H.; Azad, T.A.; Sultan, T.; Abu-Amero, K.K. Analysis of the SOD1 gene in keratoconus patients from Saudi Arabia. Ophthalmic Genet. 2015, 36, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Bykhovskaya, Y.; Rabinowitz, Y.S. Update on the genetics of keratoconus. Exp. Eye Res. 2021, 202, 108398. [Google Scholar] [CrossRef] [PubMed]
- Lema, I.; Durán, J.A. Inflammatory molecules in the tears of patients with keratoconus. Ophthalmology 2005, 112, 654–659. [Google Scholar] [CrossRef]
- Daphne Teh, A.L.; Jayapalan, J.J.; Loke, M.F.; Wan Abdul Kadir, A.J.; Subrayan, V. Identification of potential serum metabolic biomarkers for patient with keratoconus using untargeted metabolomics approach. Exp. Eye Res. 2021, 211, 108734. [Google Scholar] [CrossRef] [PubMed]
- McKay, T.B.; Hjortdal, J.; Sejersen, H.; Asara, J.M.; Wu, J.; Karamichos, D. Endocrine and Metabolic Pathways Linked to Keratoconus: Implications for the Role of Hormones in the Stromal Microenvironment. Sci. Rep. 2016, 6, 25534. [Google Scholar] [CrossRef] [PubMed]
- Jamali, H.; Heydari, M.; Masihpour, N.; Khosravi, A.; Zare, M.; Shams, M.; Omrani, G.R. Serum androgens and prolactin levels in patients with keratoconus. Clin. Exp. Optom. 2022, 1–5. [Google Scholar] [CrossRef]
- Ayan, B.; Yuksel, N.; Carhan, A.; Gumuşkaya Ocal, B.; Akcay, E.; Cagil, N.; Asik, M.D. Evaluation estrogen, progesteron and androgen receptor expressions in corneal epithelium in keratoconus. Contact Lens Anterior Eye 2019, 42, 492–496. [Google Scholar] [CrossRef]
- Karamichos, D.; Escandon, P.; Vasini, B.; Nicholas, S.E.; Van, L.; Dang, D.H.; Cunningham, R.L.; Riaz, K.M. Anterior pituitary, sex hormones, and keratoconus: Beyond traditional targets. Prog. Retin. Eye Res. 2022, 88, 101016. [Google Scholar] [CrossRef]
- Lasagni Vitar, R.M.; Bonelli, F.; Rama, P.; Ferrari, G. Nutritional and Metabolic Imbalance in Keratoconus. Nutrients 2022, 14, 913. [Google Scholar] [CrossRef]
- Karamichos, D.; Zareian, R.; Guo, X.; Hutcheon, A.E.; Ruberti, J.W.; Zieske, J.D. Novel in Vitro Model for Keratoconus Disease. J. Funct. Biomater. 2012, 3, 760–775. [Google Scholar] [CrossRef]
- Atilano, S.R.; Lee, D.H.; Fukuhara, P.S.; Chwa, M.; Nesburn, A.B.; Udar, N.; Kenney, M.C. Corneal Oxidative Damage in Keratoconus Cells due to Decreased Oxidant Elimination from Modified Expression Levels of SOD Enzymes, PRDX6, SCARA3, CPSF3, and FOXM1. J. Ophthalmic Vis. Res. 2019, 14, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ismail, S.; McGhee, J.J.; Wadhwa, H.; Noord, N.; van der Werf, B.; Sherwin, T. Differences in sphere-forming cells from keratoconic and normal corneal tissue: Implications for keratoconus pathogenesis. Exp. Eye Res. 2021, 202, 108301. [Google Scholar] [CrossRef]
- Morishige, N.; Magome, K.; Ueno, A.; Matsui, T.A.; Nishida, T. Relations Among Corneal Curvature, Thickness, and Volume in Keratoconus as Evaluated by Anterior Segment-Optical Coherence Tomography. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3794–3802. [Google Scholar] [CrossRef]
- Wojakowska, A.; Pietrowska, M.; Widlak, P.; Dobrowolski, D.; Wylęgała, E.; Tarnawska, D. Metabolomic Signature Discriminates Normal Human Cornea from Keratoconus-A Pilot GC/MS Study. Molecules 2020, 25, 2933. [Google Scholar] [CrossRef]
- McKay, T.B.; Sarker-Nag, A.; Lyon, D.; Asara, J.M.; Karamichos, D. Quercetin modulates keratoconus metabolism in vitro. Cell Biochem. Funct. 2015, 33, 341–350. [Google Scholar] [CrossRef]
- McKay, T.B.; Lyon, D.; Sarker-Nag, A.; Priyadarsini, S.; Asara, J.M.; Karamichos, D. Quercetin attenuates lactate production and extracellular matrix secretion in keratoconus. Sci. Rep. 2015, 5, 9003. [Google Scholar] [CrossRef] [PubMed]
- McKay, T.B.; Kivanany, P.B.; Nicholas, S.E.; Nag, O.K.; Elliott, M.H.; Petroll, W.M.; Karamichos, D. Quercetin Decreases Corneal Haze In Vivo and Influences Gene Expression of TGF-Beta Mediators In Vitro. Metabolites 2022, 12, 3673. [Google Scholar] [CrossRef]
- Sağlik, A.; Koyuncu, İ.; Soydan, A.; Sağlik, F.; Gönel, A. Tear Organic Acid Analysis After Corneal Collagen Crosslinking in Keratoconus. Eye Contact Lens 2020, 46 (Suppl. S2), S122–S128. [Google Scholar] [CrossRef] [PubMed]
- Sharif, R.; Sejersen, H.; Frank, G.; Hjortdal, J.; Karamichos, D. Effects of collagen cross-linking on the keratoconus metabolic network. Eye 2018, 32, 1271–1281. [Google Scholar] [CrossRef]
- Stachon, T.; Kolev, K.; Flaskó, Z.; Seitz, B.; Langenbucher, A.; Szentmáry, N. Arginase activity, urea, and hydroxyproline concentration are reduced in keratoconus keratocytes. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 91–97. [Google Scholar] [CrossRef]
- Foster, J.W.; Shinde, V.; Soiberman, U.S.; Sathe, G.; Liu, S.; Wan, J.; Qian, J.; Dauoud, Y.; Pandey, A.; Jun, A.S.; et al. Integrated Stress Response and Decreased ECM in Cultured Stromal Cells From Keratoconus Corneas. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2977–2986. [Google Scholar] [CrossRef]
- McKay, T.B.; Priyadarsini, S.; Rowsey, T.; Karamichos, D. Arginine Supplementation Promotes Extracellular Matrix and Metabolic Changes in Keratoconus. Cells 2021, 10, 2076. [Google Scholar] [CrossRef]
- Wojcik, K.A.; Kaminska, A.; Blasiak, J.; Szaflik, J.; Szaflik, J.P. Oxidative stress in the pathogenesis of keratoconus and Fuchs endothelial corneal dystrophy. Int. J. Mol. Sci. 2013, 14, 19294–19308. [Google Scholar] [CrossRef]
- Buddi, R.; Lin, B.; Atilano, S.R.; Zorapapel, N.C.; Kenney, M.C.; Brown, D.J. Evidence of oxidative stress in human corneal diseases. J. Histochem. Cytochem. 2002, 50, 341–351. [Google Scholar] [CrossRef]
- Ong Tone, S.; Kocaba, V.; Böhm, M.; Wylegala, A.; White, T.L.; Jurkunas, U.V. Fuchs endothelial corneal dystrophy: The vicious cycle of Fuchs pathogenesis. Prog. Retin. Eye Res. 2021, 80, 100863. [Google Scholar] [CrossRef]
- Lovicu, F.J.; McAvoy, J.W. Growth factor regulation of lens development. Dev. Biol. 2005, 280, 1–14. [Google Scholar] [CrossRef]
- Foster, A.; Resnikoff, S. The impact of Vision 2020 on global blindness. Eye 2005, 19, 1133–1135. [Google Scholar] [CrossRef]
- Gupta, V.B.; Rajagopala, M.; Ravishankar, B. Etiopathogenesis of cataract: An appraisal. Indian J. Ophthalmol. 2014, 62, 103–110. [Google Scholar] [CrossRef]
- Wormstone, I.M. Posterior capsule opacification: A cell biological perspective. Exp. Eye Res. 2002, 74, 337–347. [Google Scholar] [CrossRef]
- Wride, M.A. Lens fibre cell differentiation and organelle loss: Many paths lead to clarity. Philos. Trans. R Soc. Lond. B Biol. Sci. 2011, 366, 1219–1233. [Google Scholar] [CrossRef]
- Liu, P.; Edassery, S.L.; Ali, L.; Thomson, B.R.; Savas, J.N.; Jin, J. Long-lived metabolic enzymes in the crystalline lens identified by pulse-labeling of mice and mass spectrometry. eLife 2019, 8, e50170. [Google Scholar] [CrossRef] [PubMed]
- McNulty, R.; Wang, H.; Mathias, R.T.; Ortwerth, B.J.; Truscott, R.J.; Bassnett, S. Regulation of tissue oxygen levels in the mammalian lens. J. Physiol. 2004, 559, 883–898. [Google Scholar] [CrossRef]
- Lim, J.C.; Grey, A.C.; Zahraei, A.; Donaldson, P.J. Age-dependent changes in glutathione metabolism pathways in the lens: New insights into therapeutic strategies to prevent cataract formation-A review. Clin. Exp. Ophthalmol. 2020, 48, 1031–1042. [Google Scholar] [CrossRef]
- Zhao, H.; Brown, P.H.; Magone, M.T.; Schuck, P. The molecular refractive function of lens γ-crystallins. J. Mol. Biol. 2011, 411, 680–699. [Google Scholar] [CrossRef]
- Mills-Henry, I.A.; Thol, S.L.; Kosinski-Collins, M.S.; Serebryany, E.; King, J.A. Kinetic stability of long-lived human lens γ-crystallins and their isolated double Greek key domains. Biophys. J. 2019, 117, 269–280. [Google Scholar] [CrossRef]
- Truscott, R.J.W.; Friedrich, M.G. Molecular Processes Implicated in Human Age-Related Nuclear Cataract. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5007–5021. [Google Scholar] [CrossRef]
- Schey, K.L.; Wang, Z.; Friedrich, M.G.; Truscott, R.J.W. New insights into the mechanisms of age-related protein-protein crosslinking in the human lens. Exp. Eye Res. 2021, 209, 108679. [Google Scholar] [CrossRef]
- Gakamsky, A.; Duncan, R.R.; Howarth, N.M.; Dhillon, B.; Buttenschön, K.K.; Daly, D.J.; Gakamsky, D. Tryptophan and Non-Tryptophan Fluorescence of the Eye Lens Proteins Provides Diagnostics of Cataract at the Molecular Level. Sci. Rep. 2017, 7, 40375. [Google Scholar] [CrossRef]
- Modenese, A.; Gobba, F. Cataract frequency and subtypes involved in workers assessed for their solar radiation exposure: A systematic review. Acta Ophthalmol. 2018, 96, 779–788. [Google Scholar] [CrossRef]
- Cekic, O. Effect of cigarette smoking on copper, lead, and cadmium accumulation in human lens. Br. J. Ophthalmol. 1998, 82, 186–188. [Google Scholar] [CrossRef]
- Srikanthan, D.; Bateman, O.A.; Purkiss, A.G.; Slingsby, C. Sulfur in human crystallins. Exp. Eye Res. 2004, 79, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Truscott, R.J. Age-related nuclear cataract-oxidation is the key. Exp. Eye Res. 2005, 80, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J. Disulphide cross-linked protein of high molecular weight in human cataractous lens. Exp. Eye Res. 1973, 17, 377–383. [Google Scholar] [CrossRef]
- Spector, A.; Roy, D. Disulfide-linked high molecular weight protein associated with human cataract. Proc. Natl. Acad. Sci. USA 1978, 75, 3244–3248. [Google Scholar] [CrossRef]
- Truscott, R.J.; Augusteyn, R.C. Oxidative changes in human lens proteins during senile nuclear cataract formation. Biochim. Biophys. Acta 1977, 492, 43–52. [Google Scholar] [CrossRef]
- Hains, P.G.; Truscott, R.J. Post-translational modifications in the nuclear region of young, aged, and cataract human lenses. J. Proteome Res. 2007, 6, 3935–3943. [Google Scholar] [CrossRef]
- Lampi, K.J.; Ma, Z.; Hanson, S.R.; Azuma, M.; Shih, M.; Shearer, T.R.; Smith, D.L.; Smith, J.B.; David, L.L. Age-related changes in human lens crystallins identified by two-dimensional electrophoresis and mass spectrometry. Exp. Eye Res. 1998, 67, 31–43. [Google Scholar] [CrossRef]
- Norton-Baker, B.; Mehrabi, P.; Kwok, A.O.; Roskamp, K.W.; Rocha, M.A.; Sprague-Piercy, M.A.; von Stetten, D.; Miller, R.J.D.; Martin, R.W. Deamidation of the human eye lens protein gammaS-crystallin accelerates oxidative aging. Structure 2022, 30, 763–776.e4. [Google Scholar] [CrossRef]
- Vetter, C.J.; Thorn, D.C.; Wheeler, S.G.; Mundorff, C.; Halverson, K.; Wales, T.E.; Shinde, U.; Engen, J.R.; David, L.L.; Carver, J.A.; et al. Cumulative deamidations of the major lens protein γS-crystallin increase its aggregation during unfolding and oxidation. Protein Sci. 2020, 29, 1945–1963. [Google Scholar] [CrossRef]
- Zhao, W.J.; Yan, Y.B. Increasing susceptibility to oxidative stress by cataract-causing crystallin mutations. Int. J. Biol. Macromol. 2018, 108, 665–673. [Google Scholar] [CrossRef]
- Serebryany, E.; Woodard, J.C.; Adkar, B.V.; Shabab, M.; King, J.A.; Shakhnovich, E.I. An internal disulfide locks a misfolded aggregation-prone intermediate in cataract-linked mutants of human γD-crystallin. J. Biol. Chem. 2016, 291, 19172–19183. [Google Scholar] [CrossRef]
- Serebryany, E.; Thorn, D.C.; Quintanar, L. Redox chemistry of lens crystallins: A system of cysteines. Exp. Eye Res. 2021, 211, 108707. [Google Scholar] [CrossRef] [PubMed]
- Michael, R.; Bron, A.J. The ageing lens and cataract: A model of normal and pathological ageing. Philos. Trans. R Soc. Lond. B Biol. Sci. 2011, 366, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Giblin, F.J. Glutathione: A vital lens antioxidant. J. Ocul. Pharmacol. Ther. 2000, 16, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.F. Redox regulation in the lens. Prog. Retin. Eye Res. 2003, 22, 657–682. [Google Scholar] [CrossRef]
- Reddan, J.R.; Giblin, F.J.; Kadry, R.; Leverenz, V.R.; Pena, J.T.; Dziedzic, D.C. Protection from oxidative insult in glutathione depleted lens epithelial cells. Exp. Eye Res. 1999, 68, 117–127. [Google Scholar] [CrossRef]
- Sweeney, M.H.; Truscott, R.J. An impediment to glutathione diffusion in older normal human lenses: A possible precondition for nuclear cataract. Exp. Eye Res. 1998, 67, 587–595. [Google Scholar] [CrossRef]
- Fan, X.; Liu, X.; Hao, S.; Wang, B.; Robinson, M.L.; Monnier, V.M. The LEGSKO mouse: A mouse model of age-related nuclear cataract based on genetic suppression of lens glutathione synthesis. PLoS ONE 2012, 7, e50832. [Google Scholar] [CrossRef]
- Fan, X.; Zhou, S.; Wang, B.; Hom, G.; Guo, M.; Li, B.; Yang, J.; Vaysburg, D.; Monnier, V.M. Evidence of highly conserved β-crystallin disulfidome that can be mimicked by in vitro oxidation in age-related human cataract and glutathione depleted mouse lens. Mol. Cell. Proteom. 2015, 14, 3211–3223. [Google Scholar] [CrossRef]
- Wang, B.; Hom, G.; Zhou, S.; Guo, M.; Li, B.; Yang, J.; Monnier, V.M.; Fan, X. The oxidized thiol proteome in aging and cataractous mouse and human lens revealed by ICAT labeling. Aging Cell 2017, 16, 244–261. [Google Scholar] [CrossRef]
- Roskamp, K.W.; Azim, S.; Kassier, G.; Norton-Baker, B.; Sprague-Piercy, M.A.; Miller, R.J.D.; Martin, R.W. Human γS-crystallin-copper binding helps buffer against aggregation caused by oxidative damage. Biochemistry 2020, 59, 2371–2385. [Google Scholar] [CrossRef] [PubMed]
- Serebryany, E.; Yu, S.; Trauger, S.A.; Budnik, B.; Shakhnovich, E.I. Dynamic disulfide exchange in a crystallin protein in the human eye lens promotes cataract-associated aggregation. J. Biol. Chem. 2018, 293, 17997–18009. [Google Scholar] [CrossRef] [PubMed]
- Skouri-Panet, F.; Bonnete, F.; Prat, K.; Bateman, O.A.; Lubsen, N.H.; Tardieu, A. Lens crystallins and oxidation: The special case of γS. Biophys. Chem. 2001, 89, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Thorn, D.C.; Grosas, A.B.; Mabbitt, P.D.; Ray, N.J.; Jackson, C.J.; Carver, J.A. The structure and stability of the disulfide-linked γS-crystallin dimer provide insight into oxidation products associated with lens cataract formation. J. Mol. Biol. 2019, 431, 483–497. [Google Scholar] [CrossRef]
- Quinlan, R.A.; Hogg, P.J. γ-Crystallin redox-detox in the lens. J. Biol. Chem. 2018, 293, 18010–18011. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, H.; Alany, R.G.; Pierscionek, B. Age-related cataract and drug therapy: Opportunities and challenges for topical antioxidant delivery to the lens. J. Pharm. Pharmacol. 2015, 67, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Serebryany, E.; Chowdhury, S.; Woods, C.N.; Thorn, D.C.; Watson, N.E.; McClelland, A.A.; Klevit, R.E.; Shakhnovich, E.I. A native chemical chaperone in the human eye lens. eLife 2022, 11, e76923. [Google Scholar] [CrossRef]
- Fan, X.; Monnier, V.M.; Whitson, J. Lens glutathione homeostasis: Discrepancies and gaps in knowledge standing in the way of novel therapeutic approaches. Exp. Eye Res. 2017, 156, 103–111. [Google Scholar] [CrossRef]
- Shu, D.Y.; Ong, K.; Lovicu, F.J. Histopathology of Subcapsular Cataract in a Patient with Atopic Dermatitis. Optom. Vis. Sci. 2017, 94, 270–276. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Enhanced EGF receptor-signaling potentiates TGFβ-induced lens epithelial-mesenchymal transition. Exp. Eye Res. 2019, 185, 107693. [Google Scholar] [CrossRef]
- Shu, D.Y.; Wojciechowski, M.; Lovicu, F.J. ERK1/2-mediated EGFR-signaling is required for TGFβ-induced lens epithelial-mesenchymal transition. Exp. Eye Res. 2019, 178, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Wojciechowski, M.C.; Lovicu, F.J. Bone Morphogenetic Protein-7 Suppresses TGFβ2-Induced Epithelial-Mesenchymal Transition in the Lens: Implications for Cataract Prevention. Investig. Ophthalmol. Vis. Sci. 2017, 58, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, M.C.; Mahmutovic, L.; Shu, D.Y.; Lovicu, F.J. ERK1/2 signaling is required for the initiation but not progression of TGFβ-induced lens epithelial to mesenchymal transition (EMT). Exp. Eye Res. 2017, 159, 98–113. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Insights into Bone Morphogenetic Protein-(BMP-) Signaling in Ocular Lens Biology and Pathology. Cells 2021, 10, 2604. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Ng, K.; Wishart, T.F.L.; Chui, J.; Lundmark, M.; Flokis, M.; Lovicu, F.J. Contrasting roles for BMP-4 and ventromorphins (BMP agonists) in TGFbeta-induced lens EMT. Exp. Eye Res. 2021, 206, 108546. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, H.; Chen, Y.; Li, G.; Bin, Y.; Zhou, X. Moderate oxidative stress promotes epithelial-mesenchymal transition in the lens epithelial cells via the TGF-beta/Smad and Wnt/beta-catenin pathways. Mol. Cell. Biochem. 2021, 476, 1631–1642. [Google Scholar] [CrossRef]
- Chamberlain, C.G.; Mansfield, K.J.; Cerra, A. Glutathione and catalase suppress TGFbeta-induced cataract-related changes in cultured rat lenses and lens epithelial explants. Mol. Vis. 2009, 15, 895–905. [Google Scholar]
- Wei, Z.; Caty, J.; Whitson, J.; Zhang, A.D.; Srinivasagan, R.; Kavanagh, T.J.; Yan, H.; Fan, X. Reduced Glutathione Level Promotes Epithelial-Mesenchymal Transition in Lens Epithelial Cells via a Wnt/beta-Catenin-Mediated Pathway: Relevance for Cataract Therapy. Am. J. Pathol. 2017, 187, 2399–2412. [Google Scholar] [CrossRef]
- Wang, R.; Li, J.; Zhang, X.; Zhang, X.; Zhang, X.; Zhu, Y.; Chen, C.; Liu, Z.; Wu, X.; Wang, D.; et al. Extracellular vesicles promote epithelial-to-mesenchymal transition of lens epithelial cells under oxidative stress. Exp. Cell Res. 2021, 398, 112362. [Google Scholar] [CrossRef]
- Thompson, B.; Davidson, E.A.; Chen, Y.; Orlicky, D.J.; Thompson, D.C.; Vasiliou, V. Oxidative stress induces inflammation of lens cells and triggers immune surveillance of ocular tissues. Chem. Biol. Interact. 2022, 355, 109804. [Google Scholar] [CrossRef]
- Das, S.J.; Lovicu, F.J.; Collinson, E.J. Nox4 Plays a Role in TGF-beta-Dependent Lens Epithelial to Mesenchymal Transition. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3665–3673. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Wikstrom, P.; Walum, E.; Lovicu, F.J. A novel NADPH oxidase inhibitor targeting Nox4 in TGFbeta-induced lens epithelial to mesenchymal transition. Exp. Eye Res. 2019, 185, 107692. [Google Scholar] [CrossRef] [PubMed]
- McCaa, C.S. The eye and visual nervous system: Anatomy, physiology and toxicology. Environ. Health Perspect. 1982, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Masland, R.H. The fundamental plan of the retina. Nat. Neurosci. 2001, 4, 877–886. [Google Scholar] [CrossRef]
- Gollisch, T.; Meister, M. Eye smarter than scientists believed: Neural computations in circuits of the retina. Neuron 2010, 65, 150–164. [Google Scholar] [CrossRef]
- Masland, R.H. The neuronal organization of the retina. Neuron 2012, 76, 266–280. [Google Scholar] [CrossRef]
- Molday, R.S.; Moritz, O.L. Photoreceptors at a glance. J. Cell Sci. 2015, 128, 4039–4045. [Google Scholar] [CrossRef]
- Demb, J.B.; Singer, J.H. Functional Circuitry of the Retina. Annu. Rev. Vis. Sci. 2015, 1, 263–289. [Google Scholar] [CrossRef]
- Flores-Herr, N.; Protti, D.A.; Wässle, H. Synaptic currents generating the inhibitory surround of ganglion cells in the mammalian retina. J. Neurosci. 2001, 21, 4852–4863. [Google Scholar] [CrossRef]
- Bruesch, S.R.; Arey, L.B. The number of myelinated and unmyelinated fibres in the optic nerve of vertebrates. J. Comp. Neurol. 1943, 77, 631. [Google Scholar] [CrossRef]
- Garron, L.K. The ultrastructure of the retinal pigment epithelium with observations on the choriocapillaris and Bruch’s membrane. Trans. Am. Ophthalmol. Soc. 1963, 61, 545. [Google Scholar]
- Gu, X.; Neric, N.J.; Crabb, J.S.; Crabb, J.W.; Bhattacharya, S.K.; Rayborn, M.E.; Hollyfield, J.G.; Bonilha, V.L. Age-related changes in the retinal pigment epithelium (RPE). PLoS ONE 2012, 7, e38673. [Google Scholar] [CrossRef] [PubMed]
- Kur, J.; Newman, E.A.; Chan-Ling, T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Retin. Eye Res. 2012, 31, 377–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, P.; Kijlstra, A. Distribution, markers, and functions of retinal microglia. Ocul. Immunol. Inflamm. 2002, 10, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, A.; Bringmann, A. Role of Purines in Müller Glia. J. Ocul. Pharmacol. Ther. 2016, 32, 518–533. [Google Scholar] [CrossRef]
- Rathnasamy, G.; Foulds, W.S.; Ling, E.-A.; Kaur, C. Retinal microglia—A key player in healthy and diseased retina. Prog. Neurobiol. 2019, 173, 18–40. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. Glia of the human retina. Glia 2020, 68, 768–796. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Provis, J.M. Development of the Primate Retinal Vasculature. Prog. Retin. Eye Res. 2001, 20, 799–821. [Google Scholar] [CrossRef]
- Bessis, A.; Béchade, C.; Bernard, D.; Roumier, A. Microglial control of neuronal death and synaptic properties. Glia 2007, 55, 233–238. [Google Scholar] [CrossRef]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Büssow, H. The astrocytes in the retina and optic nerve head of mammals: A special glia for the ganglion cell axons. Cell Tissue Res. 1980, 206, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Distler, C.; Weigel, H.; Hoffmann, K.P. Glia cells of the monkey retina. I. Astrocytes. J. Comp. Neurol. 1993, 333, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Holländer, H.; Makarov, F.; Dreher, Z.; van Driel, D.; Chan-Ling, T.; Stone, J. Structure of the macroglia of the retina: Sharing and division of labour between astrocytes and Müller cells. J. Comp. Neurol. 1991, 313, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, H.; Seo, M.S.; Ozaki, K.; Yamada, H.; Yamada, E.; Okamoto, N.; Hofmann, F.; Wood, J.M.; Campochiaro, P.A. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am. J. Pathol. 2000, 156, 697–707. [Google Scholar] [CrossRef]
- Stone, J.; Itin, A.; Alon, T.; Pe’er, J.; Gnessin, H.; Chan-Ling, T.; Keshet, E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 1995, 15, 4738–4747. [Google Scholar] [CrossRef]
- Newman, E.; Reichenbach, A. The Müller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312. [Google Scholar] [CrossRef]
- Agte, S.; Junek, S.; Matthias, S.; Ulbricht, E.; Erdmann, I.; Wurm, A.; Schild, D.; Käs, J.A.; Reichenbach, A. Müller glial cell-provided cellular light guidance through the vital guinea-pig retina. Biophys. J. 2011, 101, 2611–2619. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. New functions of Müller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef]
- Huster, D.; Reichenbach, A.; Reichelt, W. The glutathione content of retinal Müller (glial) cells: Effect of pathological conditions. Neurochem. Int. 2000, 36, 461–469. [Google Scholar] [CrossRef]
- Coughlin, B.A.; Feenstra, D.J.; Mohr, S. Müller cells and diabetic retinopathy. Vis. Res. 2017, 139, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.E.; McCollum, G.W.; Cousins, D.B.; Penn, J.S. Linoleic Acid is a Diabetes-relevant Stimulator of Retinal Inflammation in Human Retinal Muller Cells and Microvascular Endothelial Cells. J. Diabetes Metab. 2016, 7, 718. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Y.; Cringle, S.J.; Yu, P.K.; Balaratnasingam, C.; Mehnert, A.; Sarunic, M.V.; An, D.; Su, E.N. Retinal capillary perfusion: Spatial and temporal heterogeneity. Prog. Retin. Eye Res. 2019, 70, 23–54. [Google Scholar] [CrossRef] [PubMed]
- Archer, D.B.; Gardiner, T.A.; Stitt, A.W. Functional anatomy, fine structure and basic pathology of the retinal vasculature. In Retinal Vascular Disease; Springer: Berlin/Heidelberg, Germany, 2007; pp. 3–23. [Google Scholar]
- Giannaccini, M.; Usai, A.; Chiellini, F.; Guadagni, V.; Andreazzoli, M.; Ori, M.; Pasqualetti, M.; Dente, L.; Raffa, V. Neurotrophin-conjugated nanoparticles prevent retina damage induced by oxidative stress. Cell. Mol. Life Sci. 2018, 75, 1255–1267. [Google Scholar] [CrossRef]
- Caprara, C.; Thiersch, M.; Lange, C.; Joly, S.; Samardzija, M.; Grimm, C. HIF1A is essential for the development of the intermediate plexus of the retinal vasculature. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.; Zhang, M.; Hwang, T.; Bailey, S.; Wilson, D.; Jia, Y.; Huang, D. Detailed vascular anatomy of the human retina by projection-resolved optical coherence tomography angiography. Sci. Rep. 2017, 7, 42201. [Google Scholar] [CrossRef]
- Moran, E.P.; Wang, Z.; Chen, J.; Sapieha, P.; Smith, L.E.; Ma, J.-X. Neurovascular cross talk in diabetic retinopathy: Pathophysiological roles and therapeutic implications. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H738–H749. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia–neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef]
- Wu, S.M.; Maple, B.R. Amino acid neurotransmitters in the retina: A functional overview. Vis. Res. 1998, 38, 1371–1384. [Google Scholar] [CrossRef]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal vasculature development in health and disease. Prog. Retin. Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef]
- Ames III, A. Energy requirements of CNS cells as related to their function and to their vulnerability to ischemia: A commentary based on studies on retina. Can. J. Physiol. Pharmacol. 1992, 70, S158–S164. [Google Scholar] [CrossRef] [PubMed]
- Maenhaut, N.; Boussery, K.; Delaey, C.; Van de Voorde, J. Control of retinal arterial tone by a paracrine retinal relaxing factor. Microcirculation 2007, 14, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.L.; Downie, L.E.; Ly, A.; Ward, M.M.; Batcha, A.H.; Puthussery, T.; Yee, P.; Hatzopoulos, K.M. A review of the role of glial cells in understanding retinal disease. Clin. Exp. Optom. 2008, 91, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, J.K.; Ruemmler, R.; Hartmann, E.K.; Ziebart, A.; Ludwig, M.; Patzak, A.; Xia, N.; Li, H.; Pfeiffer, N.; Gericke, A. Responses of retinal arterioles and ciliary arteries in pigs with acute respiratory distress syndrome (ARDS). Exp. Eye Res. 2019, 184, 152–161. [Google Scholar] [CrossRef]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef]
- Bellezza, I. Oxidative stress in age-related macular degeneration: Nrf2 as therapeutic target. Front. Pharmacol. 2018, 9, 1280. [Google Scholar] [CrossRef]
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2022, 9, 62–79. [Google Scholar] [CrossRef]
- Ehrlich, R.; Harris, A.; Kheradiya, N.S.; Winston, D.M.; Ciulla, T.A.; Wirostko, B. Age-related macular degeneration and the aging eye. Clin. Interv. Aging 2008, 3, 473. [Google Scholar]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef]
- Rickman, C.B.; Farsiu, S.; Toth, C.A.; Klingeborn, M. Dry age-related macular degeneration: Mechanisms, therapeutic targets, and imaging. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF68–ORSF80. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, S.D.; Pierce, K. Wet age-related macular degeneration (Wet AMD). In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Young, R.W. Pathophysiology of age-related macular degeneration. Surv. Ophthalmol. 1987, 31, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Beatty, S.; Koh, H.-H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Abokyi, S.; To, C.-H.; Lam, T.T.; Tse, D.Y. Central role of oxidative stress in age-related macular degeneration: Evidence from a review of the molecular mechanisms and animal models. Oxid. Med. Cell. Longev. 2020, 2020, 7901270. [Google Scholar] [CrossRef]
- Glickman, R.D. Ultraviolet phototoxicity to the retina. Eye Contact Lens 2011, 37, 196–205. [Google Scholar] [CrossRef]
- Totan, Y.; Yağcı, R.; Bardak, Y.; Özyurt, H.; Kendir, F.; Yılmaz, G.; Şahin, Ş.; Şahin Tığ, U. Oxidative macromolecular damage in age-related macular degeneration. Curr. Eye Res. 2009, 34, 1089–1093. [Google Scholar] [CrossRef]
- Bhutto, I.A.; Baba, T.; Merges, C.; McLeod, D.S.; Lutty, G.A. Low nitric oxide synthases (NOSs) in eyes with age-related macular degeneration (AMD). Exp. Eye Res. 2010, 90, 155–167. [Google Scholar] [CrossRef]
- Ruan, Y.; Jiang, S.; Gericke, A. Age-related macular degeneration: Role of oxidative stress and blood vessels. Int. J. Mol. Sci. 2021, 22, 1296. [Google Scholar] [CrossRef]
- Toma, C.; De Cillà, S.; Palumbo, A.; Garhwal, D.P.; Grossini, E. Oxidative and nitrosative stress in age-related macular degeneration: A review of their role in different stages of disease. Antioxidants 2021, 10, 653. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med. 2016, 14, 344. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Pang, J.-J.; Renganathan, K.; Zhan, X.; Crabb, J.W.; Kim, S.R.; Sparrow, J.R.; Hauswirth, W.W.; Lewin, A.S. SOD2 knockdown mouse model of early AMD. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4407–4420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-related retinopathy in NRF2-deficient mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Frank, S.I.; Fitch, T.C.; Karg, M.M.; Butcher, E.R.; Nnuji-John, E.; Kim, L.A.; Saint-Geniez, M. Dimethyl Fumarate Blocks Tumor Necrosis Factor-Alpha-Driven Inflammation and Metabolic Rewiring in the Retinal Pigment Epithelium. Front. Mol. Neurosci. 2022, 15, 896786. [Google Scholar] [CrossRef]
- Shimizu, H.; Takayama, K.; Yamada, K.; Suzumura, A.; Sato, T.; Nishio, Y.; Ito, M.; Ushida, H.; Nishiguchi, K.M.; Takeuchi, M.; et al. Dimethyl Fumarate Protects Retinal Pigment Epithelium from Blue Light-Induced Oxidative Damage via the Nrf2 Pathway. Antioxidants 2023, 12, 45. [Google Scholar] [CrossRef]
- Ishibashi, T.; Murata, T.; Hangai, M.; Nagai, R.; Horiuchi, S.; Lopez, P.F.; Hinton, D.R.; Ryan, S.J. Advanced glycation end products in age-related macular degeneration. Arch. Ophthalmol. 1998, 116, 1629–1632. [Google Scholar] [CrossRef]
- Kandarakis, S.A.; Piperi, C.; Topouzis, F.; Papavassiliou, A.G. Emerging role of advanced glycation-end products (AGEs) in the pathobiology of eye diseases. Prog. Retin. Eye Res. 2014, 42, 85–102. [Google Scholar] [CrossRef]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef]
- Ardeljan, D.; Tuo, J.; Chan, C.-C. Carboxyethylpyrrole plasma biomarkers in age-related macular degeneration. Drugs Future 2011, 36, 712. [Google Scholar] [CrossRef]
- Balaiya, S.; Murthy, R.K.; Brar, V.S.; Chalam, K.V. Evaluation of ultraviolet light toxicity on cultured retinal pigment epithelial and retinal ganglion cells. Clin. Ophthalmol. 2010, 4, 33. [Google Scholar] [PubMed]
- Tong, Y.; Zhang, Z.; Wang, S. Role of Mitochondria in Retinal Pigment Epithelial Aging and Degeneration. Front. Aging 2022, 3, 926627. [Google Scholar] [CrossRef]
- Alaimo, A.; Liñares, G.G.; Bujjamer, J.M.; Gorojod, R.M.; Alcon, S.P.; Martínez, J.H.; Baldessari, A.; Grecco, H.E.; Kotler, M.L. Toxicity of blue led light and A2E is associated to mitochondrial dynamics impairment in ARPE-19 cells: Implications for age-related macular degeneration. Arch. Toxicol. 2019, 93, 1401–1415. [Google Scholar] [CrossRef]
- Tao, J.-X.; Zhou, W.-C.; Zhu, X.-G. Mitochondria as potential targets and initiators of the blue light hazard to the retina. Oxid. Med. Cell. Longev. 2019, 2019, 6435364. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Różanowska, M.; Różanowski, B. Retinal photodamage. J. Photochem. Photobiol. B Biol. 2001, 64, 144–161. [Google Scholar] [CrossRef] [PubMed]
- Godley, B.F.; Shamsi, F.A.; Liang, F.-Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J. Biol. Chem. 2005, 280, 21061–21066. [Google Scholar] [CrossRef]
- Lowe, G.M.; Vlismas, K.; Young, A.J. Carotenoids as prooxidants? Mol. Asp. Med. 2003, 24, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schumann, U.; Ader, M.; Brunssen, C.; Bramke, S.; Morawietz, H.; Funk, R.H. Stress reaction in outer segments of photoreceptors after blue light irradiation. PLoS ONE 2013, 8, e71570. [Google Scholar] [CrossRef]
- Thornton, J.; Edwards, R.; Mitchell, P.; Harrison, R.; Buchan, I.; Kelly, S.P. Smoking and age-related macular degeneration: A review of association. Eye 2005, 19, 935–944. [Google Scholar] [CrossRef]
- Altuntaş, I.; Dane, Ş.; Gümüştekin, K. Effects of cigarette smoking on lipid peroxidation. J. Basic Clin. Physiol. Pharmacol. 2002, 13, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Boulton, M.E. Consequences of oxidative stress in age-related macular degeneration. Mol. Asp. Med. 2012, 33, 399–417. [Google Scholar] [CrossRef]
- Seddon, J.M.; Gensler, G.; Milton, R.C.; Klein, M.L.; Rifai, N. Association between C-reactive protein and age-related macular degeneration. JAMA 2004, 291, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.W.; Jung, I.H.; Park, E.Y.; Kim, K.H.; Kim, K.; Yeom, J.; Jung, J.; Lee, S.W. Radiation-induced C-reactive protein triggers apoptosis of vascular smooth muscle cells through ROS interfering with the STAT3/Ref-1 complex. J. Cell. Mol. Med. 2022, 26, 2104–2118. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, J.; Liu, J.; Sun, L.; Zhao, H.; Lu, Y.; Wang, J.; Li, J. C-reactive protein promotes adhesion of monocytes to endothelial cells via NADPH oxidase-mediated oxidative stress. J. Cell. Biochem. 2012, 113, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Hadzic, S.; Roxlau, E.T.; Fuehler, B.; Janise-Libawski, A.; Wimmer, T.; Lei, B.; Li, S.-W.; Weissmann, N.; Stieger, K. Retinal tissue develops an inflammatory reaction to tobacco smoke and electronic cigarette vapor in mice. J. Mol. Med. 2021, 99, 1459–1469. [Google Scholar] [CrossRef]
- Delcourt, C.; Delyfer, M.-N.; Rougier, M.-B.; Amouyel, P.; Colin, J.; Le Goff, M.; Malet, F.; Dartigues, J.-F.; Lambert, J.-C.; Korobelnik, J.-F. Associations of complement factor H and smoking with early age-related macular degeneration: The ALIENOR study. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5955–5962. [Google Scholar] [CrossRef]
- Mitter, S.K.; Rao, H.V.; Qi, X.; Cai, J.; Sugrue, A.; Dunn, W.A.; Grant, M.B.; Boulton, M.E. Autophagy in the retina: A potential role in age-related macular degeneration. Retin. Degener. Dis. 2012, 723, 83–90. [Google Scholar]
- Hyttinen, J.; Blasiak, J.; Tavi, P.; Kaarniranta, K. Therapeutic potential of PGC-1α in age-related macular degeneration (AMD)–the involvement of mitochondrial quality control, autophagy, and antioxidant response. Expert Opin. Ther. Targets 2021, 25, 773–785. [Google Scholar] [CrossRef]
- Chen, C.-L.; Chen, Y.-H.; Liang, C.-M.; Tai, M.-C.; Lu, D.-W.; Chen, J.-T. Glucosamine-induced autophagy through AMPK–mTOR pathway attenuates lipofuscin-like autofluorescence in human retinal pigment epithelial cells in vitro. Int. J. Mol. Sci. 2018, 19, 1416. [Google Scholar] [CrossRef]
- Sethna, S.; Scott, P.A.; Giese, A.P.; Duncan, T.; Jian, X.; Riazuddin, S.; Randazzo, P.A.; Redmond, T.M.; Bernstein, S.L.; Riazuddin, S. CIB2 regulates mTORC1 signaling and is essential for autophagy and visual function. Nat. Commun. 2021, 12, 3906. [Google Scholar] [CrossRef] [PubMed]
- Riazi-Esfahani, M.; Kuppermann, B.D.; Kenney, M.C. The role of mitochondria in AMD: Current knowledge and future applications. J. Ophthalmic Vis. Res. 2017, 12, 424. [Google Scholar] [PubMed]
- Sridevi Gurubaran, I.; Viiri, J.; Koskela, A.; Hyttinen, J.M.; Paterno, J.J.; Kis, G.; Antal, M.; Urtti, A.; Kauppinen, A.; Felszeghy, S. Mitophagy in the retinal pigment epithelium of dry age-related macular degeneration investigated in the NFE2l2/PGC-1α-/-mouse model. Int. J. Mol. Sci. 2020, 21, 1976. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Viiri, J.; Kaarniranta, K.; Błasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Cell. Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Bao, X.-L.; Cong, Y.-Y.; Fan, B.; Li, G.-Y. Autophagy in age-related macular degeneration: A regulatory mechanism of oxidative stress. Oxid. Med. Cell. Longev. 2020, 2020, 2896036. [Google Scholar] [CrossRef]
- Zhao, T.; Guo, X.; Sun, Y. Iron accumulation and lipid peroxidation in the aging retina: Implication of ferroptosis in age-related macular degeneration. Aging Dis. 2021, 12, 529. [Google Scholar] [CrossRef]
- Gnana-Prakasam, J.P.; Tawfik, A.; Romej, M.; Ananth, S.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Iron-mediated retinal degeneration in haemojuvelin-knockout mice. Biochem. J. 2012, 441, 599–608. [Google Scholar] [CrossRef]
- Dunaief, J.L. Iron induced oxidative damage as a potential factor in age-related macular degeneration: The Cogan Lecture. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4660–4664. [Google Scholar] [CrossRef]
- Ashok, A.; Chaudhary, S.; Wise, A.S.; Rana, N.A.; McDonald, D.; Kritikos, A.E.; Lindner, E.; Singh, N. Release of Iron-Loaded Ferritin in Sodium Iodate-Induced Model of Age Related Macular Degeneration: An In-Vitro and In-Vivo Study. Antioxidants 2021, 10, 1253. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.-F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Ying, G.; Grunwald, J.E.; Fine, S.L.; Jaffe, G.J. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011, 364, 1897–1908. [Google Scholar] [PubMed]
- Mimouni, M.; Meshi, A.; Vainer, I.; Gershoni, A.; Koren, T.; Geffen, N.; Nemet, A.Y.; Segal, O. Bevacizumab dosing every 2 weeks for neovascular age-related macular degeneration refractory to monthly dosing. Jpn. J. Ophthalmol. 2018, 62, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Damico, L.; Shams, N.; Lowman, H.; Kim, R. Development of ranibizumab, an anti–vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 2006, 26, 859–870. [Google Scholar] [CrossRef]
- Markham, A. Brolucizumab: First approval. Drugs 2019, 79, 1997–2000. [Google Scholar] [CrossRef]
- FDA. Drug Administration Center for Drug Evaluation and Research; Application Number 204042Orig1s000: Summary Review; Food and Drug Administration: Silver Spring, MD, USA, 2017. [Google Scholar]
- Wang, Y.; Wang, V.; Chan, C. The role of anti-inflammatory agents in age-related macular degeneration (AMD) treatment. Eye 2011, 25, 127–139. [Google Scholar] [CrossRef]
- De Guimaraes, T.A.C.; Varela, M.D.; Georgiou, M.; Michaelides, M. Treatments for dry age-related macular degeneration: Therapeutic avenues, clinical trials and future directions. Br. J. Ophthalmol. 2022, 106, 297–304. [Google Scholar] [CrossRef]
- Liao, D.S.; Grossi, F.V.; El Mehdi, D.; Gerber, M.R.; Brown, D.M.; Heier, J.S.; Wykoff, C.C.; Singerman, L.J.; Abraham, P.; Grassmann, F. Complement C3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: A randomized phase 2 trial. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef]
- Potilinski, M.C.; Tate, P.S.; Lorenc, V.E.; Gallo, J.E. New insights into oxidative stress and immune mechanisms involved in age-related macular degeneration tackled by novel therapies. Neuropharmacology 2021, 188, 108513. [Google Scholar] [CrossRef]
- Dziedziak, J.; Kasarełło, K.; Cudnoch-Jędrzejewska, A. Dietary antioxidants in age-related macular degeneration and glaucoma. Antioxidants 2021, 10, 1743. [Google Scholar] [CrossRef]
- Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol. 2001, 119, 1417–1436. [Google Scholar] [CrossRef] [PubMed]
- Age-Related Eye Disease Study 2 (AREDS2) Research Group. Lutein+ zeaxanthin and omega-3 fatty acids for age-related macular degeneration: The Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA 2013, 309, 2005–2015. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Olson, C.G.; Euritt, C.P.; Koulen, P. Molecular Mechanisms Underlying the Therapeutic Role of Vitamin E in Age-Related Macular Degeneration. Front. Neurosci. 2022, 16, 890021. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Igarashi, K.; Uchihara, T.; Jishage, K.-i.; Tomita, H.; Inaba, A.; Li, Y.; Arita, M.; Suzuki, H.; Mizusawa, H. Delayed-onset ataxia in mice lacking α-tocopherol transfer protein: Model for neuronal degeneration caused by chronic oxidative stress. Proc. Natl. Acad. Sci. USA 2001, 98, 15185–15190. [Google Scholar] [CrossRef]
- Kirchhof, B. Strategies to influence PVR development. Graefe’s Arch. Clin. Exp. Ophthalmol. 2004, 242, 699–703. [Google Scholar] [CrossRef]
- Mudhar, H.S. A brief review of the histopathology of proliferative vitreoretinopathy (PVR). Eye 2020, 34, 246–250. [Google Scholar] [CrossRef]
- Tseng, W.; Cortez, R.T.; Ramirez, G.; Stinnett, S.; Jaffe, G.J. Prevalence and risk factors for proliferative vitreoretinopathy in eyes with rhegmatogenous retinal detachment but no previous vitreoretinal surgery. Am. J. Ophthalmol. 2004, 137, 1105–1115. [Google Scholar] [CrossRef]
- Scott, I.U.; Flynn, H.W., Jr.; Murray, T.G.; Feuer, W.J. Outcomes of surgery for retinal detachment associated with proliferative vitreoretinopathy using perfluoro-n-octane: A multicenter study. Am. J. Ophthalmol. 2003, 136, 454–463. [Google Scholar] [CrossRef]
- Verdejo, C.; Marco, P.; Renau-Piqueras, J.; Pinazo-Duran, M.D. Lipid peroxidation in proliferative vitreoretinopathies. Eye 1999, 13, 183–188. [Google Scholar] [CrossRef]
- Cederlund, M.; Ghosh, F.; Arner, K.; Andreasson, S.; Akerstrom, B. Vitreous levels of oxidative stress biomarkers and the radical-scavenger alpha1-microglobulin/A1M in human rhegmatogenous retinal detachment. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 725–732. [Google Scholar] [CrossRef]
- Maeno, A.; Suzuki, Y.; Adachi, K.; Takahashi, S.; Yokoi, Y.; Nakazawa, M. Characterization of the biological antioxidant potential in the vitreous fluid from patients with rhegmatogenous retinal detachment. Acta Ophthalmol. 2016, 94, e515–e516. [Google Scholar] [CrossRef] [PubMed]
- Pietras-Baczewska, A.; Nowomiejska, K.; Brzozowska, A.; Toro, M.D.; Załuska, W.; Sztanke, M.; Sztanke, K.; Rejdak, R. Antioxidant Status in the Vitreous of Eyes with Rhegmatogenous Retinal Detachment with and without Proliferative Vitreoretinopathy, Macular Hole and Epiretinal Membrane. Life 2021, 11, 453. [Google Scholar] [CrossRef]
- Cicik, E.; Tekin, H.; Akar, S.; Ekmekci, O.B.; Donma, O.; Koldas, L.; Ozkan, S. Interleukin-8, nitric oxide and glutathione status in proliferative vitreoretinopathy and proliferative diabetic retinopathy. Ophthalmic Res. 2003, 35, 251–255. [Google Scholar] [CrossRef]
- Augustin, A.J.; Spitznas, M.; Koch, F.; Grus, F.; Boker, T. Indicators of oxidative tissue damage and inflammatory activity in epiretinal membranes of proliferative diabetic retinopathy, proliferative vitreoretinopathy and macular pucker. Ger. J. Ophthalmol. 1995, 4, 47–51. [Google Scholar] [PubMed]
- Delgado-Tirado, S.; Amarnani, D.; Zhao, G.; Rossin, E.J.; Eliott, D.; Miller, J.B.; Greene, W.A.; Ramos, L.; Arevalo-Alquichire, S.; Leyton-Cifuentes, D.; et al. Topical delivery of a small molecule RUNX1 transcription factor inhibitor for the treatment of proliferative vitreoretinopathy. Sci. Rep. 2020, 10, 20554. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog. Retin. Eye Res. 2017, 60, 44–65. [Google Scholar] [CrossRef]
- Roybal, C.N.; Velez, G.; Toral, M.A.; Tsang, S.H.; Bassuk, A.G.; Mahajan, V.B. Personalized Proteomics in Proliferative Vitreoretinopathy Implicate Hematopoietic Cell Recruitment and mTOR as a Therapeutic Target. Am. J. Ophthalmol. 2018, 186, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.H.; Lee, J.-J.; Wu, P.-C.; Kuo, H.-K.; Kuo, Y.-H.; Huang, H.-M. Oxidative stress enhanced the transforming growth factor-β2-induced epithelial-mesenchymal transition through chemokine ligand 1 on ARPE-19 cell. Sci. Rep. 2020, 10, 4000. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, Y.S.; Kim, J.H.; Jang, H.Y.; Ly, D.D.; Das, R.; Park, K.S. Activation of ERK1/2-mTORC1-NOX4 mediates TGF-beta1-induced epithelial-mesenchymal transition and fibrosis in retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 2020, 529, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Shi, D.P.; Chu, W.J.; Yang, L.L.; Xu, H.F. LRG1 promotes epithelial-mesenchymal transition of retinal pigment epithelium cells by activating NOX4. Int. J. Ophthalmol. 2021, 14, 349–355. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, S.J.; Jo, D.H.; Park, K.S.; Kim, J.H. Blockade of mTORC1-NOX signaling pathway inhibits TGF-beta1-mediated senescence-like structural alterations of the retinal pigment epithelium. FASEB J. 2021, 35, e21403. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, J.; Wang, Q.; Xing, Y.; Tan, Z.; Kang, Q. Novel NADPH oxidase inhibitor VAS2870 suppresses TGF-beta-dependent epithelial-to-mesenchymal transition in retinal pigment epithelial cells. Int. J. Mol. Med. 2018, 42, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Wada, I.; Sreekumar, P.G.; Spee, C.; MacKay, A.J.; Ip, M.; Kannan, R. Mechanisms of Epithelial-Mesenchymal Transition and Prevention of Dispase-Induced PVR by Delivery of an Antioxidant alphaB Crystallin Peptide. Antioxidants 2022, 11, 2080. [Google Scholar] [CrossRef]
- Cai, W.; Yu, D.; Fan, J.; Liang, X.; Jin, H.; Liu, C.; Zhu, M.; Shen, T.; Zhang, R.; Hu, W.; et al. Quercetin inhibits transforming growth factor beta1-induced epithelial-mesenchymal transition in human retinal pigment epithelial cells via the Smad pathway. Drug Des. Devel. Ther. 2018, 12, 4149–4161. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.R.; Saint-Geniez, M. Suppression of PGC-1alpha Drives Metabolic Dysfunction in TGFbeta2-Induced EMT of Retinal Pigment Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 4701. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Zhang, Y.; Chen, J.; Wu, L.D.; Chen, M.L.; Chen, C.M.; Xu, Q.H. Artesunate inhibits the development of PVR by suppressing the TGF-beta/Smad signaling pathway. Exp. Eye Res. 2021, 213, 108859. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Frank, R.N. Diabetic Retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Lieth, E.; Barber, A.J.; Xu, B.; Dice, C.; Ratz, M.J.; Tanase, D.; Strother, J.M. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes 1998, 47, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Hombrebueno, J.R.; Chen, M.; Penalva, R.G.; Xu, H. Loss of synaptic connectivity, particularly in second order neurons is a key feature of diabetic retinal neuropathy in the Ins2Akita mouse. PLoS ONE 2014, 9, e97970. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef]
- Russell, J.W.; Sullivan, K.A.; Windebank, A.J.; Herrmann, D.N.; Feldman, E.L. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol. Dis. 1999, 6, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.-Y.; Green, W.R.; Tso, M.O.M. Microglial Activation in Human Diabetic Retinopathy. Arch. Ophthalmol. 2008, 126, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Diabetes, C.; Complications Trial Research, G.; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef]
- King, P.; Peacock, I.; Donnelly, R. The UK prospective diabetes study (UKPDS): Clinical and therapeutic implications for type 2 diabetes. Br. J. Clin. Pharmacol. 1999, 48, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.H.; Timothy Greenamyre, J. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 2013, 62, 111–120. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef]
- Kowluru, R.A. Effect of Reinstitution of Good Glycemic Control on Retinal Oxidative Stress and Nitrative Stress in Diabetic Rats. Diabetes 2003, 52, 818–823. [Google Scholar] [CrossRef]
- Rohowetz, L.J.; Kraus, J.G.; Koulen, P. Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina. Int. J. Mol. Sci. 2018, 19, 3362. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Medina, J.; Zanon-Moreno, V.; Pinazo-Duran, M.; Foulquie-Moreno, E.; Rubio-Velazquez, E.; Casaroli-Marano, R.; Del-Rio-Vellosillo, M. Oxidative Stress in Diabetic Retinopathy, 2nd ed.; Preedy, V.R., Ed.; Academic Press: Cambridge, MA, USA, 2020; p. 52. [Google Scholar]
- Field, M.G.; Yang, D.; Bian, Z.-M.; Petty, H.R.; Elner, V.M. Retinal flavoprotein fluorescence correlates with mitochondrial stress, apoptosis, and chemokine expression. Exp. Eye Res. 2011, 93, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Diseases Of The Mitochondrial DNA. Annu. Rev. Biochem. 1992, 61, 1175–1212. [Google Scholar] [CrossRef] [PubMed]
- Trumpower, B.L. The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J. Biol. Chem. 1990, 265, 11409–11412. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787. [Google Scholar] [CrossRef]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, D.; Ceriello, A.; Paolisso, G. Oxidative Stress and Diabetic Vascular Complications. Diabetes Care 1996, 19, 257–267. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Du, X.; Carratú, A.; Gerfen, G.J.; D’Apolito, M.; Giardino, I.; Rasola, A.; Marin, O.; Divakaruni, A.S.; Murphy, A.N.; et al. GLP-1 Cleavage Product Reverses Persistent ROS Generation After Transient Hyperglycemia by Disrupting an ROS-Generating Feedback Loop. Diabetes 2015, 64, 3273–3284. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.P.; Toro, A.L.; Barber, A.J.; Dennis, M.D. REDD1 Activates a ROS-Generating Feedback Loop in the Retina of Diabetic Mice REDD1 Promotes Oxidative Stress in Retina. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Mokdad, A.H.; Giles, W.H.; Brown, D.W. The Metabolic Syndrome and Antioxidant Concentrations. Diabetes 2003, 52, 2346–2352. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kern, T.S.; Engerman, R.L.; Armstrong, D. Abnormalities of Retinal Metabolism in Diabetes or Experimental Galactosemia. III. Effects of Antioxidants. Diabetes 1996, 45, 1233–1237. [Google Scholar] [CrossRef]
- Wohaieb, S.A.; Godin, D.V. Alterations in Free Radical Tissue-Defense Mechanisms in Streptozocin-Induced Diabetes in Rat: Effects of Insulin Treatment. Diabetes 1987, 36, 1014–1018. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Haskins, K.; Bradley, B.; Powers, K.; Fadok, V.; Flores, S.; Ling, X.; Pugazhenthi, S.; Reusch, J.; Kench, J. Oxidative Stress in Type 1 Diabetes. Ann. N. Y. Acad. Sci. 2003, 1005, 43–54. [Google Scholar] [CrossRef]
- Takahashi, T.; Harris, R.C. Role of endothelial nitric oxide synthase in diabetic nephropathy: Lessons from diabetic eNOS knockout mice. J. Diabetes Res. 2014, 2014, 590541. [Google Scholar] [CrossRef]
- Felaco, M.; Grilli, A.; De Lutiis, M.A.; Patruno, A.; Libertini, N.; Taccardi, A.A.; Di Napoli, P.; Di Giulio, C.; Barbacane, R.; Conti, P. Endothelial Nitric Oxide Synthase (eNOS) Expression and Localization in Healthy and Diabetic Rat Hearts. Ann. Clin. Lab. Sci. 2001, 31, 179–186. [Google Scholar]
- Carmo, A.; Cunha-Vaz, J.G.; Carvalho, A.P.; Lopes, M.C. Nitric Oxide Synthase Activity in Retinas from Non-Insulin-Dependent Diabetic Goto-Kakizaki Rats: Correlation with Blood–Retinal Barrier Permeability. Nitric Oxide 2000, 4, 590–596. [Google Scholar] [CrossRef]
- Martínez, M.C.; Andriantsitohaina, R. Reactive Nitrogen Species: Molecular Mechanisms and Potential Significance in Health and Disease. Antioxid. Redox. Signal. 2008, 11, 669–702. [Google Scholar] [CrossRef] [PubMed]
- Afanas’ev, I. Signaling of reactive oxygen and nitrogen species in Diabetes mellitus. Oxid. Med. Cell. Longev. 2010, 3, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Stockklauser-Färber, K.; Rösen, P. Generation of reactive oxygen intermediates, activation of NF-κB, and induction of apoptosis in human endothelial cells by glucose: Role of nitric oxide synthase? Free Radic. Biol. Med. 1999, 27, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Chough, E.; Daley, J.; Oates, P.; Tornheim, K.; Ruderman, N.B.; Keaney, J.F. Hyperglycemia increases endothelial superoxide that impairs smooth muscle cell Na+-K+-ATPase activity. Am. J. Physiol. Cell Physiol. 2002, 282, C560–C566. [Google Scholar] [CrossRef]
- Maritim, A.C.; Sanders, R.A.; Watkins Iii, J.B. Diabetes, oxidative stress, and antioxidants: A review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. [Google Scholar] [CrossRef]
- Golbidi, S.; Alireza Ebadi, S.; Laher, I. Antioxidants in the Treatment of Diabetes. Curr. Diabetes Rev. 2011, 7, 106–125. [Google Scholar] [CrossRef]
- Sheikh-Ali, M.; Chehade, J.M.; Mooradian, A.D. The Antioxidant Paradox in Diabetes Mellitus. Am. J. Ther. 2011, 18, 266–278. [Google Scholar] [CrossRef]
- Miller, W.P.; Sunilkumar, S.; Giordano, J.F.; Toro, A.L.; Barber, A.J.; Dennis, M.D. The stress response protein REDD1 promotes diabetes-induced oxidative stress in the retina by Keap1-independent Nrf2 degradation. J. Biol. Chem. 2020, 295, 7350–7361. [Google Scholar] [CrossRef]
- Marra, G.; Cotroneo, P.; Pitocco, D.; Manto, A.; Di Leo, M.A.S.; Ruotolo, V.; Caputo, S.; Giardina, B.; Ghirlanda, G.; Santini, S.A. Early Increase of Oxidative Stress and Reduced Antioxidant Defenses in Patients With Uncomplicated Type 1 Diabetes. Diabetes Care 2002, 25, 370–375. [Google Scholar] [CrossRef]
- Martín-Gallán, P.; Carrascosa, A.; Gussinye, M.; Domínguez, C. Estimation of lipoperoxidative damage and antioxidant status in diabetic children: Relationship with individual antioxidants. Free Radic. Res. 2005, 39, 933–942. [Google Scholar] [CrossRef]
- Bhatia, S.; Shukla, R.; Venkata Madhu, S.; Kaur Gambhir, J.; Madhava Prabhu, K. Antioxidant status, lipid peroxidation and nitric oxide end products in patients of type 2 diabetes mellitus with nephropathy. Clin. Biochem. 2003, 36, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, H.; Pan, H.; Zhou, X.; Ruan, Q.; Kong, D.; Chu, Z.; Li, H.; Huang, J.; Huang, X.; et al. Nrf2 Suppression Delays Diabetic Wound Healing Through Sustained Oxidative Stress and Inflammation. Front. Pharmacol. 2019, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chen, X.; Niu, C.; Huang, X.; An, N.; Sun, J.; Huang, S.; Ye, W.; Li, S.; Shen, Y.; et al. Baicalin alleviates hyperglycemia-induced endothelial impairment via Nrf2. J. Endocrinol. 2019, 240, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Shivarudrappa, A.H.; Ponesakki, G. Lutein reverses hyperglycemia-mediated blockage of Nrf2 translocation by modulating the activation of intracellular protein kinases in retinal pigment epithelial (ARPE-19) cells. J. Cell Commun. Signal. 2020, 14, 207–221. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Wong-Riley, M.T. Energy metabolism of the visual system. Eye Brain 2010, 2, 99–116. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Rinaldi, C.; Aragona, P.; Briuglia, S.; D’Ascola, A.; D’Angelo, R.; Sidoti, A. Stargardt Phenotype Associated With Two ELOVL4 Promoter Variants and ELOVL4 Downregulation: New Possible Perspective to Etiopathogenesis? Investig. Ophthalmol. Vis. Sci. 2018, 59, 843–857. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Abbas, S.N. Diabetes-Induced Mitochondrial Dysfunction in the Retina. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5327–5334. [Google Scholar] [CrossRef]
- Ellis, E.A.; Guberski, D.L.; Somogyi-Mann, M.; Grant, M.B. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/WOR diabetic rat. Free Radic. Biol. Med. 2000, 28, 91–101. [Google Scholar] [CrossRef]
- Miller, W.P.; Sha, C.M.; Sunilkumar, S.; Toro, A.L.; VanCleave, A.M.; Kimball, S.R.; Dokholyan, N.V.; Dennis, M.D. Activation of Disulfide Redox Switch in REDD1 Promotes Oxidative Stress Under Hyperglycemic Conditions. Diabetes 2022, 71, 2764–2776. [Google Scholar] [CrossRef]
- Zheng, L.; Du, Y.; Miller, C.; Gubitosi-Klug, R.A.; Kern, T.S.; Ball, S.; Berkowitz, B.A. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 2007, 50, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Smith, M.A.; Miller, C.M.; Kern, T.S. Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J. Neurochem. 2002, 80, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kanwar, M.; Kennedy, A. Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp. Diabetes Res. 2007, 2007, 21976. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Liang, H.L.; Wong-Riley, M.T. Transcriptional coupling of synaptic transmission and energy metabolism: Role of nuclear respiratory factor 1 in co-regulating neuronal nitric oxide synthase and cytochrome c oxidase genes in neurons. Biochim. Biophys. Acta 2009, 1793, 1604–1613. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Barber, A.J.; Antonetti, D.A.; LaNoue, K.F.; Robinson, K.A.; Buse, M.G.; Gardner, T.W. Excessive Hexosamines Block the Neuroprotective Effect of Insulin and Induce Apoptosis in Retinal Neurons. J. Biol. Chem. 2001, 276, 43748–43755. [Google Scholar] [CrossRef] [PubMed]
- Hangai, M.; He, S.; Hoffmann, S.; Lim, J.I.; Ryan, S.J.; Hinton, D.R. Sequential induction of angiogenic growth factors by TNF-alpha in choroidal endothelial cells. J. Neuroimmunol. 2006, 171, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Koppolu, P. Diabetes-induced activation of caspase-3 in retina: Effect of antioxidant therapy. Free Radic. Res. 2002, 36, 993–999. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Koppolu, P.; Chakrabarti, S.; Chen, S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic. Res. 2003, 37, 1169–1180. [Google Scholar] [CrossRef]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Müller cell changes in human diabetic retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef]
- Phipps, J.A.; Fletcher, E.L.; Vingrys, A.J. Paired-flash identification of rod and cone dysfunction in the diabetic rat. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4592–4600. [Google Scholar] [CrossRef]
- Ali, T.K.; Al-Gayyar, M.M.; Matragoon, S.; Pillai, B.A.; Abdelsaid, M.A.; Nussbaum, J.J.; El-Remessy, A.B. Diabetes-induced peroxynitrite impairs the balance of pro-nerve growth factor and nerve growth factor, and causes neurovascular injury. Diabetologia 2011, 54, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Mohammad, G.; Kowluru, Q.Z.; Renu, A. Diabetic Retinopathy, Superoxide Damage and Antioxidants. Curr. Pharm. Biotechnol. 2011, 12, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yanoff, M.; Jian, B.; He, Z. Altered mRNA Levels of Antioxidant Enzymes in Pre-Apoptotic Pericytes From Human Diabetic Retinas. Cell. Mol. Biol. 1999, 45, 59–66. [Google Scholar] [PubMed]
- Zhong, Q.; Mishra, M.; Kowluru, R.A. Transcription Factor Nrf2-Mediated Antioxidant Defense System in the Development of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3941–3948. [Google Scholar] [CrossRef]
- Yue, K.K.M.; Chung, W.-S.; Leung, A.W.N.; Cheng, C.H.K. Redox changes precede the occurrence of oxidative stress in eyes and aorta, but not in kidneys of diabetic rats. Life Sci. 2003, 73, 2557–2570. [Google Scholar] [CrossRef]
- Miranda, M.; Muriach, M.; Roma, J.; Bosch-Morell, F.; Genovés, J.M.; Barcia, J.; Araiz, J.; Díaz-Llopis, M.; Romero, F.J. Oxidative Stress In a Model of Experimental Diabetic Retinopathy: The Utility of Peroxynitrite Scavengers. Arch. Soc. Esp. Oftalmol. 2006, 81, 27–32. [Google Scholar] [CrossRef]
- Chen, B.H.; Jiang, D.Y.; Tang, L.S. Advanced glycation end-products induce apoptosis involving the signaling pathways of oxidative stress in bovine retinal pericytes. Life Sci. 2006, 79, 1040–1048. [Google Scholar] [CrossRef]
- Kowluru, R.A. Effect of advanced glycation end products on accelerated apoptosis of retinal capillary cells under in vitro conditions. Life Sci. 2005, 76, 1051–1060. [Google Scholar] [CrossRef]
- Millen, A.E.; Klein, R.; Folsom, A.R.; Stevens, J.; Palta, M.; Mares, J.A. Relation between intake of vitamins C and E and risk of diabetic retinopathy in the Atherosclerosis Risk in Communities Study. Am. J. Clin. Nutr. 2004, 79, 865–873. [Google Scholar] [CrossRef]
- Mayer-Davis, E.J.; Bell, R.A.; Reboussin, B.A.; Rushing, J.; Marshall, J.A.; Hamman, R.F. Antioxidant nutrient intake and diabetic retinopathy: The San Luis Valley Diabetes Study. Ophthalmology 1998, 105, 2264–2270. [Google Scholar] [CrossRef]
- Millen, A.E.; Gruber, M.; Klein, R.; Klein, B.E.; Palta, M.; Mares, J.A. Relations of serum ascorbic acid and alpha-tocopherol to diabetic retinopathy in the Third National Health and Nutrition Examination Survey. Am. J. Epidemiol. 2003, 158, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Coleman, A.L.; Miglior, S. Risk factors for glaucoma onset and progression. Surv. Ophthalmol. 2008, 53 (Suppl. S1), S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.K.; Kee, C. Population-based glaucoma prevalence studies in Asians. Surv. Ophthalmol. 2014, 59, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Esporcatte, B.L.; Tavares, I.M. Normal-tension glaucoma: An update. Arq. Bras. Oftalmol. 2016, 79, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Evelson, P.A.; Llesuy, S.F. Oxidative stress markers in aqueous humor of glaucoma patients. Am. J. Ophthalmol. 2004, 137, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Kaidzu, S.; Takai, Y.; Ohira, A. Correlation between Systemic Oxidative Stress and Intraocular Pressure Level. PLoS ONE 2015, 10, e0133582. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef]
- Casson, R.J.; Chidlow, G.; Wood, J.P.M. Estimate of the adenosine triphosphate requirement of human retinal ganglion cells. Clin. Exp. Ophthalmol. 2019, 47, 683–684. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog. Retin. Eye Res. 2021, 83, 100916. [Google Scholar] [CrossRef]
- Tsai, J. Should we measure (and treat) ocular perfusion pressure in glaucoma patients? Glaucoma Today 2009, 5, 31–35. [Google Scholar]
- Zhao, J.; Wang, S.; Zhong, W.; Yang, B.; Sun, L.; Zheng, Y. Oxidative stress in the trabecular meshwork (Review). Int. J. Mol. Med. 2016, 38, 995–1002. [Google Scholar] [CrossRef]
- Mozaffarieh, M.; Grieshaber, M.C.; Flammer, J. Oxygen and blood flow: Players in the pathogenesis of glaucoma. Mol. Vis. 2008, 14, 224–233. [Google Scholar]
- Iyama, T.; Wilson, D.M., III. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Cuchra, M.; Markiewicz, L.; Mucha, B.; Pytel, D.; Szymanek, K.; Szemraj, J.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. The role of base excision repair in the development of primary open angle glaucoma in the Polish population. Mutat. Res. 2015, 778, 26–40. [Google Scholar] [CrossRef]
- Sacca, S.C.; Pascotto, A.; Camicione, P.; Capris, P.; Izzotti, A. Oxidative DNA damage in the human trabecular meshwork: Clinical correlation in patients with primary open-angle glaucoma. Arch. Ophthalmol. 2005, 123, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, K.; Dada, R.; Dada, T. Oxidative DNA damage and reduced expression of DNA repair genes: Role in primary open angle glaucoma (POAG). Ophthalmic Genet. 2017, 38, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Caballero, M.; Liton, P.B.; Epstein, D.L.; Gonzalez, P. Proteasome inhibition by chronic oxidative stress in human trabecular meshwork cells. Biochem. Biophys. Res. Commun. 2003, 308, 346–352. [Google Scholar] [CrossRef]
- Fatma, N.; Kubo, E.; Toris, C.B.; Stamer, W.D.; Camras, C.B.; Singh, D.P. PRDX6 attenuates oxidative stress- and TGFbeta-induced abnormalities of human trabecular meshwork cells. Free Radic. Res. 2009, 43, 783–795. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, P.G.; Hinton, D.R.; Kannan, R. The Emerging Role of Senescence in Ocular Disease. Oxid. Med. Cell Longev. 2020, 2020, 2583601. [Google Scholar] [CrossRef] [PubMed]
- Flammer, J.; Konieczka, K.; Flammer, A.J. The primary vascular dysregulation syndrome: Implications for eye diseases. EPMA J. 2013, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B(3) modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.H.; Romano, G.L.; Amato, R.; Porciatti, V. Nicotinamide-Rich Diet in DBA/2J Mice Preserves Retinal Ganglion Cell Metabolic Function as Assessed by PERG Adaptation to Flicker. Nutrients 2020, 12, 1910. [Google Scholar] [CrossRef] [PubMed]
- Kouassi Nzoughet, J.; Chao de la Barca, J.M.; Guehlouz, K.; Leruez, S.; Coulbault, L.; Allouche, S.; Bocca, C.; Muller, J.; Amati-Bonneau, P.; Gohier, P.; et al. Nicotinamide Deficiency in Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2509–2514. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D. Oxidative stress: An evolving definition. Fac. Rev. 2021, 10, 13. [Google Scholar] [CrossRef]
- Canizales, L.; Rodriguez, L.; Rivera, C.; Martinez, A.; Mendez, F.; Castillo, A. Low-level expression of SOD1 in peripheral blood samples of patients diagnosed with primary open-angle glaucoma. Biomark. Med. 2016, 10, 1218–1223. [Google Scholar] [CrossRef]
- Hashizume, K.; Hirasawa, M.; Imamura, Y.; Noda, S.; Shimizu, T.; Shinoda, K.; Kurihara, T.; Noda, K.; Ozawa, Y.; Ishida, S.; et al. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am. J. Pathol. 2008, 172, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Mitochondria: Are they the seat of senescence? Aging Cell 2004, 3, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Chidlow, G.; Wood, J.P.M.; Casson, R.J. Investigations into Hypoxia and Oxidative Stress at the Optic Nerve Head in a Rat Model of Glaucoma. Front. Neurosci. 2017, 11, 478. [Google Scholar] [CrossRef] [PubMed]
- Fan Gaskin, J.C.; Shah, M.H.; Chan, E.C. Oxidative Stress and the Role of NADPH Oxidase in Glaucoma. Antioxidants 2021, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef]
- Song, H.; Vijayasarathy, C.; Zeng, Y.; Marangoni, D.; Bush, R.A.; Wu, Z.; Sieving, P.A. NADPH Oxidase Contributes to Photoreceptor Degeneration in Constitutively Active RAC1 Mice. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2864–2875. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. Nitric oxide, mitochondria, and cell death. IUBMB Life 2001, 52, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Durango, R.; Fernandez-Martinez, A.; Garcia-Feijoo, J.; Castillo, A.; de la Casa, J.M.; Garcia-Bueno, B.; Perez-Nievas, B.G.; Fernandez-Cruz, A.; Leza, J.C. Expression of nitrotyrosine and oxidative consequences in the trabecular meshwork of patients with primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci. 2008, 49, 2506–2511. [Google Scholar] [CrossRef]
- Ghanem, A.A.; Elewa, A.M.; Arafa, L.F. Endothelin-1 and nitric oxide levels in patients with glaucoma. Ophthalmic Res. 2011, 46, 98–102. [Google Scholar] [CrossRef]
- Noske, W.; Hensen, J.; Wiederholt, M. Endothelin-like immunoreactivity in aqueous humor of patients with primary open-angle glaucoma and cataract. Graefe’s Arch. Clin. Exp. Ophthalmol. 1997, 235, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Choritz, L.; Machert, M.; Thieme, H. Correlation of endothelin-1 concentration in aqueous humor with intraocular pressure in primary open angle and pseudoexfoliation glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7336–7342. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; Calkins, D.J. The Neurovascular Unit in Glaucomatous Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Jansen, E.H.; Ruskovska, T. Comparative Analysis of Serum (Anti)oxidative Status Parsmall a, Cyrillicmeters in Healthy Persons. Int. J. Mol. Sci. 2013, 14, 6106–6115. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Kondkar, A.A.; Mousa, A.; Osman, E.A.; Al-Obeidan, S.A. Decreased total antioxidants in patients with primary open angle glaucoma. Curr. Eye Res. 2013, 38, 959–964. [Google Scholar] [CrossRef]
- Ruskovska, T.; Jansen, E.H.; Antarorov, R. Evaluation of assays for measurement of serum (anti)oxidants in hemodialysis patients. BioMed Res. Int. 2014, 2014, 843157. [Google Scholar] [CrossRef]
- Tanito, M.; Kaidzu, S.; Takai, Y.; Ohira, A. Association between systemic oxidative stress and visual field damage in open-angle glaucoma. Sci. Rep. 2016, 6, 25792. [Google Scholar] [CrossRef]
- Kilic, R.; Bayraktar, A.C.; Bayraktar, S.; Kurt, A.; Kavutcu, M. Evaluation of Serum Superoxide Dismutase Activity, Malondialdehyde, and Zinc and Copper Levels in Patients With Keratoconus. Cornea 2016, 35, 1512–1515. [Google Scholar] [CrossRef]
- Nucci, C.; Di Pierro, D.; Varesi, C.; Ciuffoletti, E.; Russo, R.; Gentile, R.; Cedrone, C.; Pinazo Duran, M.D.; Coletta, M.; Mancino, R. Increased malondialdehyde concentration and reduced total antioxidant capacity in aqueous humor and blood samples from patients with glaucoma. Mol. Vis. 2013, 19, 1841–1846. [Google Scholar]
- Mousa, A.; Kondkar, A.A.; Al-Obeidan, S.A.; Azad, T.A.; Sultan, T.; Osman, E.; Abu-Amero, K.K. Association of total antioxidants level with glaucoma type and severity. Saudi Med. J. 2015, 36, 671–677. [Google Scholar] [CrossRef]
- Bosco, A.; Steele, M.R.; Vetter, M.L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011, 519, 599–620. [Google Scholar] [CrossRef]
- Tribble, J.R.; Harder, J.M.; Williams, P.A.; John, S.W.M. Ocular hypertension suppresses homeostatic gene expression in optic nerve head microglia of DBA/2 J mice. Mol. Brain 2020, 13, 81. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, J.; Wax, M.B. Heat shock proteins, immunity and glaucoma. Brain Res. Bull. 2004, 62, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 2018, 9, 3209. [Google Scholar] [CrossRef]
- Tsai, T.; Grotegut, P.; Reinehr, S.; Joachim, S.C. Role of Heat Shock Proteins in Glaucoma. Int. J. Mol. Sci. 2019, 20, 5160. [Google Scholar] [CrossRef]
- Soto, I.; Howell, G.R. The complex role of neuroinflammation in glaucoma. Cold Spring Harb. Perspect. Med. 2014, 4, a017269. [Google Scholar] [CrossRef]
- Chen, F.; Pandey, D.; Chadli, A.; Catravas, J.D.; Chen, T.; Fulton, D.J. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid. Redox Signal. 2011, 14, 2107–2119. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Cho, K.S.; Thee, E.F.; Jager, M.J.; Chen, D.F. Neuroinflammation and microglia in glaucoma: Time for a paradigm shift. J. Neurosci. Res. 2019, 97, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; Lopez-Cuenca, I.; Rojas, P.; Trivino, A.; Ramirez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Guo, Y.; Shrestha, M.; Sun, D.; Gregory-Ksander, M.; Jakobs, T.C. Microglia depletion exacerbates retinal ganglion cell loss in a mouse model of glaucoma. Exp. Eye Res. 2022, 225, 109273. [Google Scholar] [CrossRef]
- Tanaka-Gonome, T.; Xie, Y.; Yamauchi, K.; Maeda-Monai, N.; Tanabu, R.; Kudo, T.; Nakazawa, M. The protective effect of astaxanthin on the ganglion cell complex in glutamate/aspartate transporter deficient mice, a model of normal tension glaucoma, analyzed by spectral domain-optical coherence tomography. Biochem. Biophys. Rep. 2020, 23, 100777. [Google Scholar] [CrossRef]
- Angel Aillegas, N.; Tartara, L.I.; Caballero, G.; Campana, V.; Allemandi, D.A.; Palma, S.D. Antioxidant status in rabbit aqueous humor after instillation of ascorbyl laurate-based nanostructures. Pharmacol. Rep. 2019, 71, 794–797. [Google Scholar] [CrossRef]
- Harris, A.; Gross, J.; Moore, N.; Do, T.; Huang, A.; Gama, W.; Siesky, B. The effects of antioxidants on ocular blood flow in patients with glaucoma. Acta Ophthalmol. 2018, 96, e237–e241. [Google Scholar] [CrossRef]
- Ozates, S.; Elgin, K.U.; Yilmaz, N.S.; Demirel, O.O.; Sen, E.; Yilmazbas, P. Evaluation of oxidative stress in pseudo-exfoliative glaucoma patients treated with and without topical coenzyme Q10 and vitamin E. Eur. J. Ophthalmol. 2019, 29, 196–201. [Google Scholar] [CrossRef]
- Romeo Villadoniga, S.; Rodriguez Garcia, E.; Sagastagoia Epelde, O.; Alvarez Diaz, M.D.; Domingo Pedrol, J.C. Effects of Oral Supplementation with Docosahexaenoic Acid (DHA) plus Antioxidants in Pseudoexfoliative Glaucoma: A 6-Month Open-Label Randomized Trial. J. Ophthalmol. 2018, 2018, 8259371. [Google Scholar] [CrossRef]
- Sinclair, D.A.; LaPlante, M.D. Lifespan: Why We Age—And Why We Don’t Have To; Simon and Schuster: New York, NY, USA, 2019. [Google Scholar]
- Barzilai, N. Age Later: Health Span, Life Span, and the New Science of Longevity; St. Martin’s Press: New York, NY, USA, 2020. [Google Scholar]
- Mitnitski, A.B.; Graham, J.E.; Mogilner, A.J.; Rockwood, K. Frailty, fitness and late-life mortality in relation to chronological and biological age. BMC Geriatr. 2002, 2, 1. [Google Scholar] [CrossRef]
- Schultz, M.B.; Kane, A.E.; Mitchell, S.J.; MacArthur, M.R.; Warner, E.; Vogel, D.S.; Mitchell, J.R.; Howlett, S.E.; Bonkowski, M.S.; Sinclair, D.A. Age and life expectancy clocks based on machine learning analysis of mouse frailty. Nat. Commun. 2020, 11, 4618. [Google Scholar] [CrossRef]
- Rowe, J.W.; Kahn, R.L. Successful aging. Gerontologist 1997, 37, 433–440. [Google Scholar] [CrossRef]
- Sinha, M.; Jang, Y.C.; Oh, J.; Khong, D.; Wu, E.Y.; Manohar, R.; Miller, C.; Regalado, S.G.; Loffredo, F.S.; Pancoast, J.R. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014, 344, 649–652. [Google Scholar] [CrossRef]
- Katsimpardi, L.; Litterman, N.K.; Schein, P.A.; Miller, C.M.; Loffredo, F.S.; Wojtkiewicz, G.R.; Chen, J.W.; Lee, R.T.; Wagers, A.J.; Rubin, L.L. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 2014, 344, 630–634. [Google Scholar] [CrossRef]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Bordone, L.; Guarente, L. Calorie restriction, SIRT1 and metabolism: Understanding longevity. Nat. Rev. Mol. Cell Biol. 2005, 6, 298–305. [Google Scholar] [CrossRef]
- Garay, R.P. Investigational drugs and nutrients for human longevity. Recent clinical trials registered in ClinicalTrials.gov and clinicaltrialsregister.eu. Expert Opin. Investig. Drugs 2021, 30, 749–758. [Google Scholar] [CrossRef]
- Bailey, C.J.; Turner, R.C. Metformin. N. Engl. J. Med. 1996, 334, 574–579. [Google Scholar] [CrossRef]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef]
- Hurley, D.J.; Irnaten, M.; O’Brien, C. Metformin and Glaucoma—Review of Anti-Fibrotic Processes and Bioenergetics. Cells 2021, 10, 2131. [Google Scholar] [CrossRef]
- Brown, E.E.; Lewin, A.S.; Ash, J.D. Mitochondria: Potential targets for protection in age-related macular degeneration. Retin. Degener. Dis. 2018, 1074, 11–17. [Google Scholar]
- Amin, S.V.; Khanna, S.; Parvar, S.P.; Shaw, L.T.; Dao, D.; Hariprasad, S.M.; Skondra, D. Metformin and retinal diseases in preclinical and clinical studies: Insights and review of literature. Exp. Biol. Med. 2022, 247, 317–329. [Google Scholar] [CrossRef]
- Romdhoniyyah, D.F.; Harding, S.P.; Cheyne, C.P.; Beare, N.A. Metformin, a potential role in age-related macular degeneration: A systematic review and meta-analysis. Ophthalmol. Ther. 2021, 10, 245–260. [Google Scholar] [CrossRef]
- Onyango, P.; Celic, I.; McCaffery, J.M.; Boeke, J.D.; Feinberg, A.P. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. USA 2002, 99, 13653–13658. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.; Liu, T.; Hwang, Y.J.; Hyeon, S.J.; Im, H.; Lee, K.; Alvarez, V.E.; McKee, A.C.; Um, S.-J.; et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 2018, 17, e12679. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Wang, J.; Ghena, N.; Zhao, Q.; Perone, I.; King, T.M.; Veech, R.L.; Gorospe, M.; Wan, R.; Mattson, M.P. SIRT3 Haploinsufficiency Aggravates Loss of GABAergic Interneurons and Neuronal Network Hyperexcitability in an Alzheimer’s Disease Model. J. Neurosci. 2020, 40, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Bhawal, R.; Xu, H.; Chen, H.; Anderson, E.T.; Haroutunian, V.; Cross, A.C.; Zhang, S.; Gibson, G.E. The human brain acetylome reveals that decreased acetylation of mitochondrial proteins associates with Alzheimer’s disease. J. Neurochem. 2021, 158, 282–296. [Google Scholar] [CrossRef]
- Tyagi, A.; Nguyen, C.U.; Chong, T.; Michel, C.R.; Fritz, K.S.; Reisdorph, N.; Knaub, L.; Reusch, J.E.B.; Pugazhenthi, S. SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain. Sci. Rep. 2018, 8, 17547. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Y.-M.; Huang, H.; Chen, C.; Wan, J.; Ma, L.-H.; Sun, Y.-Y.; Miao, H.-H.; Wu, Y.-Q. Sirtuin 3 protects against anesthesia/surgery-induced cognitive decline in aged mice by suppressing hippocampal neuroinflammation. J. Neuroinflamm. 2021, 18, 41. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Forman, H.J.; Fukuto, J.M.; Torres, M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol. Cell Physiol. 2004, 287, C246–C256. [Google Scholar] [CrossRef]
- Torres, M. Mitogen-activated protein kinase pathways in redox signaling. Front. Biosci. 2003, 8, d369–d391. [Google Scholar] [CrossRef]
- Alvarez-Barrios, A.; Alvarez, L.; Garcia, M.; Artime, E.; Pereiro, R.; Gonzalez-Iglesias, H. Antioxidant Defenses in the Human Eye: A Focus on Metallothioneins. Antioxidants 2021, 10, 89. [Google Scholar] [CrossRef] [PubMed]




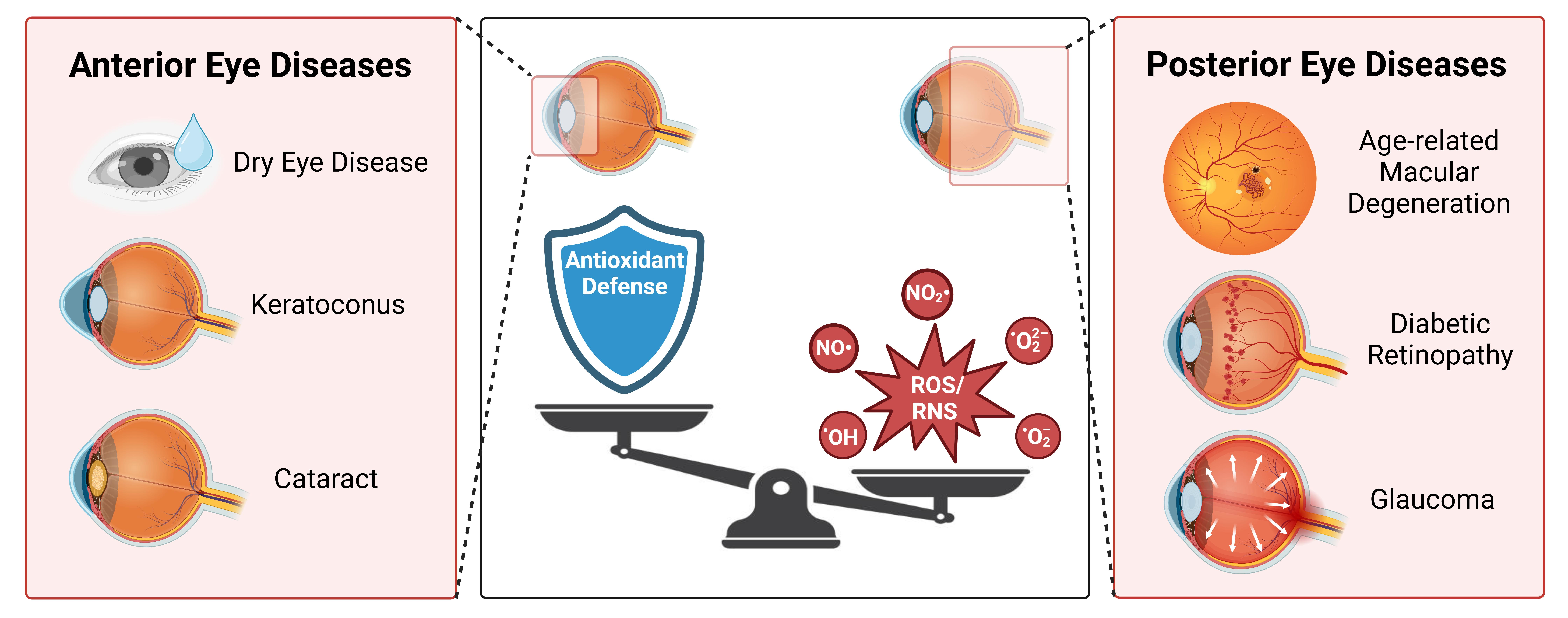{kind=link}
{kind=link}
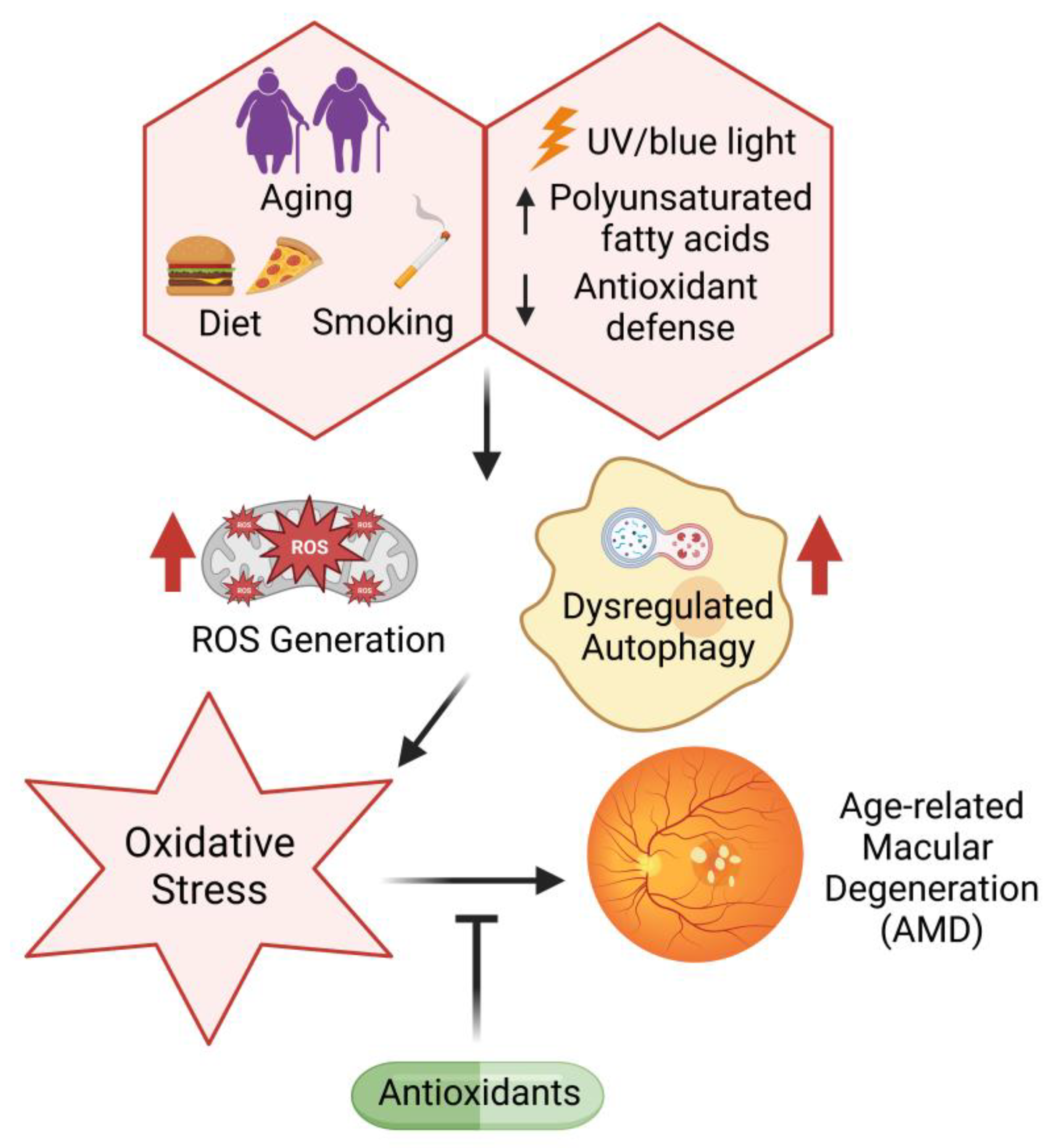{kind=link}
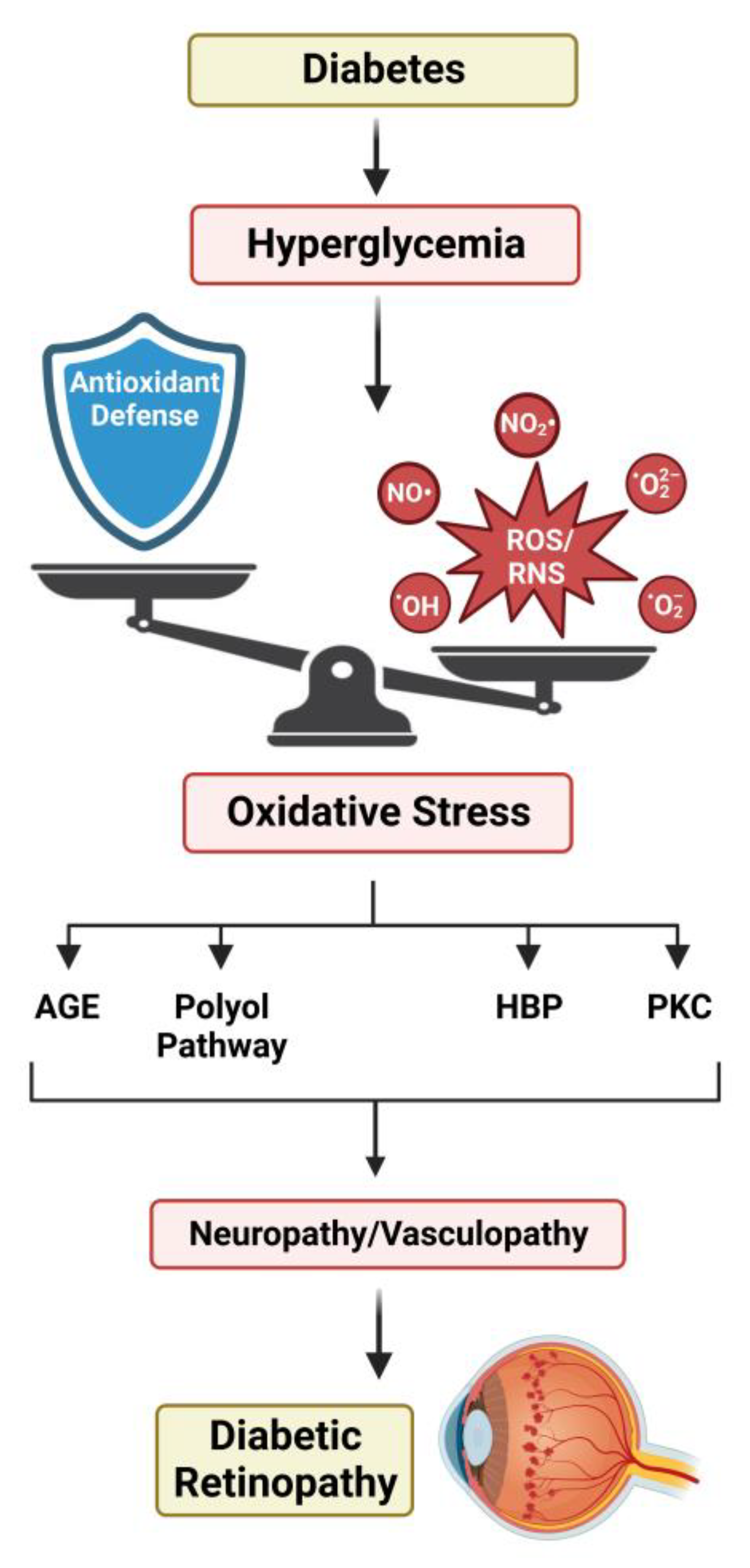{kind=link}
| Chemical Species | Source(s) | Downstream Reaction(s) | Ref. |
|---|---|---|---|
| Superoxide (O2·−) | The reaction of O2 with enzymes in the electron transport chain in the mitochondria generated via a single-electron transfer; enzymatic and non-enzymatic biosynthetic pathways; produced by neutrophils | Reacts with O2·− and H2O to generate H2O2 and O2 | [13,14,15] |
| Hydroxyl radical (·OH) | The reaction of H2O2 with iron or copper (Fenton reaction); may also be generated as a byproduct of the exposure of water molecules to ionizing radiation | Reacts with deoxyguanosine residues of DNA to form 8-hydroxy-2-deoxyguanosine; also reacts with deoxycytidine and deoxyadenosine, among others | [16,17] |
| Hydrogen peroxide (H2O2) | The reaction of O2·− molecules mediated via superoxide dismutase and non-enzymatically; may also be generated as a byproduct of normal catalytic oxidative processes mediated via oxidases | May be converted by myeloperoxidase or other enzymes containing Fe2+ or react with UV light to form hydroxyl radical (OH·); involved in downstream signaling pathways, such as platelet-derived growth factor signaling | [18,19] |
| Malondialdehyde (MDA) (CH2(CHO)2) | Produced by lipid peroxidation of polyunsaturated fatty acids | Reacts with deoxyguanosine of DNA to form 8-hydroxy-2-deoxyguanosine; also reacts with deoxyadenosine residues; may also react with lysine residues on proteins to form secondary oxidation products | [20,21] |
| 4-Hydroxynonenal (4-HNE) (CH3(CH2)4CH(OH)CH=CH(CHO)) | Produced by lipid peroxidation of polyunsaturated fatty acids or linoleic or arachidonic side chains | Reacts with lysine on proteins to form carbonylated side chains, increasing the hydrophobicity of modified proteins; also involved in downstream signaling pathways, including activation of glutamate-cysteine ligase expression | [22,23] |
| Condition | Clinical Manifestation(s) | Identified Markers of Oxidative Stress | Ref. |
|---|---|---|---|
| Dry eye disease | Instability of the lipid layer, decreased tear secretion, ocular irritation | Elevated 8-OHdG, 4-HNE, and MDA and higher immune infiltration in dry eye animal models; elevated 4-HNE and hexanoyl-lysine in the conjunctiva of patients with dry eye and Sjögren’s syndrome | [42,43,45] |
| Keratoconus | Thinning of the corneal stroma leading to bulging of the central cornea | Elevated lipid peroxidation, MDA, and proinflammatory cytokines in tears of patients with keratoconus; increased lactate production and altered glycolytic and TCA cycle metabolite levels in corneal fibroblasts; decreased NRF2 expression | [64,65,66,67] |
| Cataract | Opacification of the lens | Oxidation, deamidation, and other chemical modifications of crystallin proteins, leading to protein aggregation and precipitation; decreased GSH in lens epithelial cells | [117,121,138] |
| Age-related macular degeneration | Drusen formation and photoreceptor degeneration; choroidal neovascularization, hemorrhage, and retinal fibrosis | Elevated MDA and 8-OHdG; decreased SOD in RPE cells; elevated carboxyethylpyrrole in drusen | [221,225,233] |
| Proliferative vitreoretinopathy | Formation of fibrotic membranes on the retinal surface | Reduced SOD and catalase in vitreous; decreased GSH in vitreous and blood | [283,284] |
| Diabetic retinopathy | Vascular abnormalities in the retina; microaneurysms, hemorrhaging, and angiogenesis | Increased ROS production by ETC with hyperglycemia; decreased SOD, catalase, and GSH production | [321,322,329] |
| Glaucoma | Loss of RGCs in the retina | Reduced total reactive antioxidant potential and increased MDA in the aqueous humor and blood | [381,382,424] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, D.Y.; Chaudhary, S.; Cho, K.-S.; Lennikov, A.; Miller, W.P.; Thorn, D.C.; Yang, M.; McKay, T.B. Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites 2023, 13, 187. https://doi.org/10.3390/metabo13020187
Shu DY, Chaudhary S, Cho K-S, Lennikov A, Miller WP, Thorn DC, Yang M, McKay TB. Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites. 2023; 13(2):187. https://doi.org/10.3390/metabo13020187
Chicago/Turabian StyleShu, Daisy Y., Suman Chaudhary, Kin-Sang Cho, Anton Lennikov, William P. Miller, David C. Thorn, Menglu Yang, and Tina B. McKay. 2023. "Role of Oxidative Stress in Ocular Diseases: A Balancing Act" Metabolites 13, no. 2: 187. https://doi.org/10.3390/metabo13020187
APA StyleShu, D. Y., Chaudhary, S., Cho, K.-S., Lennikov, A., Miller, W. P., Thorn, D. C., Yang, M., & McKay, T. B. (2023). Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites, 13(2), 187. https://doi.org/10.3390/metabo13020187