
Molecular and Biological Investigation of Isolated Marine Fungal Metabolites as Anticancer Agents: A Multi-Target Approach

 ,
,  ,
,  ,
,  , , , , ,
, , , , ,
Abstract
1. Introduction
2. Experimental Design
2.1. Fungal Materials
2.2. Fermentation, Extraction, Isolation, and Purification of Compounds 1–3
2.3. In Vitro Activity
2.4. Molecular Docking
2.5. Molecular Dynamics Ensembles
2.6. Drug-Likeness and Pharmacokinetic Profiling
3. Results and Discussion
3.1. Isolation, Purification, and Structural Identification of Compounds 1–3
3.2. In Vitro Activity of Isolated Compounds 1–3
3.3. Multi-Target Docking Analysis on Cancer-Associated Molecular Biotargets
3.3.1. Docking Simulation on Cdc-25A
3.3.2. Docking Simulation on PTP-1B
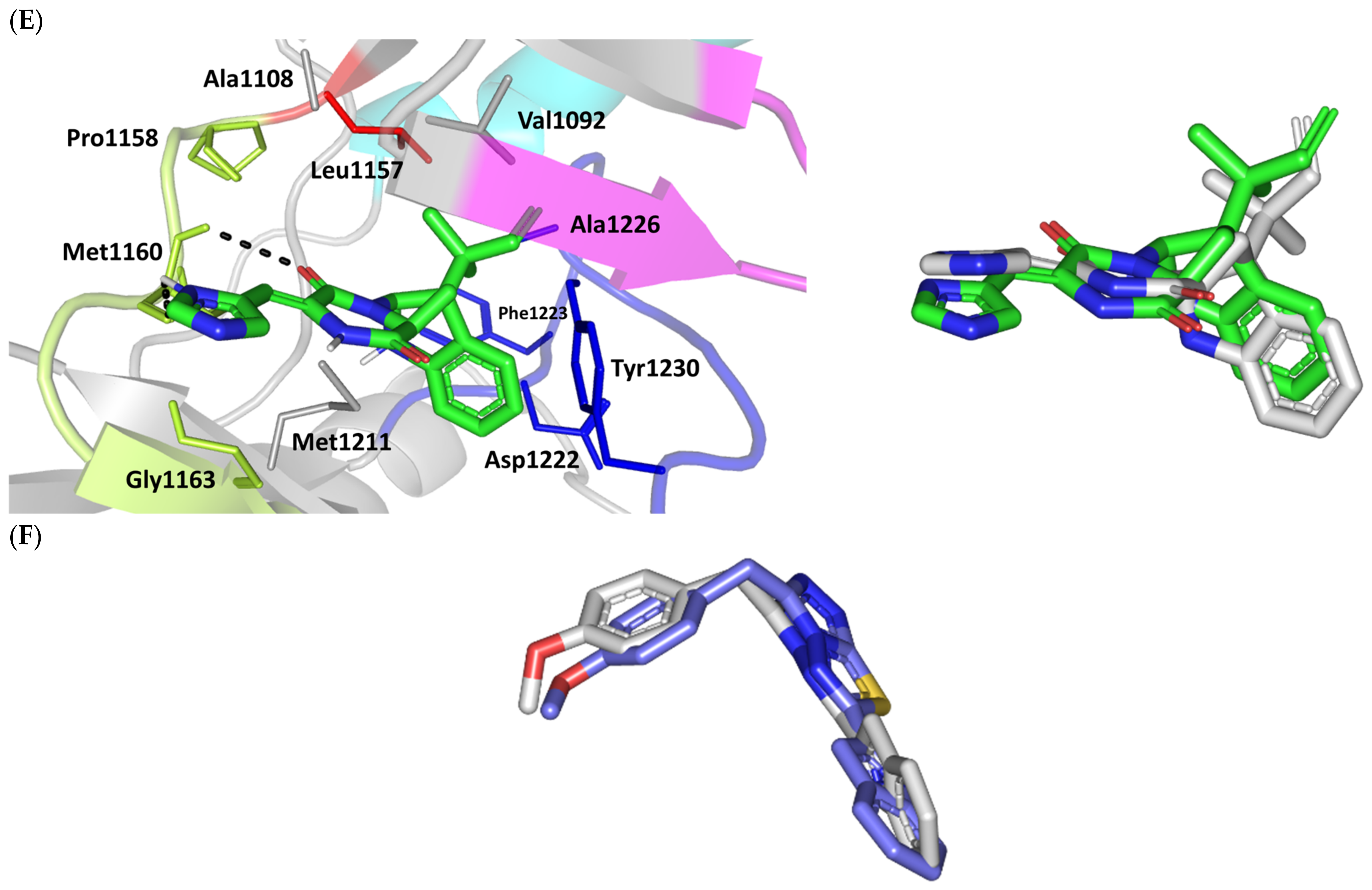
3.3.3. Docking Simulation on c-Met Kinase
3.4. Molecular Dynamics Simulations
3.4.1. Analysis on Cdc-25A
3.4.2. Analysis on PTP-1B
3.4.3. Analysis on c-Met Kinase
3.5. Drug-Likeness and Pharmacokinetic Profiling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uzma, F.; Mohan, C.D.; Hashem, A.; Konappa, N.M.; Rangappa, S.; Kamath, P.V.; Singh, B.P.; Mudili, V.; Gupta, V.K.; Siddaiah, C.N. Endophytic fungi—Alternative sources of cytotoxic compounds: A review. Front. Pharmacol. 2018, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO FactSheets: Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 15 May 2022).
- Chavda, V.P. Chapter 4-Nanobased Nano Drug Delivery: A Comprehensive Review. In Applications of Targeted Nano Drugs and Delivery Systems; Mohapatra, S.S., Ranjan, S., Dasgupta, N., Mishra, R.K., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 69–92. [Google Scholar] [CrossRef]
- Chavda, V.P.; Ertas, Y.N.; Walhekar, V.; Modh, D.; Doshi, A.; Shah, N.; Anand, K.; Chhabria, M. Advanced Computational Methodologies Used in the Discovery of New Natural Anticancer Compounds. Front. Pharmacol. 2021, 12, 702611. [Google Scholar] [CrossRef] [PubMed]
- Tran, G.; Zafar, S.Y. Financial toxicity and implications for cancer care in the era of molecular and immune therapies. Ann. Transl. Med. 2018, 6, 166. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.; Imperatore, C.; Casertano, M.; Aiello, A.; Balestri, F.; Piazza, L.; Menna, M.; Del Corso, A.; Paoli, P. Dual Targeting of PTP1B and Aldose Reductase with Marine Drug Phosphoeleganin: A Promising Strategy for Treatment of Type 2 Diabetes. Mar. Drugs 2021, 19, 535. [Google Scholar] [CrossRef]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar] [CrossRef]
- Remesh, A. Toxicities of Anticancer Drugs and Its Management. Int. J. Basic Clin. Pharmacol. 2012, 1, 2–12. [Google Scholar] [CrossRef]
- Kumar, M.S.; Adki, K.M. Marine natural products for multi-targeted cancer treatment: A future insight. Biomed. Pharmacother. 2018, 105, 233–245. [Google Scholar] [CrossRef]
- Taylor, W.F.; Moghadam, S.E.; Moridi Farimani, M.; Ebrahimi, S.N.; Tabefam, M.; Jabbarzadeh, E. A multi-targeting natural compound with growth inhibitory and anti-angiogenic properties re-sensitizes chemotherapy resistant cancer. PLoS ONE 2019, 14, e0218125. [Google Scholar] [CrossRef]
- Schulz, B.; Boyle, C.; Draeger, S.; Römmert, A.-K.; Krohn, K. Endophytic fungi: A source of novel biologically active secondary metabolites. Mycol. Res. 2002, 106, 996–1004. [Google Scholar] [CrossRef]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Mady, M.; Haggag, E. Review on Fungi of Genus Penicillium As a Producers of Biologically Active Polyketides. J. Adv. Pharm. Res. 2020, 4, 33–45. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, Z.-X.; Song, Y.-L.; Dong, D.; Zheng, J.; Liu, T.; Zhao, Y.-F.; Ferreira, D.; Zjawiony, J.K.; Tu, P.-F. Nitric oxide inhibitory meroterpenoids from the fungus Penicillium purpurogenum MHZ 111. J. Nat. Prod. 2016, 79, 1415–1422. [Google Scholar] [CrossRef]
- Samson, R.; Houbraken, J.; Thrane, U.; Frisvad, J.; Andersen, B. Food and Indoor Fungi, CBS Laboratory Manual Series 2; CBS-Fungal Biodiversity Centre: Utrecht, The Netherlands, 2010. [Google Scholar]
- Song, Q.-Y.; Cheng, F.-S.; Sun, M.; Gao, K.; Nan, Z.-B. Metabolites from Epichloë bromicola Obtained by Co-Culture with Pestalotiopsis microspore as Inhibitors of Cdc25A Phosphatases, Plant Pathogens, and Grasses. Chem. Nat. Compd. 2021, 57, 382–384. [Google Scholar] [CrossRef]
- Han, W.; Cai, J.; Zhong, W.; Xu, G.; Wang, F.; Tian, X.; Zhou, X.; Liu, Q.; Liu, Y.; Wang, J. Protein tyrosine phosphatase 1B (PTP1B) inhibitorsfrom the deep-sea fungus Penicillium chrysogenum SCSIO 07007. Bioorg. Chem. 2020, 96, 103646. [Google Scholar] [CrossRef]
- Fauman, E.B.; Cogswell, J.P.; Lovejoy, B.; Rocque, W.J.; Holmes, W.; Montana, V.G.; Piwnica-Worms, H.; Rink, M.J.; Saper, M.A. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell 1998, 93, 617–625. [Google Scholar] [CrossRef]
- Sur, S.; Agrawal, D.K. Phosphatases and kinases regulating CDC25 activity in the cell cycle: Clinical implications of CDC25 overexpression and potential treatment strategies. Mol. Cell. Biochem. 2016, 416, 33–46. [Google Scholar] [CrossRef]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, G.; Kong, C. High expression of Cdc25B and low expression of 14-3-3σ is associated with the development and poor prognosis in urothelial carcinoma of bladder. Tumor Biol. 2014, 35, 2503–2512. [Google Scholar] [CrossRef]
- Julien, S.G.; Dubé, N.; Read, M.; Penney, J.; Paquet, M.; Han, Y.; Kennedy, B.P.; Muller, W.J.; Tremblay, M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 2007, 39, 338–346. [Google Scholar] [CrossRef]
- Bentires-Alj, M.; Neel, B.G. Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 2007, 67, 2420–2424. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Bjorge, J.D.; Fujita, D.J. PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Res. 2007, 67, 10129–10137. [Google Scholar] [CrossRef] [PubMed]
- Cortesio, C.L.; Chan, K.T.; Perrin, B.J.; Burton, N.O.; Zhang, S.; Zhang, Z.Y.; Huttenlocher, A. Calpain 2 and PTP1B function in a novel pathway with Src to regulate invadopodia dynamics and breast cancer cell invasion. J. Cell Biol. 2008, 180, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y. Protein tyrosine phosphatases: Structure and function, substrate specificity, and inhibitor development. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 209–234. [Google Scholar] [CrossRef] [PubMed]
- Liu, G. Protein tyrosine phosphatase 1B inhibition: Opportunities and challenges. Curr. Med. Chem. 2003, 10, 1407–1421. [Google Scholar] [CrossRef]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Z.Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef]
- Mady, M.S.; Mohyeldin, M.M.; Ebrahim, H.Y.; Elsayed, H.E.; Houssen, W.E.; Haggag, E.G.; Soliman, R.F.; El Sayed, K.A. The indole alkaloid meleagrin, from the olive tree endophytic fungus Penicillium chrysogenum, as a novel lead for the control of c-Met-dependent breast cancer proliferation, migration and invasion. Bioorg. Med. Chem. 2016, 24, 113–122. [Google Scholar] [CrossRef]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef]
- Ma, P.C.; Tretiakova, M.S.; MacKinnon, A.C.; Ramnath, N.; Johnson, C.; Dietrich, S.; Seiwert, T.; Christensen, J.G.; Jagadeeswaran, R.; Krausz, T.; et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer 2008, 47, 1025–1037. [Google Scholar] [CrossRef]
- Cui, J.J. Targeting receptor tyrosine kinase MET in cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453. [Google Scholar] [CrossRef]
- Furlan, A.; Kherrouche, Z.; Montagne, R.; Copin, M.C.; Tulasne, D. Thirty years of research on met receptor to move a biomarker from bench to bedside. Cancer Res. 2014, 74, 6737–6744. [Google Scholar] [CrossRef]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef]
- Asfour, H.Z.; Awan, Z.A.; Bagalagel, A.A.; Elfaky, M.A.; Abdelhameed, R.F.A.; Elhady, S.S. Large-Scale Production of Bioactive Terrein by Aspergillus terreus Strain S020 Isolated from the Saudi Coast of the Red Sea. Biomolecules 2019, 9, 480. [Google Scholar] [CrossRef]
- Alahdal, A.M.; Asfour, H.Z.; Ahmed, S.A.; Noor, A.O.; Al-Abd, A.M.; Elfaky, M.A.; Elhady, S.S. Anti-Helicobacter, Antitubercular and Cytotoxic Activities of Scalaranes from the Red Sea Sponge Hyrtios erectus. Molecules 2018, 23, 978. [Google Scholar] [CrossRef]
- Papazisis, K.T.; Geromichalos, G.D.; Dimitriadis, K.A.; Kortsaris, A.H. Optimization of the sulforhodamine B colorimetric assay. J. Immunol. Methods 1997, 208, 151–158. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Liu, X.; Russell, P.; Li, J.; Pan, W.; Fu, J.; Zhang, A. Evaluation of the binding performance of flavonoids to estrogen receptor alpha by Autodock, Autodock Vina and Surflex-Dock. Ecotoxicol. Environ. Saf. 2022, 233, 113323. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. The PyMOL Molecular Graphics System; Version 2.0.6; Schrödinger LLC: New York, NY, USA, 2016. [Google Scholar]
- Elhady, S.S.; Abdelhameed, R.F.A.; Malatani, R.T.; Alahdal, A.M.; Bogari, H.A.; Almalki, A.J.; Mohammad, K.A.; Ahmed, S.A.; Khedr, A.I.M.; Darwish, K.M. Molecular Docking and Dynamics Simulation Study of Hyrtios erectus Isolated Scalarane Sesterterpenes as Potential SARS-CoV-2 Dual Target Inhibitors. Biology 2021, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale; Markidis, S., Laure, E., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–27. [Google Scholar]
- Saleh, A.H.; Abdelwaly, A.; Darwish, K.M.; Eissa, A.; Chittiboyina, A.; Helal, M.A. Deciphering the molecular basis of the kappa opioid receptor selectivity: A Molecular Dynamics study. J. Mol. Graph. Model. 2021, 106, 107940. [Google Scholar] [CrossRef]
- Ross, G.A.; Rustenburg, A.S.; Grinaway, P.B.; Fass, J.; Chodera, J.D. Biomolecular Simulations under Realistic Macroscopic Salt Conditions. J. Phys. Chem. B 2018, 122, 5466–5486. [Google Scholar] [CrossRef]
- Zaki, A.A.; Ashour, A.; Elhady, S.S.; Darwish, K.M.; Al-Karmalawy, A.A. Calendulaglycoside A Showing Potential Activity Against SARS-CoV-2 Main Protease: Molecular Docking, Molecular Dynamics, and SAR Studies. J. Tradit. Complement. Med. 2021, 12, 16–34. [Google Scholar] [CrossRef]
- Tuble, S.C.; Anwar, J.; Gale, J.D. An Approach to Developing a Force Field for Molecular Simulation of Martensitic Phase Transitions between Phases with Subtle Differences in Energy and Structure. J. Am. Chem. Soc. 2004, 126, 396–405. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Comm. 2013, 184, 2641–2650. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef]
- Lipinski, C.A. Chris Lipinski discusses life and chemistry after the Rule of Five. Drug Discov. Today 2003, 8, 12–16. [Google Scholar]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from Monte Carlo simulations. Bioorg. Med. Chem. Lett. 2000, 10, 1155–1158. [Google Scholar] [CrossRef]
- Colmenarejo, G.; Alvarez-Pedraglio, A.; Lavandera, J.-L. Cheminformatic Models To Predict Binding Affinities to Human Serum Albumin. J. Med. Chem. 2001, 44, 4370–4378. [Google Scholar] [CrossRef]
- Yazdanian, M.; Glynn, S.L.; Wright, J.L.; Hawi, A. Correlating Partitioning and Caco-2 Cell Permeability of Structurally Diverse Small Molecular Weight Compounds. Pharm. Res. 1998, 15, 1490–1494. [Google Scholar] [CrossRef]
- Duffy, E.M.; Jorgensen, W.L. Prediction of Properties from Simulations: Free Energies of Solvation in Hexadecane, Octanol, and Water. J. Am. Chem. Soc. 2000, 122, 2878–2888. [Google Scholar] [CrossRef]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Poluzzi, E.; De Ponti, F.; Recanatini, M. Toward a pharmacophore for drugs inducing the long QT syndrome: Insights from a CoMFA study of HERG K(+) channel blockers. J. Med. Chem. 2002, 45, 3844–3853. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.-L.; Zhang, D.; Xia, J.-M.; Hu, C.-C.; Lin, T.; Lin, Y.-K.; Wang, G.-H.; Tian, W.-J.; Li, Z.-P.; Zhang, X.-K.; et al. Steroids from the Deep-Sea-Derived Fungus Penicillium granulatum MCCC 3A00475 Induced Apoptosis via Retinoid X Receptor (RXR)-α Pathway. Mar. Drugs 2019, 17, 178. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Han, Z.; Peng, J.; Qian, P.Y.; Qi, S.H. Antifouling indole alkaloids from two marine derived fungi. Nat. Prod. Commun. 2013, 8, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. The FSSP database of structurally aligned protein fold families. Nucleic Acids Res. 1994, 22, 3600–3609. [Google Scholar]
- Su, X.D.; Taddei, N.; Stefani, M.; Ramponi, G.; Nordlund, P. The crystal structure of a low-molecular-weight phosphotyrosine protein phosphatase. Nature 1994, 370, 575–578. [Google Scholar] [CrossRef]
- Rudolph, J. Cdc25 phosphatases: Structure, specificity, and mechanism. Biochemistry 2007, 46, 3595–3604. [Google Scholar] [CrossRef]
- Denu, J.M.; Dixon, J.E. A catalytic mechanism for the dual-specific phosphatases. Proc. Natl. Acad. Sci. USA 1995, 92, 5910–5914. [Google Scholar] [CrossRef]
- Borgne, A.; Meijer, L. Sequential dephosphorylation of p34(cdc2) on Thr-14 and Tyr-15 at the prophase/metaphase transition. J. Biol. Chem. 1996, 271, 27847–27854. [Google Scholar] [CrossRef]
- Russo, A.A.; Jeffrey, P.D.; Pavletich, N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996, 3, 696–700. [Google Scholar] [CrossRef]
- McCain, D.F.; Catrina, I.E.; Hengge, A.C.; Zhang, Z.Y. The catalytic mechanism of Cdc25A phosphatase. J. Biol. Chem. 2002, 277, 11190–11200. [Google Scholar] [CrossRef]
- Arantes, G.M. Free-energy profiles for catalysis by dual-specificity phosphatases. Biochem. J. 2006, 399, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.X.; Hassell, A.M.; Vanderwall, D.; Lambert, M.H.; Holmes, W.D.; Luther, M.A.; Rocque, W.J.; Milburn, M.V.; Zhao, Y.; Ke, H.; et al. Atomic structure of PDE4: Insights into phosphodiesterase mechanism and specificity. Science 2000, 288, 1822–1825. [Google Scholar] [CrossRef]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef]
- Albuquerque, S.O.; Barros, T.G.; Dias, L.R.S.; Lima, C.; Azevedo, P.; Flores-Junior, L.A.P.; Dos Santos, E.G.; Loponte, H.F.; Pinheiro, S.; Dias, W.B.; et al. Biological evaluation and molecular modeling of peptidomimetic compounds as inhibitors for O-GlcNAc transferase (OGT). Eur. J. Pharm. Sci. 2020, 154, 105510. [Google Scholar] [CrossRef]
- De Souza, A.S.; Pacheco, B.D.C.; Pinheiro, S.; Muri, E.M.F.; Dias, L.R.S.; Lima, C.H.S.; Garrett, R.; de Moraes, M.B.M.; de Souza, B.E.G.; Puzer, L. 3-Acyltetramic acids as a novel class of inhibitors for human kallikreins 5 and 7. Bioorg. Med. Chem. Lett. 2019, 29, 1094–1098. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Hassan, A.M.A.; Elkaeed, E.B.; Alesawy, M.S.; Al-Karmalawy, A.A. Design, synthesis, and SAR studies of novel 4-methoxyphenyl pyrazole and pyrimidine derivatives as potential dual tyrosine kinase inhibitors targeting both EGFR and VEGFR-2. Bioorg. Chem. 2022, 123, 105770. [Google Scholar] [CrossRef]
- Park, H.; Bahn, Y.J.; Jung, S.K.; Jeong, D.G.; Lee, S.H.; Seo, I.; Yoon, T.S.; Kim, S.J.; Ryu, S.E. Discovery of novel Cdc25 phosphatase inhibitors with micromolar activity based on the structure-based virtual screening. J. Med. Chem. 2008, 51, 5533–5541. [Google Scholar] [CrossRef]
- Hwang-Seo, P.; Suk-Kyeong, J.; Young-Jae, B.; Seong Eon, R.; Seung-Jun, K. Discovery of Novel and Potent Cdc25 Phosphatase Inhibitors Based on the Structure-Based De Novo Design. Bull. Korean Chem. Soc. 2009, 30, 1313–1316. [Google Scholar] [CrossRef]
- Kabakci, Z.; Käppeli, S.; Cantù, C.; Jensen, L.D.; König, C.; Toggweiler, J.; Gentili, C.; Ribaudo, G.; Zagotto, G.; Basler, K.; et al. Pharmacophore-guided discovery of CDC25 inhibitors causing cell cycle arrest and tumor regression. Sci. Rep. 2019, 9, 1335. [Google Scholar] [CrossRef]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal structure of human protein tyrosine phosphatase 1B. Science 1994, 263, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y. Protein-tyrosine phosphatases: Biological function, structural characteristics, and mechanism of catalysis. Crit. Rev. Biochem. Mol. Biol. 1998, 33, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Brandão, T.A.S.; Hengge, A.C.; Johnson, S.J. Insights into the reaction of protein-tyrosine phosphatase 1B: Crystal structures for transition state analogs of both catalytic steps. J. Biol. Chem. 2010, 285, 15874–15883. [Google Scholar] [CrossRef]
- Shen, K.; Keng, Y.F.; Wu, L.; Guo, X.L.; Lawrence, D.S.; Zhang, Z.Y. Acquisition of a specific and potent PTP1B inhibitor from a novel combinatorial library and screening procedure. J. Biol. Chem. 2001, 276, 47311–47319. [Google Scholar] [CrossRef]
- Reddy, M.V.; Ghadiyaram, C.; Panigrahi, S.K.; Krishnamurthy, N.R.; Hosahalli, S.; Chandrasekharappa, A.P.; Manna, D.; Badiger, S.E.; Dubey, P.K.; Mangamoori, L.N. X-ray structure of PTP1B in complex with a new PTP1B inhibitor. Protein Pept. Lett. 2014, 21, 90–93. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]
- Liu, W.S.; Wang, R.R.; Yue, H.; Zheng, Z.H.; Lu, X.H.; Wang, S.Q.; Dong, W.L.; Wang, R.L. Design, synthesis, biological evaluation and molecular dynamics studies of 4-thiazolinone derivatives as protein tyrosine phosphatase 1B (PTP1B) inhibitors. J. Biomol. Struct. Dyn. 2020, 38, 3814–3824. [Google Scholar] [CrossRef]
- Mahapatra, M.K.; Bera, K.; Singh, D.V.; Kumar, R.; Kumar, M. In silico modelling and molecular dynamics simulation studies of thiazolidine based PTP1B inhibitors. J. Biomol. Struct. Dyn. 2018, 36, 1195–1211. [Google Scholar] [CrossRef]
- Manasa, B.; Manandhar, S.; Hari, G.; Priya, K.; Kumar, B.H.; Pai, K.S.R. Virtual structure-based docking, WaterMap, and molecular dynamics guided identification of the potential natural compounds as inhibitors of protein-tyrosine phosphatase 1B. J. Mol. Struct. 2021, 1226, 129396. [Google Scholar] [CrossRef]
- Cai, J.; Zhao, L.; Tao, W. Potent protein tyrosine phosphatase 1B (PTP1B) inhibiting constituents from Anoectochilus chapaensis and molecular docking studies. Pharm. Biol. 2015, 53, 1030–1034. [Google Scholar] [CrossRef]
- Peach, M.L.; Tan, N.; Choyke, S.J.; Giubellino, A.; Athauda, G.; Burke, T.R., Jr.; Nicklaus, M.C.; Bottaro, D.P. Directed discovery of agents targeting the Met tyrosine kinase domain by virtual screening. J. Med. Chem. 2009, 52, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.J.; Li, P.; Wu, C.L.; Zhou, X.Y.; Lu, H.J.; Zhou, T. Response and acquired resistance to crizotinib in Chinese patients with lung adenocarcinomas harboring MET Exon 14 splicing alternations. Lung Cancer 2016, 102, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Sequist, L.V.; Borger, D.; Gainor, J.F.; Arellano, R.S.; Le, L.P.; Dias-Santagata, D.; Clark, J.W.; Engelman, J.A.; Shaw, A.T.; et al. Acquired Resistance to Crizotinib in NSCLC with MET Exon 14 Skipping. J. Thorac. Oncol. 2016, 11, 1242–1245. [Google Scholar] [CrossRef]
- Reungwetwattana, T.; Liang, Y.; Zhu, V.; Ou, S.I. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The Why, the How, the Who, the Unknown, and the Inevitable. Lung Cancer 2017, 103, 27–37. [Google Scholar] [CrossRef]
- Kang, J.; Chen, H.J.; Wang, Z.; Liu, J.; Li, B.; Zhang, T.; Yang, Z.; Wu, Y.L.; Yang, J.J. Osimertinib and Cabozantinib Combinatorial Therapy in an EGFR-Mutant Lung Adenocarcinoma Patient with Multiple MET Secondary-Site Mutations after Resistance to Crizotinib. J. Thorac. Oncol. 2018, 13, e49–e53. [Google Scholar] [CrossRef]
- Parikh, P.K.; Ghate, M.D. Recent advances in the discovery of small molecule c-Met Kinase inhibitors. Eur. J. Med. Chem. 2018, 143, 1103–1138. [Google Scholar] [CrossRef]
- Nishii, H.; Chiba, T.; Morikami, K.; Fukami, T.A.; Sakamoto, H.; Ko, K.; Koyano, H. Discovery of 6-benzyloxyquinolines as c-Met selective kinase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1405–1409. [Google Scholar] [CrossRef]
- Ye, L.; Wu, J.; Yang, J.; Chen, W.; Luo, Y.; Zhang, Y. Design, synthesis and molecular docking analysis of some novel 7-[(quinolin-6-yl)methyl] purines as potential c-Met inhibitors. Med. Chem. Res. 2015, 24, 3327–3333. [Google Scholar] [CrossRef]
- Ye, L.; Ou, X.; Tian, Y.; Yu, B.; Luo, Y.; Feng, B.; Lin, H.; Zhang, J.; Wu, S. Indazoles as potential c-Met inhibitors: Design, synthesis and molecular docking studies. Eur. J. Med. Chem. 2013, 65, 112–118. [Google Scholar] [CrossRef]
- Cui, H.; Peng, X.; Liu, J.; Ma, C.; Ji, Y.; Zhang, W.; Geng, M.; Li, Y. Design, synthesis and biological evaluation of c-Met kinase inhibitors bearing 2-oxo-1,2-dihydroquinoline scaffold. Bioorg. Med. Chem. Lett. 2016, 26, 4483–4486. [Google Scholar] [CrossRef]
- El-Wakil, M.H.; Ashour, H.M.; Saudi, M.N.; Hassan, A.M.; Labouta, I.M. Design, synthesis and molecular modeling studies of new series of antitumor 1,2,4-triazines with potential c-Met kinase inhibitory activity. Bioorg. Chem. 2018, 76, 154–165. [Google Scholar] [CrossRef]
- El-Wakil, M.H.; Teleb, M. Transforming Type II to Type I c-Met kinase inhibitors via combined scaffold hopping and structure-guided synthesis of new series of 1,3,4-thiadiazolo[2,3-c]-1,2,4-triazin-4-one derivatives. Bioorg. Chem. 2021, 116, 105304. [Google Scholar] [CrossRef]
- Yuan, H.; Zhuang, J.; Hu, S.; Li, H.; Xu, J.; Hu, Y.; Xiong, X.; Chen, Y.; Lu, T. Molecular modeling of exquisitely selective c-Met inhibitors through 3D-QSAR and molecular dynamics simulations. J. Chem. Inf. Model. 2014, 54, 2544–2554. [Google Scholar] [CrossRef]
- Balasubramanian, P.K.; Balupuri, A.; Bhujbal, S.P.; Cho, S.J. 3D-QSAR-aided design of potent c-Met inhibitors using molecular dynamics simulation and binding free energy calculation. J. Biomol. Struct. Dyn. 2019, 37, 2165–2178. [Google Scholar] [CrossRef]
- Damghani, T.; Sedghamiz, T.; Sharifi, S.; Pirhadi, S. Critical c-Met-inhibitor interactions resolved from molecular dynamics simulations of different c-Met complexes. J. Mol. Struct. 2020, 1203, 127456. [Google Scholar] [CrossRef]
- Karplus, M.; Petsko, G.A. Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef]
- Gad, M.M.; Abdelwaly, A.; Helal, M.A. Structural basis for the selectivity of 3rd generation EGFR inhibitors: A molecular dynamics study. J. Biomol. Struct. Dyn. 2022, 29, 1–11. [Google Scholar] [CrossRef]
- Soltan, M.A.; Eldeen, M.A.; Elbassiouny, N.; Kamel, H.L.; Abdelraheem, K.M.; El-Gayyed, H.A.; Gouda, A.M.; Sheha, M.F.; Fayad, E.; Ali, O.A.A.; et al. In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi. Cells 2021, 10, 3014. [Google Scholar] [CrossRef]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure Of Biomolecules Through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bhatt, D.; Zuckerman, D.M. Automated sampling assessment for molecular simulations using the effective sample size. J. Chem. Theory Comput. 2010, 6, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, A.; Blass, B.E.; Colussi, D.J.; Almi, I.; Abou-Gharbia, M.; Klein, M.L.; Elokely, K.M. Targeting SARS-CoV-2 M3CLpro by HCV NS3/4a Inhibitors: In Silico Modeling and In Vitro Screening. J. Chem. Inf. Model. 2021, 61, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Almalki, A.J.; Ibrahim, T.S.; Elhady, S.S.; Hegazy, W.A.H.; Darwish, K.M. Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents. Pharmaceuticals 2022, 15, 110. [Google Scholar] [CrossRef]
- Benson, N.C.; Daggett, V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 8722–8731. [Google Scholar] [CrossRef]
- Helal, M.A.; Chittiboyina, A.G.; Avery, M.A. New insights into the binding mode of melanin concentrating hormone receptor-1 antagonists: Homology modeling and explicit membrane molecular dynamics simulation study. J. Chem. Inf. Model. 2011, 51, 635–646. [Google Scholar] [CrossRef]
- Yuvaniyama, J.; Denu, J.M.; Dixon, J.E.; Saper, M.A. Crystal structure of the dual specificity protein phosphatase VHR. Science 1996, 272, 1328–1331. [Google Scholar] [CrossRef]
- Gliubich, F.; Gazerro, M.; Zanotti, G.; Delbono, S.; Bombieri, G.; Berni, R. Active site structural features for chemically modified forms of rhodanese. J. Biol. Chem. 1996, 271, 21054–21061. [Google Scholar] [CrossRef]
- Zhang, M.; Van Etten, R.L.; Stauffacher, C.V. Crystal structure of bovine heart phosphotyrosyl phosphatase at 2.2-A resolution. Biochemistry 1994, 33, 11097–11105. [Google Scholar] [CrossRef]
- Cavasotto, C.N. Binding Free Energy Calculation Using Quantum Mechanics Aimed for Drug Lead Optimization. Methods Mol. Biol. 2020, 2114, 257–268. [Google Scholar] [CrossRef]
- Singh, W.; Karabencheva-Christova, T.G.; Black, G.W.; Ainsley, J.; Dover, L.; Christov, C.Z. Conformational Dynamics, Ligand Binding and Effects of Mutations in NirE an S-Adenosyl-L-Methionine Dependent Methyltransferase. Sci. Rep. 2016, 6, 20107. [Google Scholar] [CrossRef]
- Fatriansyah, J.F.; Rizqillah, R.K.; Yandi, M.Y.; Fadilah; Sahlan, M. Molecular docking and dynamics studies on propolis sulabiroin-A as a potential inhibitor of SARS-CoV-2. J. King Saud Univ. Sci. 2022, 34, 101707. [Google Scholar] [CrossRef]
- Sun, J.P.; Fedorov, A.A.; Lee, S.Y.; Guo, X.L.; Shen, K.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Crystal structure of PTP1B complexed with a potent and selective bidentate inhibitor. J. Biol. Chem. 2003, 278, 12406–12414. [Google Scholar] [CrossRef]
- Scapin, G.; Patel, S.B.; Becker, J.W.; Wang, Q.; Desponts, C.; Waddleton, D.; Skorey, K.; Cromlish, W.; Bayly, C.; Therien, M.; et al. The structural basis for the selectivity of benzotriazole inhibitors of PTP1B. Biochemistry 2003, 42, 11451–11459. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, G.; Xin, Z.; Serby, M.D.; Pei, Z.; Szczepankiewicz, B.G.; Hajduk, P.J.; Abad-Zapatero, C.; Hutchins, C.W.; Lubben, T.H.; et al. Isoxazole carboxylic acids as protein tyrosine phosphatase 1B (PTP1B) inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 5543–5546. [Google Scholar] [CrossRef]
- Eathiraj, S.; Palma, R.; Volckova, E.; Hirschi, M.; France, D.S.; Ashwell, M.A.; Chan, T.C. Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal-epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197. J. Biol. Chem. 2011, 286, 20666–20676. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J. Med. Chem. 2008, 51, 7921–7932. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Harmange, J.C.; Bauer, D.; Berry, L.; Bode, C.; Boezio, A.A.; Chen, A.; Choquette, D.; Dussault, I.; Fridrich, C.; et al. Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase. J. Med. Chem. 2008, 51, 2879–2882. [Google Scholar] [CrossRef]
- Bellon, S.F.; Kaplan-Lefko, P.; Yang, Y.; Zhang, Y.; Moriguchi, J.; Rex, K.; Johnson, C.W.; Rose, P.E.; Long, A.M.; O’Connor, A.B.; et al. c-Met inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations. J. Biol. Chem. 2008, 283, 2675–2683. [Google Scholar] [CrossRef]
- Johnson, L.N.; Noble, M.E.; Owen, D.J. Active and inactive protein kinases: Structural basis for regulation. Cell 1996, 85, 149–158. [Google Scholar] [CrossRef]
- Kornev, A.P.; Haste, N.M.; Taylor, S.S.; Eyck, L.F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 17783–17788. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Niv, M.Y. Molecular Dynamics Simulations and Elastic Network Analysis of Protein Kinase B (Akt/PKB) Inactivation. J. Chem. Inf. Model. 2010, 50, 1602–1610. [Google Scholar] [CrossRef] [PubMed]
- Elmaaty, A.A.; Darwish, K.M.; Chrouda, A.; Boseila, A.A.; Tantawy, M.A.; Elhady, S.S.; Shaik, A.B.; Mustafa, M.; Al-karmalawy, A.A. In Silico and In Vitro Studies for Benzimidazole Anthelmintics Repurposing as VEGFR-2 Antagonists: Novel Mebendazole-Loaded Mixed Micelles with Enhanced Dissolution and Anticancer Activity. ACS Omega 2022, 7, 875–899. [Google Scholar] [CrossRef] [PubMed]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Cell Line | MEL | ROC | ISO | DOX |
|---|---|---|---|---|---|
| Lung cancer | A-549 | 3.66 ± 0.10 | 18.70 ± 1.06 | 20.30 ± 1.06 | 0.01 ± 0.03 |
| Cervical cancer | HeLa | 2.90 ± 0.19 | 46.97 ± 2.01 | 53.00 ± 1.36 | 0.05 ± 0.01 |
| Prostate cancer | DU-145 | 0.03 ± 0.02 | 4.80 ± 0.14 | 17.40 ± 0.44 | 0.34 ± 0.10 |
| Hepatocellular carcinoma | HepG2 | 0.10 ± 0.03 | 7.80 ± 0.69 | 13.20 ± 0.69 | 0.92 ± 0.09 |
| Key Structural Motifs | Residues | MEL | ROC | ISO | |
|---|---|---|---|---|---|
| CH2A | Asp383 | 0.38 | 0.28 | 0.40 | |
| Cys384 | 0.40 | 0.37 | 0.42 | ||
| Arg385 | 0.53 | 0.44 | 0.49 | ||
| Tyr386 | 0.59 | 0.65 | 0.62 | ||
| Pro387 | 0.57 | 0.68 | 0.65 | ||
| Tyr388 | 0.70 | 0.89 | 0.81 | ||
| Glu389 | 0.64 | 0.69 | 0.64 | ||
| Tyr390 | 0.49 | 0.49 | 0.47 | ||
| Glu391 | 0.47 | 0.50 | 0.53 | ||
| Gly392 | 0.47 | 0.43 | 0.49 | ||
| Gly393 | 0.33 | 0.31 | 0.34 | ||
| Val399 | 0.33 | 0.28 | 0.40 | ||
| Asn400 | 0.44 | 0.40 | 0.46 | ||
| Leu401 | 0.45 | 0.36 | 0.42 | ||
| His402 | 0.54 | 0.46 | 0.47 | ||
| Met403 | 0.55 | 0.43 | 0.30 | ||
| Catalytic site | Phe428 | 0.28 | 0.22 | 0.27 | |
| P-loop | His429 | 0.29 | 0.33 | 0.42 | |
| Cys430 | 0.39 | 0.38 | 0.43 | ||
| Glu431 | 0.42 | 0.48 | 0.51 | ||
| Phe432 | 0.42 | 0.49 | 0.44 | ||
| Ser433 | 0.83 | 0.49 | 0.44 | ||
| Ser434 | 0.72 | 0.66 | 0.49 | ||
| Glu435 | 0.73 | 0.70 | 0.58 | ||
| Arg436 | 0.57 | 0.60 | 0.52 | ||
| Gly437 | 0.40 | 0.45 | 0.42 | ||
| Pro438 | 0.38 | 0.50 | 0.39 | ||
| Arg439 | 0.30 | 0.45 | 0.32 | ||
| CH2B | Leu465 | 0.34 | 0.32 | 0.24 | |
| Lys466 | 0.35 | 0.37 | 0.33 | ||
| Gly467 | 0.40 | 0.44 | 0.41 | ||
| Gly468 | 0.36 | 0.36 | 0.28 | ||
| Tyr469 | 0.38 | 0.31 | 0.35 | ||
| Lys470 | 0.50 | 0.36 | 0.44 | ||
| Glu471 | 0.43 | 0.38 | 0.31 | ||
| Energy (kJ/mol ± SE) | MEL | ROC | ISO |
|---|---|---|---|
| van der Waal | −59.43 ± 26.86 | −60.84 ± 38.50 | −76.11 ± 29.79 |
| Electrostatic | −87.18 ± 39.97 | −78.03 ± 28.62 | −40.20 ± 15.01 |
| Solvation; Polar | 70.41 ± 36.218 | 69.12 ± 41.72 | 59.05 ± 49.09 |
| Solvation; SASA | −8.88 ± 4.77 | −7.43 ± 4.82 | −10.84 ± 4.45 |
| Binding energy | −85.08 ± 17.39 | −77.18 ± 11.47 | −68.10 ± 13.79 |
| Active Site and Vicinal Structural Loops | Residues | MEL | ROC | ISO | INC |
|---|---|---|---|---|---|
| PTR-loop | Asn40 | 0.87 | 0.90 | 0.82 | 0.97 |
| Lys41 | 0.52 | 0.96 | 0.65 | 0.70 | |
| Asn42 | 0.51 | 0.75 | 0.54 | 0.79 | |
| Arg43 | 0.74 | 0.52 | 0.63 | 0.80 | |
| Asn44 | 0.79 | 0.72 | 0.71 | 0.79 | |
| Arg45 | 0.78 | 0.69 | 0.73 | 0.80 | |
| Tyr46 | 0.75 | 0.85 | 0.72 | 0.87 | |
| Arg47 | 0.81 | 0.97 | 0.69 | 0.95 | |
| R-loop | Leu110 | 0.20 | 0.21 | 0.19 | 0.19 |
| Asn111 | 0.35 | 0.23 | 0.38 | 0.38 | |
| Arg112 | 0.57 | 0.46 | 0.61 | 0.63 | |
| Val113 | 0.72 | 0.60 | 0.72 | 0.73 | |
| Met114 | 0.78 | 0.78 | 0.85 | 0.87 | |
| Glu115 | 0.66 | 0.63 | 0.70 | 0.82 | |
| Lys116 | 0.64 | 0.52 | 0.53 | 0.70 | |
| Gly117 | 0.84 | 0.66 | 0.75 | 0.85 | |
| Ser118 | 0.73 | 0.55 | 0.79 | 0.85 | |
| Leu119 | 0.75 | 0.67 | 0.79 | 0.91 | |
| Lys120 | 0.56 | 0.52 | 0.65 | 0.72 | |
| Cys121 | 0.48 | 0.45 | 0.52 | 0.51 | |
| WPD-loop | Thr177 | 0.33 | 0.54 | 0.67 | 0.52 |
| Thr178 | 0.36 | 0.40 | 0.71 | 0.67 | |
| Trp179 | 0.40 | 0.44 | 0.53 | 0.45 | |
| Pro180 | 0.18 | 0.38 | 0.44 | 0.51 | |
| Asp181 | 0.30 | 0.40 | 0.34 | 0.38 | |
| Phe182 | 0.32 | 0.30 | 0.28 | 0.33 | |
| Gly183 | 0.34 | 0.20 | 0.21 | 0.37 | |
| Val184 | 0.39 | 0.24 | 0.32 | 0.50 | |
| Pro185 | 0.33 | 0.03 | 0.33 | 0.46 | |
| Catalytic P-loop | His214 | 0.03 | −0.01 | −0.03 | −0.03 |
| Cys215 | 0.21 | 0.17 | 0.13 | 0.21 | |
| Ser216 | 0.41 | 0.41 | 0.37 | 0.44 | |
| Ala217 | 0.35 | 0.47 | 0.34 | 0.44 | |
| Gly218 | 0.10 | 0.12 | 0.13 | 0.19 | |
| Ile219 | 0.21 | 0.13 | 0.20 | 0.25 | |
| Gly220 | 0.14 | 0.13 | 0.02 | 0.13 | |
| Arg221 | 0.34 | 0.34 | 0.27 | 0.38 | |
| His214 | 0.03 | −0.01 | −0.03 | −0.03 | |
| Q-loop | Gln262 | 0.37 | 0.29 | 0.25 | 0.36 |
| Thr263 | 0.41 | 0.31 | 0.32 | 0.41 | |
| Ala264 | 0.43 | 0.32 | 0.32 | 0.40 | |
| Asp265 | 0.40 | 0.31 | 0.33 | 0.41 | |
| Gln266 | 0.27 | 0.24 | 0.19 | 0.29 | |
| Leu267 | 0.35 | 0.33 | 0.23 | 0.34 | |
| Arg268 | 0.47 | 0.43 | 0.38 | 0.47 | |
| Phe269 | 0.45 | 0.42 | 0.39 | 0.46 | |
| Tyr-P cleft | Tyr20 | 0.57 | 0.40 | 0.34 | 0.61 |
| Arg24 | 0.74 | 0.55 | 0.13 | 0.82 | |
| His25 | 0.88 | 0.80 | 0.21 | 1.03 | |
| Ala27 | 0.93 | 0.60 | −0.93 | 1.02 | |
| Phe52 | 1.12 | 1.01 | 0.78 | 1.13 | |
| Arg254 | 0.77 | 0.64 | 0.51 | 0.81 | |
| Met258 | 0.61 | 0.68 | 0.65 | 0.68 | |
| Gly259 | 0.37 | 0.50 | 0.41 | 0.47 |
| Energy (kJ/mol ± SE) | MEL | ROC | ISO | INC |
|---|---|---|---|---|
| van der Waal | −131.05 ± 12.67 | −120.20 ± 10.53 | −94.19 ± 18.09 | −129.63 ± 19.96 |
| Electrostatic | −36.77 ± 24.82 | −35.34 ± 15.79 | −40.20 ± 31.41 | −69.59 ± 18.72 |
| Solvation; Polar | 129.37 ± 39.09 | 124.63 ± 17.16 | 140.39 ± 47.46 | 154.38 ± 18.77 |
| Solvation; SASA | −12.74 ± 1.27 | −13.81 ± 1.367 | −11.02 ± 2.00 | −15.84 ± 1.27 |
| Binding energy | −51.19 ± 31.56 | −44.72 ± 26.39 | −15.02 ± 12.78 | −60.68 ± 25.99 |
| Active Site and Vicinal Structural Loops | Residues | MEL | ROC | ISO | INC | |
|---|---|---|---|---|---|---|
| P-loop | Gly1085 | 1.30 | 1.01 | 0.67 | 0.57 | |
| Arg1086 | 1.51 | 1.10 | 0.47 | 0.82 | ||
| Gly1087 | 2.10 | 1.55 | 1.04 | 1.29 | ||
| His1088 | 2.29 | 1.78 | 1.38 | 1.57 | ||
| Phe1089 | 2.84 | 2.31 | 2.43 | 1.53 | ||
| Gly1090 | 2.82 | 2.27 | 2.35 | 2.17 | ||
| 1α/C-helix | Ile1115 | 1.94 | 1.27 | 1.48 | 0.23 | |
| Thr1116 | 2.74 | 1.68 | 2.07 | 0.81 | ||
| Asp1117 | 2.60 | 2.68 | 2.63 | 0.33 | ||
| Ile1118 | 3.47 | 4.05 | 3.77 | 0.83 | ||
| Gly1119 | 2.74 | 3.36 | 3.15 | 0.72 | ||
| Glu1120 | 2.78 | 3.26 | 3.14 | 1.30 | ||
| Val1121 | 2.37 | 2.71 | 2.69 | 1.31 | ||
| Ser1122 | 2.33 | 2.60 | 2.53 | 1.32 | ||
| Gln1123 | 2.46 | 2.74 | 2.63 | 1.17 | ||
| Phe1124 | 2.27 | 2.54 | 2.49 | 0.89 | ||
| Leu1125 | 2.22 | 2.36 | 2.34 | 0.95 | ||
| Thr1126 | 2.14 | 2.33 | 2.18 | 0.92 | ||
| Glu1127 | 1.87 | 2.15 | 1.96 | 0.44 | ||
| Gly1128 | 1.98 | 2.07 | 2.00 | 0.21 | ||
| Ile1129 | 1.91 | 1.88 | 1.72 | 1.13 | ||
| Ile1130 | 1.52 | 1.69 | 1.40 | 1.56 | ||
| Met1131 | 0.34 | 1.48 | 1.39 | 1.28 | ||
| Lys1132 | 1.85 | 1.78 | 1.69 | 1.83 | ||
| Asp1133 | 1.71 | 1.97 | 1.67 | 1.73 | ||
| Leu1142 | 1.66 | 1.79 | 1.73 | 1.57 | ||
| GK | Leu1157 | 1.43 | 1.55 | 1.49 | 1.28 | |
| Hinge region | Pro1158 | 1.26 | 1.39 | 1.34 | 1.32 | |
| Tyr1159 | 1.12 | 1.21 | 1.12 | 1.10 | ||
| Met1160 | 0.68 | 0.86 | 0.65 | 0.88 | ||
| Lys1161 | 0.61 | 0.85 | 0.46 | 0.89 | ||
| His1162 | 0.53 | 0.67 | 0.54 | 0.73 | ||
| Gly1163 | 0.42 | 0.52 | 0.33 | 0.58 | ||
| Asp1164 | 0.28 | 0.43 | 0.23 | 0.43 | ||
| Leu1165 | 0.22 | 0.34 | 0.14 | 0.28 | ||
| His1202 | 0.62 | 0.81 | 0.66 | 0.55 | ||
| Ala1221 | 0.33 | 0.86 | 0.68 | 0.67 | ||
| DFG motif | Asp1222 | −0.10 | 1.01 | 0.70 | 0.71 | |
| Phe1223 | −0.01 | 1.07 | 0.63 | 0.68 | ||
| Gly1224 | 0.11 | 1.49 | 0.51 | 1.13 | ||
| A-loop | Leu1225 | −0.97 | 1.57 | 0.90 | 1.04 | |
| Ala1226 | −0.66 | 1.37 | 0.75 | 0.77 | ||
| Arg1227 | 0.36 | 0.98 | 0.31 | 0.30 | ||
| Asp1228 | 0.77 | 0.92 | −0.01 | 0.57 | ||
| Met1229 | 0.72 | 0.70 | −1.23 | 0.34 | ||
| Tyr1230 | 0.26 | 0.20 | −1.99 | −0.33 | ||
| Asp1231 | 0.30 | −0.12 | −3.24 | −0.39 | ||
| Lys1232 | 0.49 | 0.22 | −2.82 | −0.19 | ||
| Glu1233 | 0.63 | −1.44 | −3.44 | −0.85 | ||
| Tyr1234 | 0.70 | −1.36 | −4.15 | −0.50 | ||
| Tyr1235 | 0.81 | −1.65 | −3.48 | 0.23 | ||
| Ser1236 | 1.04 | −1.40 | −2.88 | −0.64 | ||
| Val1237 | 0.83 | −2.16 | −3.18 | −1.58 | ||
| His1238 | 0.31 | −2.42 | −3.17 | −2.32 | ||
| Asn1239 | −0.04 | −0.99 | −1.49 | −3.95 | ||
| Lys1240 | −0.45 | −1.37 | −1.71 | −4.22 | ||
| Thr1241 | 0.06 | −1.09 | −2.59 | −3.97 | ||
| Gly1242 | 0.20 | −0.63 | −2.69 | −3.55 | ||
| Ala1243 | 0.56 | −0.32 | −1.84 | −2.23 | ||
| Lys1244 | 0.61 | −0.62 | −0.68 | −1.22 | ||
| Leu1245 | 0.60 | −0.48 | 0.21 | −0.22 | ||
| Pro1246 | 0.49 | 0.37 | −0.13 | 0.43 | ||
| Val1247 | 0.84 | 0.69 | 0.56 | 0.70 | ||
| Lys1248 | 0.91 | 0.69 | 0.73 | 0.85 | ||
| Trp1249 | 0.52 | 0.37 | 0.43 | 0.48 | ||
| Met1250 | 0.47 | 0.50 | 0.41 | 0.35 | ||
| Ala1251 | 0.50 | 0.72 | 0.50 | 0.30 | ||
| Energy (kJ/mol ± SE) | MEL | ROC | ISO | 6TD |
|---|---|---|---|---|
| van der Waal | −146.39 ± 19.06 | −171.95 ± 15.78 | −173.08 ± 14.38 | −175.90 ± 19.17 |
| Electrostatic | −16.02 ± 11.72 | −15.25 ± 9.46 | −31.70 ± 3.82 | −30.22 ± 9.00 |
| Solvation; Polar | 103.31 ± 22.21 | 133.68 ± 15.86 | 147.32 ± 12.43 | 145.35 ± 16.59 |
| Solvation; SASA | −17.03 ± 2.32 | −17.79 ± 1.18 | −17.90 ± 1.10 | −17.57 ± 0.82 |
| Binding energy | −76.13 ± 10.33 | −71.31 ± 22.66 | −75.36 ± 17.63 | −78.34 ± 11.13 |
| Ligand | “R-O5” HBD HBA θ SASA MW Violation | QPlogP −2.0 → 6.5 | QPlogS mol/dm3 −6.5 → 0.5 | QPPCaco nm/s <25 Poor >500 Great | QPPMDCK nm/s <25 Poor >500 Great | QPlogBB −3.0 → 1.2 | QPlogKHSA −1.5 → 1.5 | %HOA <25% Poor >80% Great | QPlogHERG >−5.0 | Oral Rat LD50 μg/Kg | AMES Mutagensis |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MEL | 3 5 4 629.64 433.47 0 | 1.75 | −2.15 | 39.75 | 16.76 | −1.05 | −0.22 | 61.05 | −5.48 | 7.89 | Negative (0.16) |
| ROC | 3 3 3 646.57 389.45 0 | 2.27 | −3.39 | 65.75 | 28.88 | −0.80 | 0.29 | 72.77 | −5.93 | 16.23 | Negative (0.26) |
| ISO | 3 3 3 630.24 389.45 0 | 2.36 | −3.66 | 69.44 | 30.63 | −0.84 | 0.30 | 73.73 | −6.33 | 16.23 | Negative (0.26) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogari, H.A.; Elhady, S.S.; Darwish, K.M.; Refaey, M.S.; Mohamed, R.A.; Abdelhameed, R.F.A.; Almalki, A.J.; Aldurdunji, M.M.; Lashkar, M.O.; Alshehri, S.O.; et al. Molecular and Biological Investigation of Isolated Marine Fungal Metabolites as Anticancer Agents: A Multi-Target Approach. Metabolites 2023, 13, 162. https://doi.org/10.3390/metabo13020162
Bogari HA, Elhady SS, Darwish KM, Refaey MS, Mohamed RA, Abdelhameed RFA, Almalki AJ, Aldurdunji MM, Lashkar MO, Alshehri SO, et al. Molecular and Biological Investigation of Isolated Marine Fungal Metabolites as Anticancer Agents: A Multi-Target Approach. Metabolites. 2023; 13(2):162. https://doi.org/10.3390/metabo13020162
Chicago/Turabian StyleBogari, Hanin A., Sameh S. Elhady, Khaled M. Darwish, Mohamed S. Refaey, Radi A. Mohamed, Reda F. A. Abdelhameed, Ahmad J. Almalki, Mohammed M. Aldurdunji, Manar O. Lashkar, Samah O. Alshehri, and et al. 2023. "Molecular and Biological Investigation of Isolated Marine Fungal Metabolites as Anticancer Agents: A Multi-Target Approach" Metabolites 13, no. 2: 162. https://doi.org/10.3390/metabo13020162
APA StyleBogari, H. A., Elhady, S. S., Darwish, K. M., Refaey, M. S., Mohamed, R. A., Abdelhameed, R. F. A., Almalki, A. J., Aldurdunji, M. M., Lashkar, M. O., Alshehri, S. O., Malatani, R. T., Yamada, K., & Khedr, A. I. M. (2023). Molecular and Biological Investigation of Isolated Marine Fungal Metabolites as Anticancer Agents: A Multi-Target Approach. Metabolites, 13(2), 162. https://doi.org/10.3390/metabo13020162