
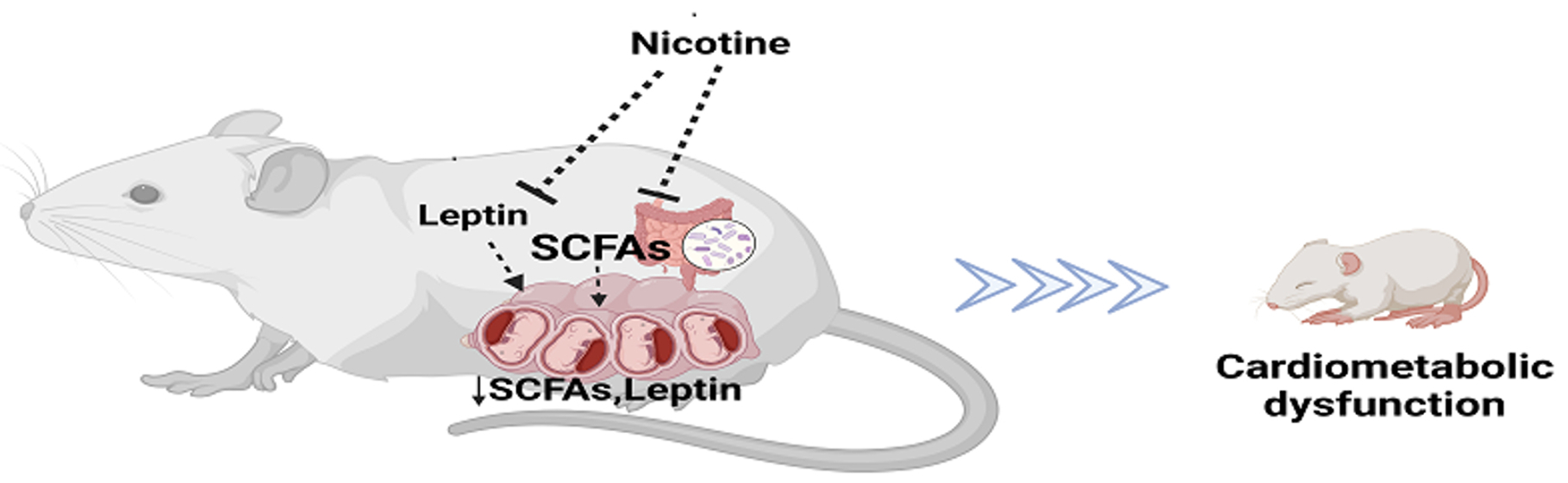
Nicotine Exposure during Rodent Pregnancy Alters the Composition of Maternal Gut Microbiota and Abundance of Maternal and Amniotic Short Chain Fatty Acids

 , ,
, , 
Abstract

1. Introduction
2. Methods
Animals
3. Exposure to Nicotine
3.1. Endpoint Collection of Tissues, Plasma, Amniotic Fluid and Cecal Content
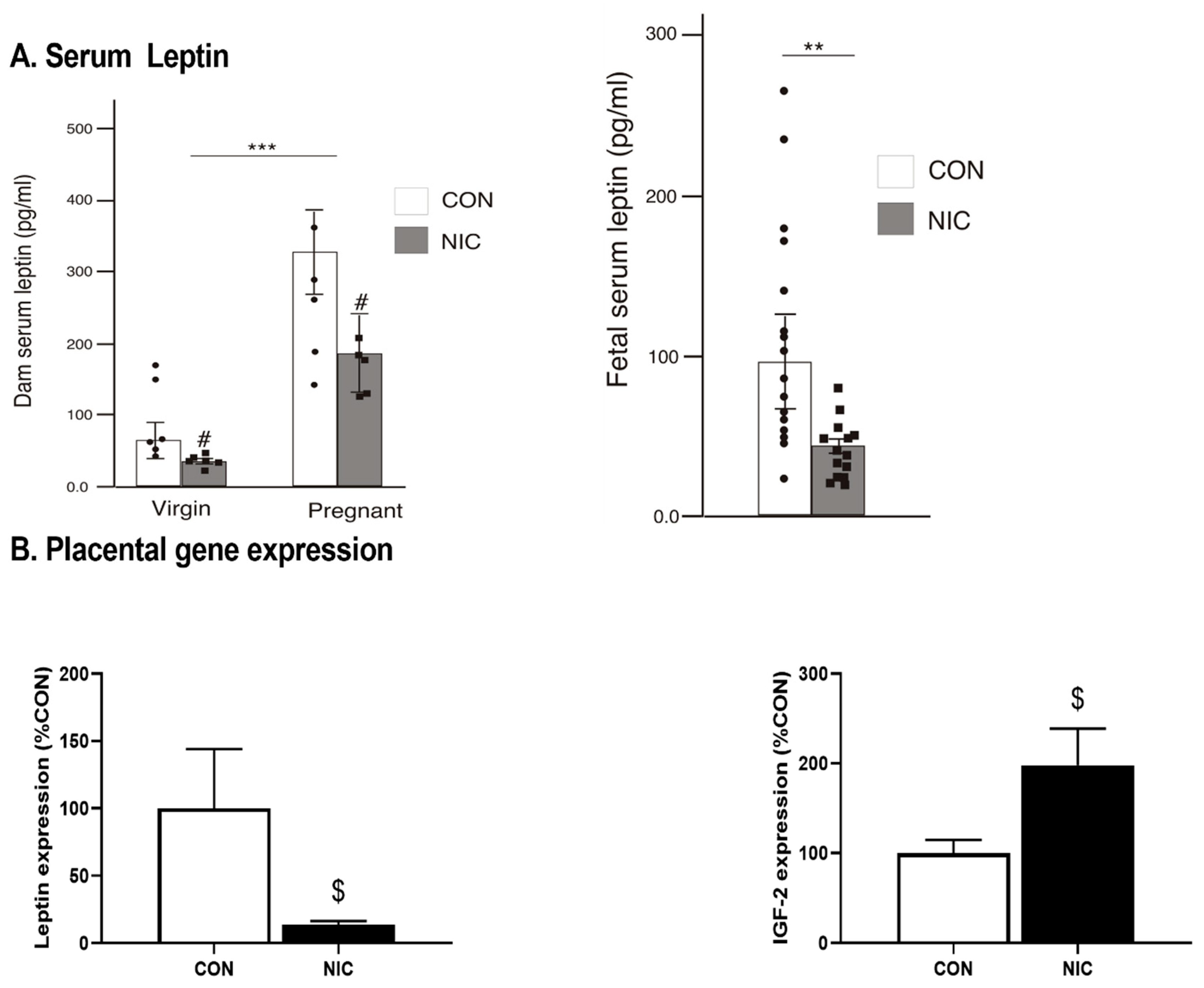
3.2. Leptin Analysis in Maternal and Virgin Female Rat Serum, Fetal Serum and Amniotic Fluid
3.3. Cecal SCFA Extraction and Analysis
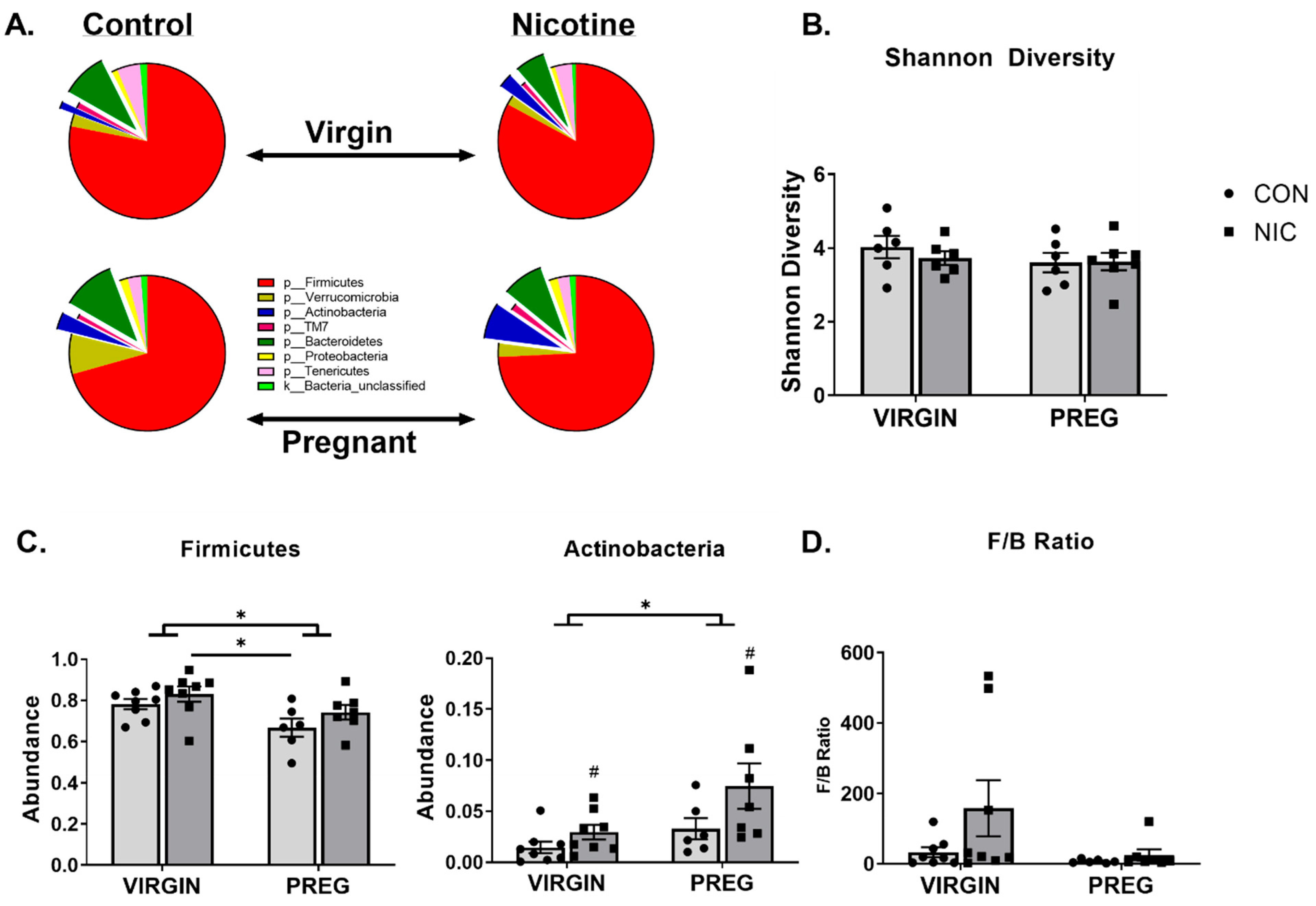
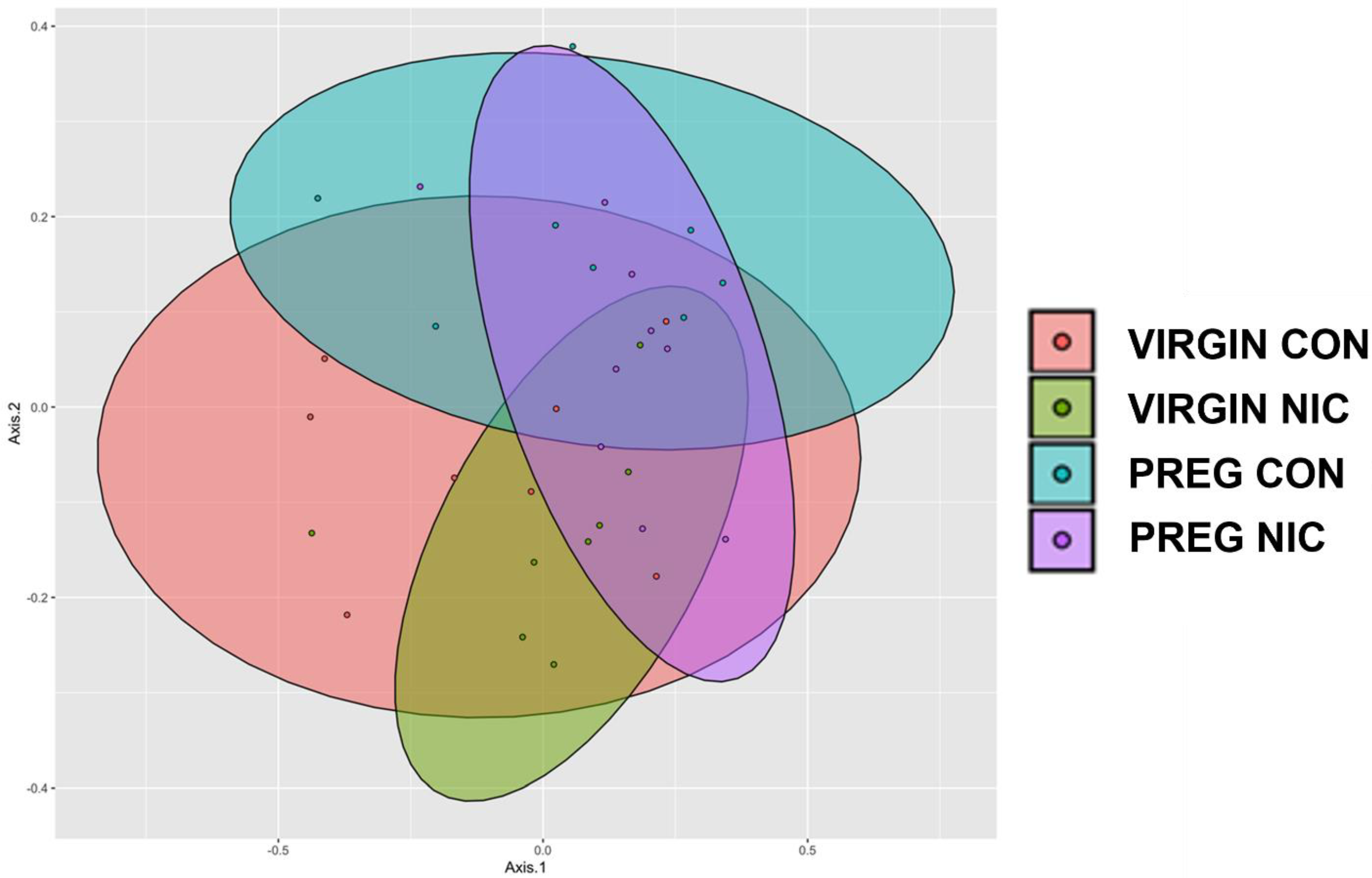
3.4. 16S Bacterial Sequencing and Analysis
3.5. SCFA Extraction and Analysis in Maternal and Virgin Female Rat Serum and Fetal Amniotic Fluid
4. Gene Expression Analysis
5. Statistical Analysis
6. Results
6.1. Impact of NIC on Body Weight, Fetal Weight, and Fetal Number
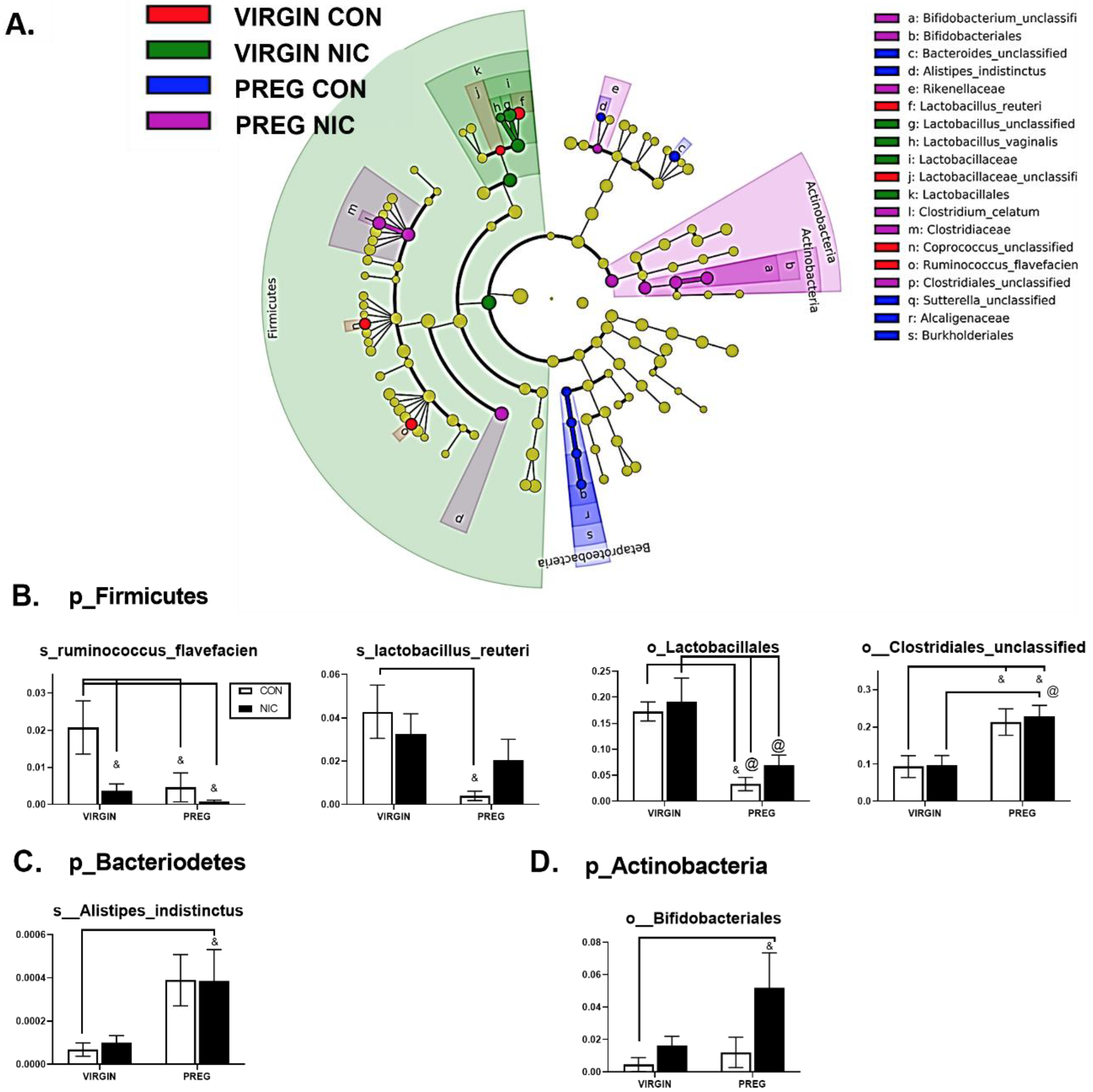
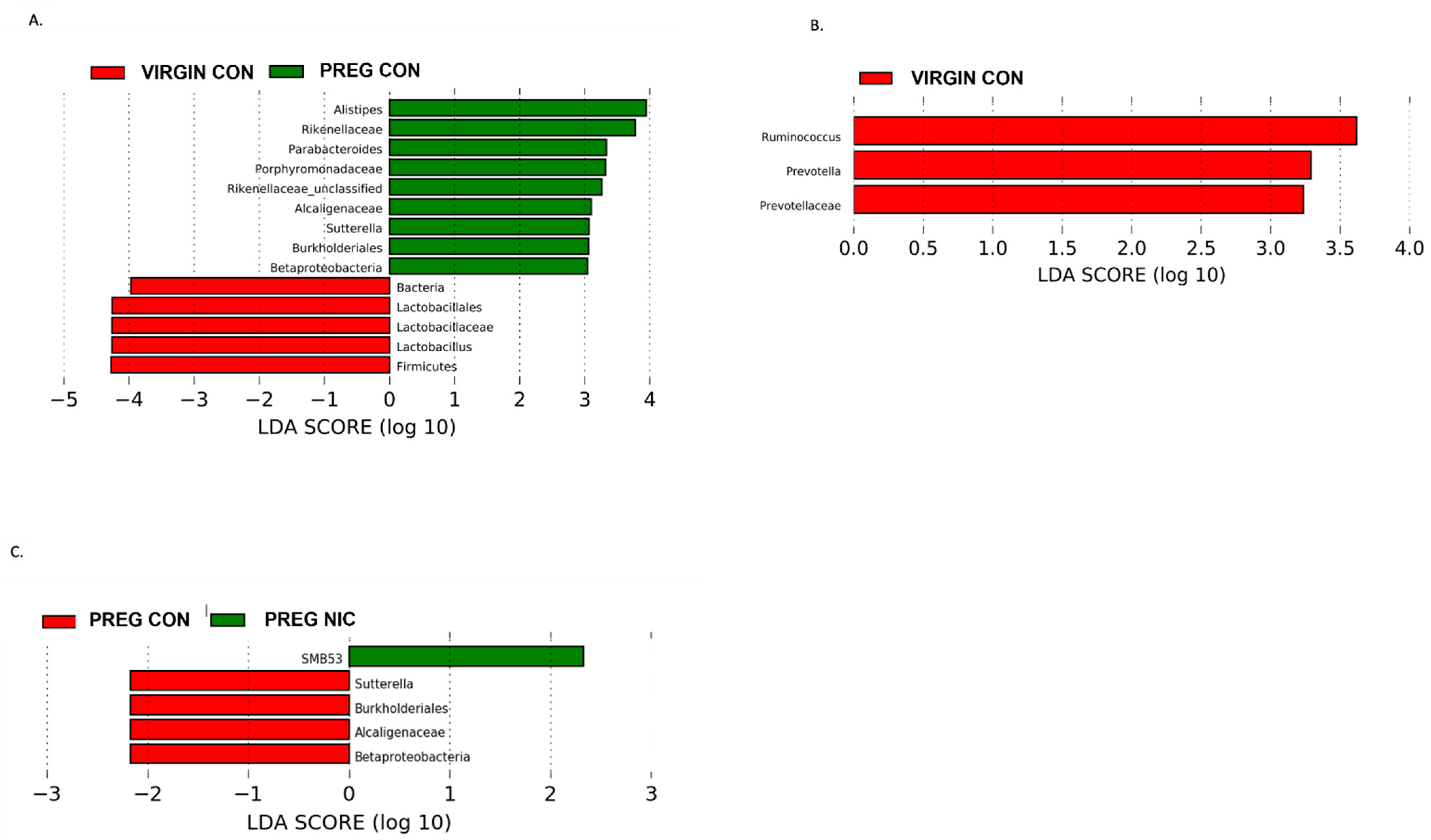
6.2. Effect of NIC and Pregnancy on the Female Gut Microbiome
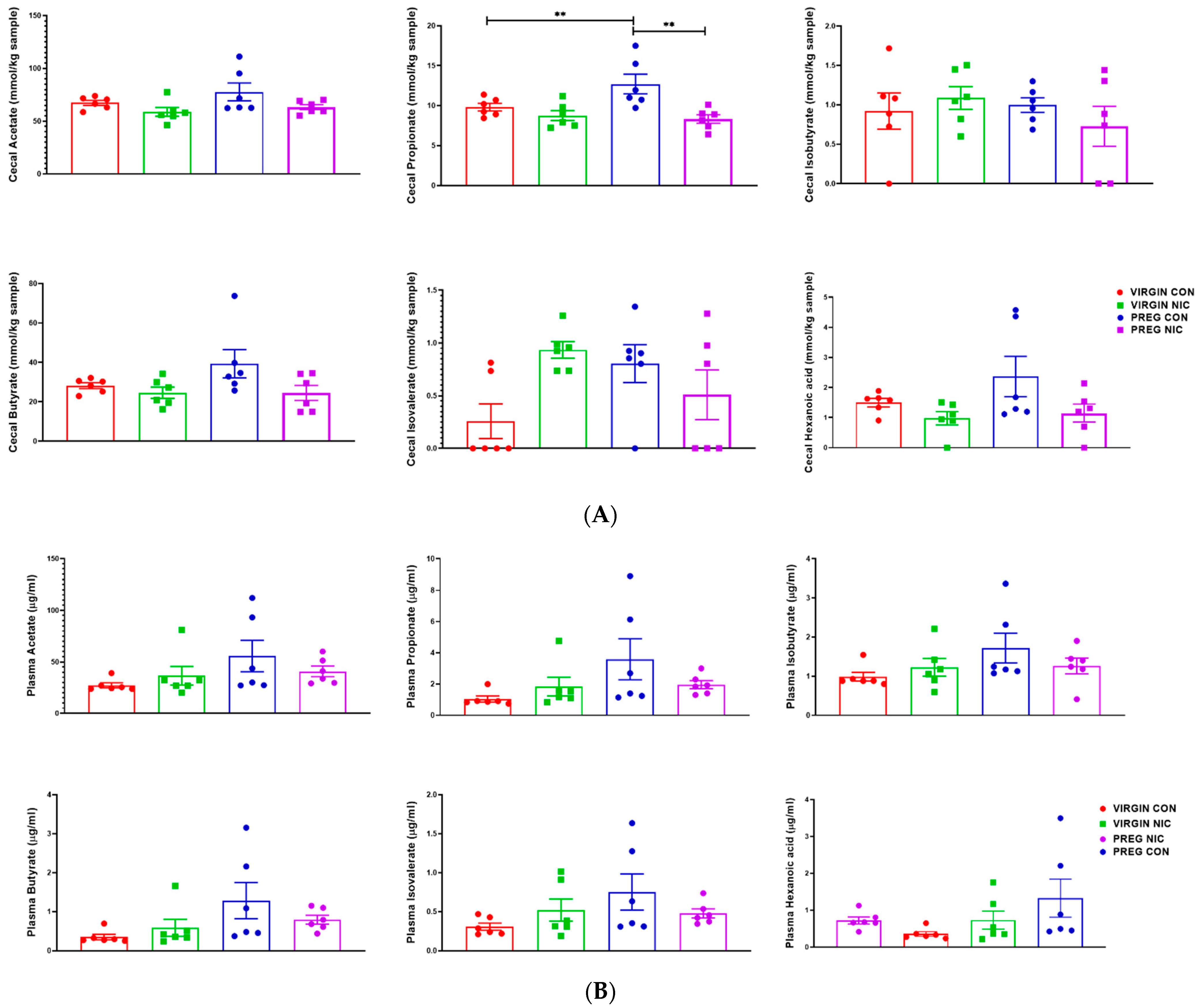
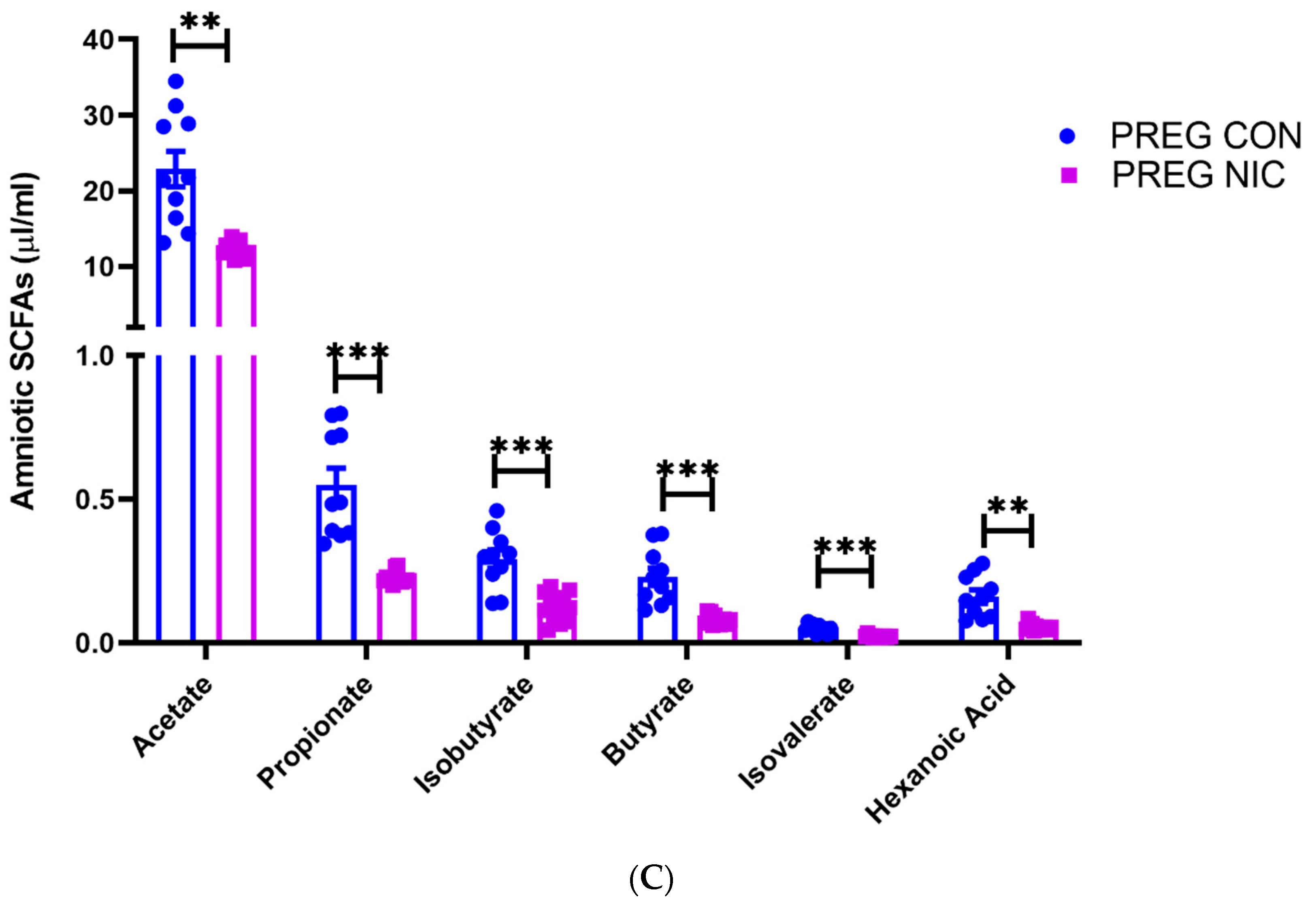
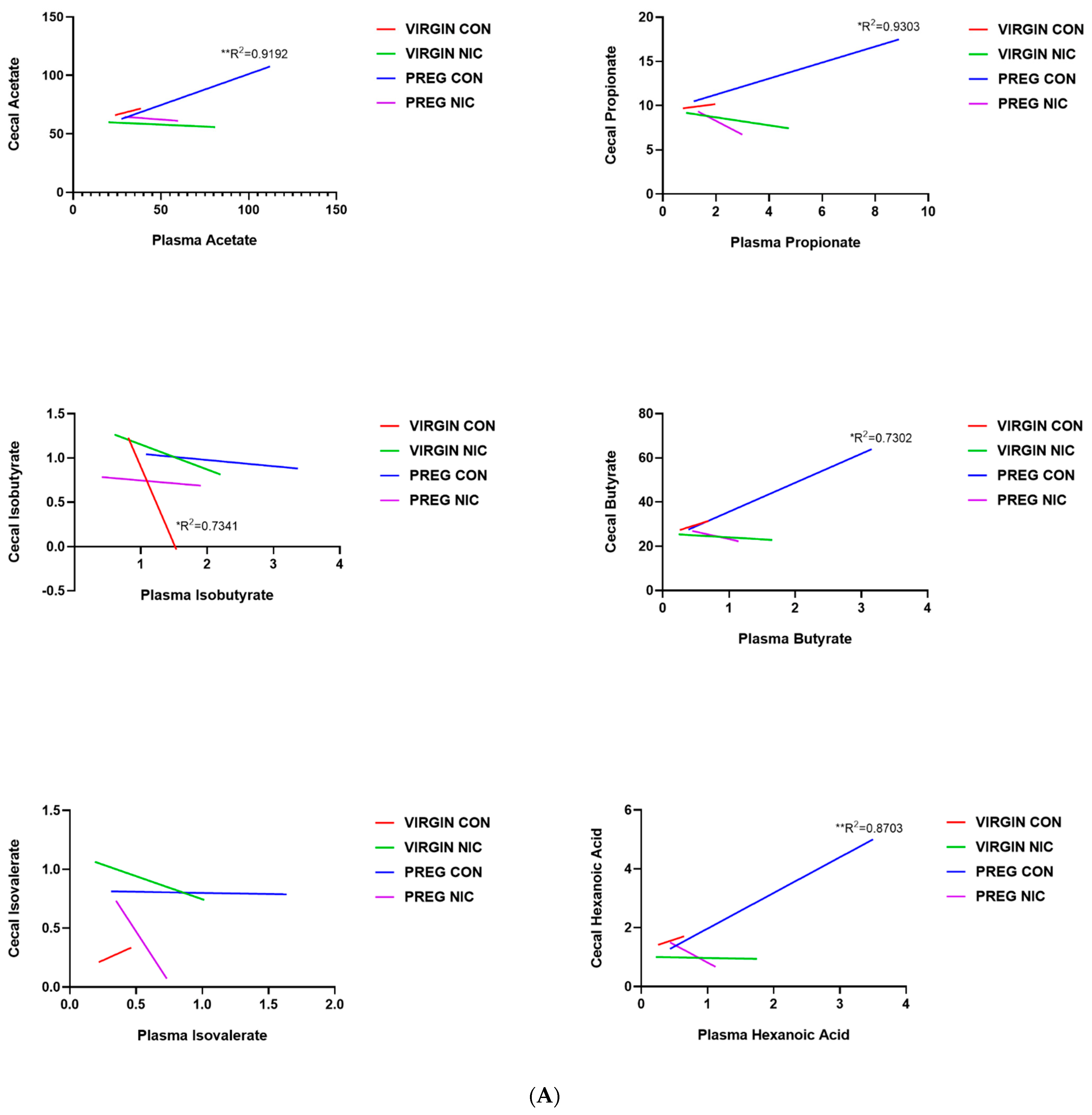
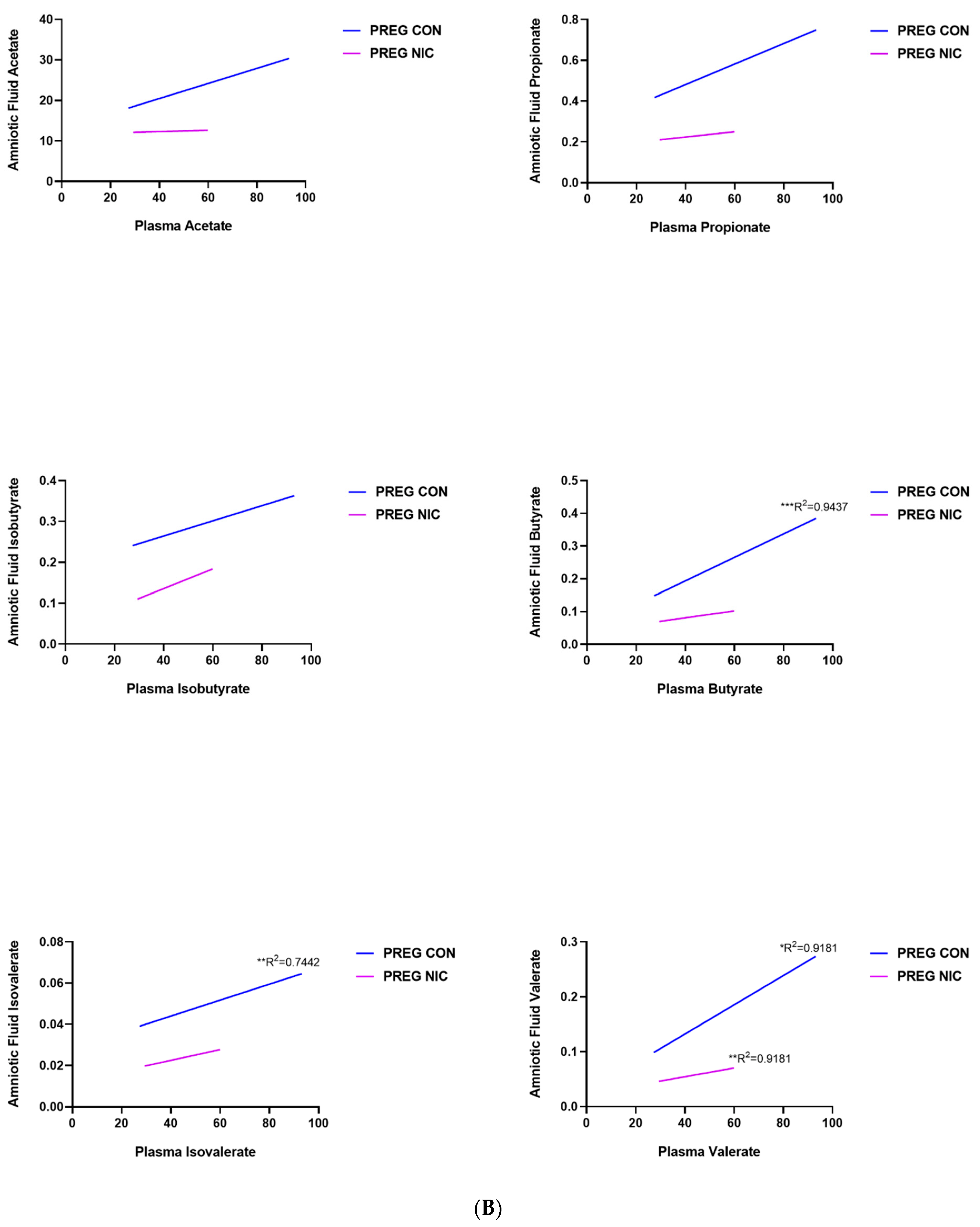
6.3. Effect of NIC and Pregnancy on SCFAs in the Female SD Rats
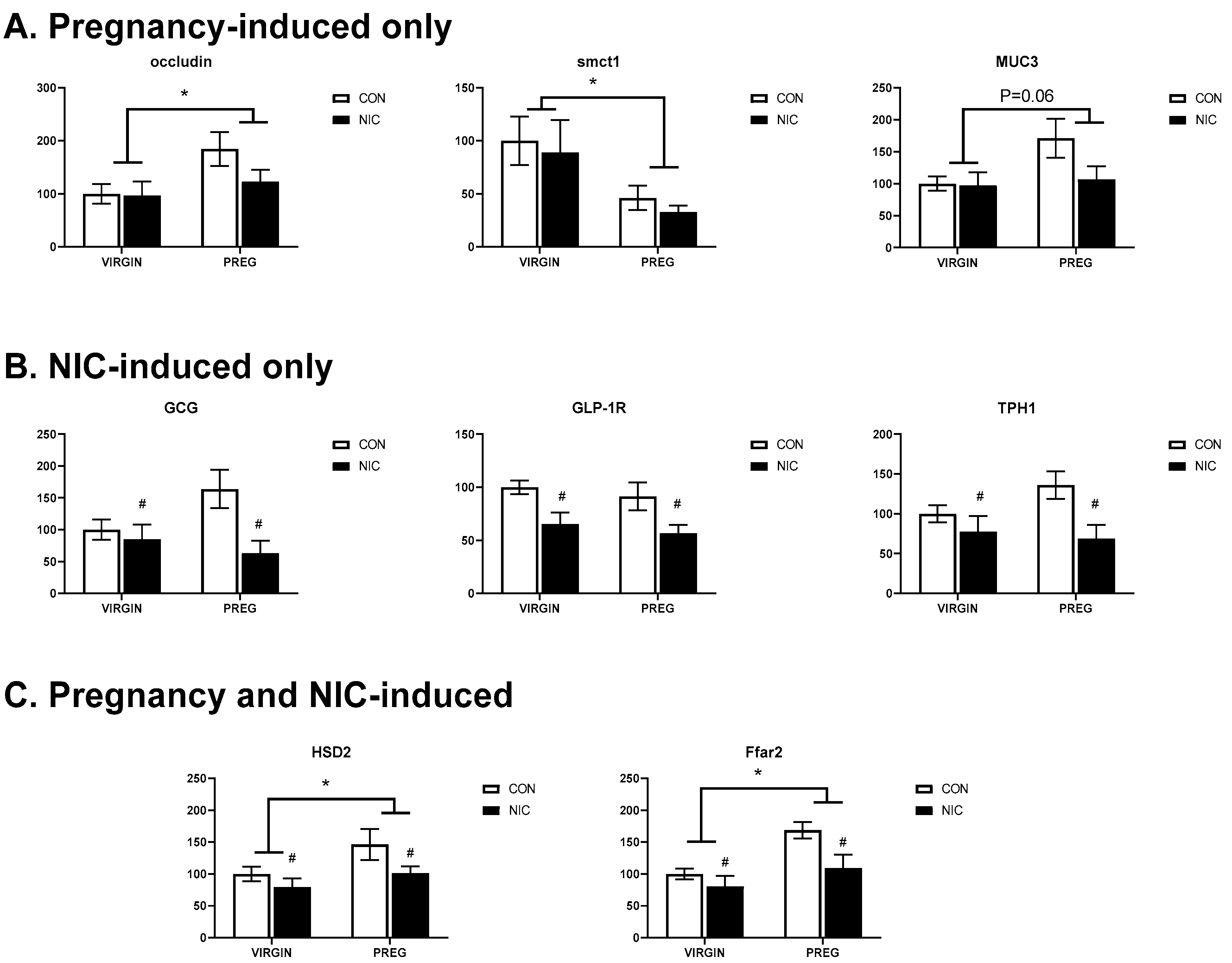
6.4. Pregnancy and NIC-Induced Changes in Cecal Tissue Relative Gene Expression
7. Discussion
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pineles, B.L.; Park, E.; Samet, J.M. Systematic review and meta-analysis of miscarriage and maternal exposure to tobacco smoke during pregnancy. Am. J. Epidemiol. 2014, 179, 807–823. [Google Scholar] [CrossRef]
- Shah, N.R.; Bracken, M.B. A systematic review and meta-analysis of prospective studies on the association between maternal cigarette smoking and preterm delivery. Am. J. Obstet. Gynecol. 2000, 182, 465–472. [Google Scholar] [CrossRef]
- Dahlin, S.; Gunnerbeck, A.; Wikström, A.-K.; Cnattingius, S.; Bonamy, A.-K.E. Maternal tobacco use and extremely premature birth—A population-based cohort study. BJOG 2016, 123, 1938–1946. [Google Scholar] [CrossRef]
- Pereira, P.P.D.S.; Da Mata, F.A.F.; Figueiredo, A.C.G.; De Andrade, K.R.C.; Pereira, M.G. Maternal Active Smoking during Pregnancy and Low Birth Weight in the Americas: A Systematic Review and Meta-analysis. Nicotine Tob. Res. 2017, 19, 497–505. [Google Scholar] [CrossRef]
- Jamshed, L.; Perono, G.A.; Jamshed, S.; Holloway, A.C. Early Life Exposure to Nicotine: Postnatal Metabolic, Neurobehavioral and Respiratory Outcomes and the Development of Childhood Cancers. Toxicol. Sci. 2020, 178, 3–15. [Google Scholar] [CrossRef]
- Shah, T.; Sullivan, K.; Carter, J. Sudden infant death syndrome and reported maternal smoking during pregnancy. Am. J. Public Health 2006, 96, 1757–1759. [Google Scholar] [CrossRef]
- Anderson, T.M.; Ferres, J.M.L.; Ren, S.Y.; Moon, R.Y.; Goldstein, R.D.; Ramirez, J.-M.; Mitchell, E.A. Maternal Smoking before and during Pregnancy and the Risk of Sudden Unexpected Infant Death. Pediatrics 2019, 143, e20183325. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, X. Maternal smoking and increased risk of sudden infant death syndrome: A meta-analysis. Leg. Med. 2012, 15, 115–121. [Google Scholar] [CrossRef]
- Stick, S. The effects of in-utero tobacco-toxin exposure on the respiratory system in children. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 312–316. [Google Scholar] [CrossRef]
- Spindel, E.R.; McEvoy, C.T. The Role of Nicotine in the Effects of Maternal Smoking during Pregnancy on Lung Development and Childhood Respiratory Disease. Implications for Dangers of E-Cigarettes. Am. J. Respir. Crit. Care Med. 2016, 193, 486–494. [Google Scholar] [CrossRef]
- Rayfield, S.; Plugge, E. Systematic review and meta-analysis of the association between maternal smoking in pregnancy and childhood overweight and obesity. J. Epidemiol. Community Health 2016, 71, 162–173. [Google Scholar] [CrossRef]
- Somm, E.; Schwitzgebel, V.; Vauthay, D.M.; Aubert, M.L.; Hüppi, P. Prenatal nicotine exposure and the programming of metabolic and cardiovascular disorders. Mol. Cell. Endocrinol. 2009, 304, 69–77. [Google Scholar] [CrossRef]
- Banderali, G.; Martelli, A.; Landi, M.; Moretti, F.; Betti, F.; Radaelli, G.; Lassandro, C.; Verduci, E. Short and long term health effects of parental tobacco smoking during pregnancy and lactation: A descriptive review. J. Transl. Med. 2015, 13, 327. [Google Scholar] [CrossRef]
- Vaglenova, J.; Birru, S.; Pandiella, N.M.; Breese, C.R. An assessment of the long-term developmental and behavioral teratogenicity of prenatal nicotine exposure. Behav. Brain Res. 2004, 150, 159–170. [Google Scholar] [CrossRef]
- SanMiguel, C.P.; Gupta, A.; Mayer, E.A. Gut Microbiome and Obesity: A Plausible Explanation for Obesity. Curr. Obes. Rep. 2015, 4, 250–261. [Google Scholar] [CrossRef]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra65. [Google Scholar] [CrossRef]
- Jašarević, E.; Howerton, C.L.; Howard, C.D.; Bale, T.L. Alterations in the Vaginal Microbiome by Maternal Stress Are Associated with Metabolic Reprogramming of the Offspring Gut and Brain. Endocrinology 2015, 156, 3265–3276. [Google Scholar] [CrossRef]
- Hu, Y.; Peng, J.; Tai, N.; Hu, C.; Zhang, X.; Wong, F.S.; Wen, L. Maternal Antibiotic Treatment Protects Offspring from Diabetes Development in Nonobese Diabetic Mice by Generation of Tolerogenic APCs. J. Immunol. 2015, 195, 4176–4184. [Google Scholar] [CrossRef]
- Foley, K.A.; MacFabe, D.F.; Kavaliers, M.; Ossenkopp, K.-P. Sexually dimorphic effects of prenatal exposure to lipopolysaccharide, and prenatal and postnatal exposure to propionic acid, on acoustic startle response and prepulse inhibition in adolescent rats: Relevance to autism spectrum disorders. Behav. Brain Res. 2015, 278, 244–256. [Google Scholar] [CrossRef]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef]
- Allais, L.; Kerckhof, F.-M.; Verschuere, S.; Bracke, K.; De Smet, R.; Laukens, D.; Abbeele, P.V.D.; De Vos, M.; Boon, N.; Brusselle, G.; et al. Chronic cigarette smoke exposure induces microbial and inflammatory shifts and mucin changes in the murine gut. Environ. Microbiol. 2015, 18, 1352–1363. [Google Scholar] [CrossRef]
- Biedermann, L.; Zeitz, J.; Mwinyi, J.; Sutter-Minder, E.; Rehman, A.; Ott, S.J.; Steurer-Stey, C.; Frei, A.; Frei, P.; Scharl, M.; et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS ONE 2013, 8, e59260. [Google Scholar] [CrossRef]
- Aagaard, K.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.-A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Veyver, I.V.D.; Milosavljevic, A.; et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef]
- Labrecque, M.T.; Caldwell, K.E.; Allan, A.M. Impact of Ethanol and Saccharin on Fecal Microbiome in Pregnant and Non-Pregnant Mice. J. Pregnancy Child Health 2015, 2, 100193. [Google Scholar] [CrossRef]
- Naqvi, S.; Asar, T.O.; Kumar, V.; Al-Abbasi, F.A.; Alhayyani, S.; Kamal, M.A.; Anwar, F. A cross-talk between gut microbiome, salt and hypertension. Biomed. Pharmacother. 2021, 134, 111156. [Google Scholar] [CrossRef]
- Ahmad, A.F.; Dwivedi, G.; O’Gara, F.; Caparros-Martin, J.A.; Ward, N.C. The gut microbiome and cardiovascular disease: Current knowledge and clinical potential. Am. J. Physiol.-Heart Circ. Physiol. 2019, 317, H923–H938. [Google Scholar] [CrossRef]
- Soliman, M.M.; Ahmed, M.M.; Salah-Eldin, A.-E.; Abdel-Aal, A.A.-A. Butyrate regulates leptin expression through different signaling pathways in adipocytes. J. Vet. Sci. 2011, 12, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Zeltser, L.M. Developmental influences on circuits programming susceptibility to obesity. Front. Neuroendocr. 2015, 39, 17–27. [Google Scholar] [CrossRef]
- Kirk, S.L.; Samuelsson, A.-M.; Argenton, M.; Dhonye, H.; Kalamatianos, T.; Poston, L.; Taylor, P.D.; Coen, C.W. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS ONE 2009, 4, e5870. [Google Scholar] [CrossRef]
- Choi, J.S. Effects of Maternal and Post-Weaning High-Fat Diet on Leptin Resistance and Hypothalamic Appetite Genes in Sprague Dawley Rat Offspring. Clin. Nutr. Res. 2018, 7, 276–290. [Google Scholar] [CrossRef]
- Behl, M.; Rao, D.; Aagaard, K.; Davidson, T.; Levin, E.D.; Slotkin, T.A.; Srinivasan, S.; Wallinga, D.; White, M.F.; Walker, V.R.; et al. Evaluation of the Association between Maternal Smoking, Childhood Obesity, and Metabolic Disorders: A National Toxicology Program Workshop Review. Environ. Health Perspect. 2013, 121, 170–180. [Google Scholar] [CrossRef]
- Ginzel, K.; Maritz, G.S.; Marks, D.F.; Neuberger, M.; Pauly, J.R.; Polito, J.R.; Schulte-Hermann, R.; Slotkin, T.A. Critical Review: Nicotine for the fetus, the infant and the adolescent? J. Health Psychol. 2007, 12, 215–224. [Google Scholar] [CrossRef]
- Slotkin, T.A.; MacKillop, E.A.; Rudder, C.L.; Ryde, I.T.; Tate, C.A.; Seidler, F.J. Permanent sex-selective effects of prenatal or adolescent nicotine exposure, separately or sequentially, in rat brain regions: Indices of cholinergic and serotinergic synaptic function, cell signaling, and neural cell number and size at 6 months of age. Neuropsychopharmacology 2007, 32, 1082–1097. [Google Scholar] [CrossRef]
- Fewell, J.; Smith, F.G.; Ng, V.K.Y.; Wong, V.H.; Wang, Y. Perinatal nicotine exposure impairs ability of newborn rats to autoresuscitate from apnea. J. Appl. Physiol. 2000, 85, 2066–2074. [Google Scholar] [CrossRef]
- Fewell, J.E.; Eliason, H.L.; Crisanti, K.C. Prenatal exposure to nicotine attenuates stress-induced hyperthermia in 7- to 8-week-old rats upon exposure to a novel environment. Physiol. Behav. 2001, 74, 595–601. [Google Scholar] [CrossRef]
- Fewell, J.E.; Smith, F.G.; Ng, V.K. Prenatal exposure to nicotine impairs protective responses of rat pups to hypoxia in an age-dependent manner. Respir. Physiol. 2001, 127, 61–73. [Google Scholar] [CrossRef]
- Boychuk, C.; Hayward, L.F. Prenatal nicotine exposure alters postnatal cardiorespriatory integration in young male but not female rats. Exp. Neurol. 2011, 232, 212–221. [Google Scholar] [CrossRef]
- Chang, G.-Q.; Karatayev, O.; Leibowitz, S.F. Prenatal exposure to nicotine stimulates neurogenesis of orexigenic peptide-expressing neurons in hypothalamus and amygdala. J. Neurosci. 2013, 33, 13600–13611. [Google Scholar] [CrossRef]
- Fewell, J.E.; Smith, F.G.; Ng, V.K.Y. Threshold levels of maternal nicotine impairing protective responses of newborn rats to intermittent hypoxia. J. Appl. Physiol. 2001, 90, 1968–1976. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Seidler, F.J. Mimicking maternal smoking and pharmacotherapy of preterm labor: Fetal nicotine exposure enhances the effect of late gestational dexamethasone treatment on noradrenergic circuits. Brain Res. Bull. 2011, 86, 435–440. [Google Scholar] [CrossRef]
- Mantella, N.M.; Kent, P.F.; Youngentob, S.L. Fetal nicotine exposure increases preference for nicotine odor in early postnatal and adolescent, but not adult, rats. PLoS ONE 2013, 8, e84989. [Google Scholar] [CrossRef]
- Von Chamier, M.; Reyes, L.; Hayward, L.F.; Brown, M.B. Impact of gestational nicotine exposure on intrauterine and fetal infection in a rodent model. Biol. Reprod. 2017, 96, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Celadilla, L.F.; Rueda, M.C.; Rodríguez, M.M. Intrauterine growth restriction in spontaneously hypertensive rats. Hypertens. Pregnancy 2004, 23, 275–283. [Google Scholar] [CrossRef]
- Pankevich, D.E.; Mueller, B.R.; Brockel, B.; Bale, T.L. Prenatal stress programming of offspring feeding behavior and energy balance begins early in pregnancy. Physiol. Behav. 2009, 98, 94–102. [Google Scholar] [CrossRef]
- Glover, V.; O’Connor, T.; O’Donnell, K. Prenatal stress and the programming of the HPA axis. Neurosci. Biobehav. Rev. 2010, 35, 17–22. [Google Scholar] [CrossRef]
- Tzschoppe, A.; Struwe, E.; Blessing, H.; Fahlbusch, F.; Liebhaber, G.; Doerr, H.G.; Rauh, M.; Rascher, W.; Goecke, T.W.; Schild, R.L.; et al. Placental 11beta-HSD2 gene expression at birth is inversely correlated with growth velocity in the first year of life after intrauterine growth restriction. Pediatr. Res. 2009, 65, 647–653. [Google Scholar] [CrossRef]
- Zhao, G.; Nyman, M.; Jönsson, J. Rapid determination of short-chain fatty acids in colonic contents and faeces of humans and rats by acidified water-extraction and direct-injection gas chromatography. Biomed. Chromatogr. 2005, 20, 674–682. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Boychuk, C.R.; Fuller, D.D.; Hayward, L.F. Sex differences in heart rate variability during sleep following prenatal nicotine exposure in rat pups. Behav. Brain Res. 2011, 219, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, J.R.; Chetrite, G.S. The formation and transformation of hormones in maternal, placental and fetal compartments: Biological implications. Horm. Mol. Biol. Clin. Investig. 2016, 27, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Magee, K.L.; Colon-Perez, L.M.; Larkin, R.; Liao, Y.-S.; Balazic, E.; Cowart, J.R.; Arocha, R.; Redler, T.; Febo, M.; et al. Impaired butyrate absorption in the proximal colon, low serum butyrate and diminished central effects of butyrate on blood pressure in spontaneously hypertensive rats. Acta Physiol. 2019, 226, e13256. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef]
- Petriz, B.A.; Castro, A.P.; Almeida, J.A.; Gomes, C.P.; Fernandes, G.R.; Kruger, R.H.; Pereira, R.W.; Franco, O.L. Exercise induction of gut microbiota modifications in obese, non-obese and hypertensive rats. BMC Genom. 2014, 15, 511. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in Lean and Overweight Healthy Subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Chakraborti, C.K. New-found link between microbiota and obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut Dysbiosis Is Linked to Hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Pindjakova, J.; Sartini, C.; Re, O.L.; Rappa, F.; Coupe, B.; Lelouvier, B.; Pazienza, V.; Vinciguerra, M. Gut Dysbiosis and Adaptive Immune Response in Diet-induced Obesity vs. Systemic Inflammation. Front. Microbiol. 2017, 8, 1157. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Ziętek, M.; Celewicz, Z.; Szczuko, M. Short-Chain Fatty Acids, Maternal Microbiota and Metabolism in Pregnancy. Nutrients 2021, 13, 1244. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, M.; Thomas, A.; Reisetter, A.C.; Scholtens, D.M.; Wolever, T.M.; Josefson, J.; Layden, B.T. Maternal short-chain fatty acids are associated with metabolic parameters in mothers and newborns. Transl. Res. 2014, 164, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Wyss, M.T.; Magistretti, P.J.; Buck, A.; Weber, B. Labeled acetate as a marker of astrocytic metabolism. J. Cereb. Blood Flow Metab. 2011, 31, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-E.; Hashimoto-Hill, S.; Woo, V.; Eshleman, E.M.; Whitt, J.; Engleman, L.; Karns, R.; Denson, L.A.; Haslam, D.B.; Alenghat, T. Microbiota-derived metabolite promotes HDAC3 activity in the gut. Nature 2020, 586, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Cupul-Uicab, L.A.; Skjaerven, R.; Haug, K.; Melve, K.K.; Engel, S.M.; Longnecker, M.P. In utero exposure to maternal tobacco smoke and subsequent obesity, hypertension, and gestational diabetes among women in the MoBa cohort. Environ. Health Perspect. 2012, 120, 355–360. [Google Scholar] [CrossRef]
- Xiao, D.; Xu, Z.; Huang, X.; Longo, L.D.; Yang, S.; Zhang, L. Prenatal Gender-Related Nicotine Exposure Increases Blood Pressure Response to Angiotensin II in Adult Offspring. Hypertension 2008, 51, 1239–1247. [Google Scholar] [CrossRef]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef]
- Boro, P.; Kumaresan, A.; Singh, A.; Gupta, D.; Kumar, S.; Manimaran, A.; Mohanty, A.; Pathak, R.; Attupuram, N.; Baithalu, R.; et al. Expression of short chain fatty acid receptors and pro-inflammatory cytokines in utero-placental tissues is altered in cows developing retention of fetal membranes. Placenta 2014, 35, 455–460. [Google Scholar] [CrossRef]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-Chain Fatty Acids Activate GPR41 and GPR43 on Intestinal Epithelial Cells to Promote Inflammatory Responses in Mice. Gastroenterology 2013, 145, 396–406.e1-10. [Google Scholar] [CrossRef]
- Kaji, I.; Karaki, S.-I.; Kuwahara, A. Short-Chain Fatty Acid Receptor and Its Contribution to Glucagon-Like Peptide-1 Release. Digestion 2014, 89, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Heinritz, S.N.; Weiss, E.; Eklund, M.; Aumiller, T.; Louis, S.; Rings, A.; Messner, S.; Camarinha-Silva, A.; Seifert, J.; Bischoff, S.C.; et al. Intestinal Microbiota and Microbial Metabolites Are Changed in a Pig Model Fed a High-Fat/Low-Fiber or a Low-Fat/High-Fiber Diet. PLoS ONE 2016, 11, e0154329. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Srinivasan, M.; Thamadilok, S.; Patel, M.S. Hypothalamic alterations in fetuses of high fat diet-fed obese female rats. J. Endocrinol. 2008, 200, 293–300. [Google Scholar] [CrossRef]
- García, M.C.G.; Casanueva, F.F.; Diéguez, C.; Señaris, R. Gestational Profile of Leptin Messenger Ribonucleic Acid (mRNA) Content in the Placenta and Adipose Tissue in the Rat, and Regulation of the mRNA Levels of the Leptin Receptor Subtypes in the Hypothalamus during Pregnancy and Lactation1. Biol. Reprod. 2000, 62, 698–703. [Google Scholar] [CrossRef]
- Karakosta, P.; Chatzi, L.; Plana, E.; Margioris, A.; Castanas, E.; Kogevinas, M. Leptin levels in cord blood and anthropometric measures at birth: A systematic review and meta-analysis. Paediatr. Périnat. Epidemiol. 2010, 25, 150–163. [Google Scholar] [CrossRef]
- Tessier, D.; Ferraro, Z.; Gruslin, A. Role of leptin in pregnancy: Consequences of maternal obesity. Placenta 2013, 34, 205–211. [Google Scholar] [CrossRef]
- Steinbrekera, B.; Roghair, R. Modeling the impact of growth and leptin deficits on the neuronal regulation of blood pressure. J. Endocrinol. 2016, 231, R47–R60. [Google Scholar] [CrossRef]
- Glahn, A.; Rhein, M.; Frieling, H.; Dette, F.; Bleich, S.; Hillemacher, T.; Muschler, M. Smoking-induced changes in leptin serum levels and c/EBPalpha-related methylation status of the leptin core promotor during smoking cessation. Psychoneuroendocrinology 2019, 100, 106–112. [Google Scholar] [CrossRef]
- Tomoda, K.; Kubo, K.; Nishii, Y.; Yamamoto, Y.; Yoshikawa, M.; Kimura, H. Changes of ghrelin and leptin levels in plasma by cigarette smoke in rats. J. Toxicol. Sci. 2012, 37, 131–138. [Google Scholar] [CrossRef]
- Nicklas, B.J.; Tomoyasu, N.; Muir, J.; Goldberg, A.P. Effects of cigarette smoking and its cessation on body weight and plasma leptin levels. Metabolism 1999, 48, 804–808. [Google Scholar] [CrossRef]
- Helland, I.B.; Reseland, J.E.; Saugstad, O.D.; Drevon, C.A. Smoking related to plasma leptin concentration in pregnant women and their newborn infants. Acta Paediatr. 2001, 90, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Chełchowska, M.; Ambroszkiewicz, J.; Mazur, J.; Lewandowski, L.; Maciejewski, T.M.; Oltarzewski, M.; Gajewski, J. Effect of tobacco smoking on the maternal and fetal adipokine axis in relation to newborn birth weight and length. Prz. Lek. 2014, 71, 567–571. [Google Scholar]
- Kayemba-Kay’S, S.; Geary, M.P.P.; Pringle, J.; Rodeck, C.H.; Kingdom, J.C.P.; Hindmarsh, P.C. Gender, smoking during pregnancy and gestational age influence cord leptin concentrations in newborn infants. Eur. J. Endocrinol. 2008, 159, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Reseland, J.E.; Mundal, H.H.; Hollung, K.; Haugen, F.; Zahid, N.; Anderssen, S.A.; Drevon, C.A. Cigarette smoking may reduce plasma leptin concentration via catecholamines. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 43–49. [Google Scholar] [CrossRef]
- Lisboa, P.C.; de Oliveira, E.; de Moura, E.G. Obesity and endocrine dysfunction programmed by maternal smoking in pregnancy and lactation. Front. Physiol. 2012, 3, 437. [Google Scholar] [CrossRef]
- Xu, D.; Liang, G.; Yan, Y.; He, W.; Liu, Y.; Chen, L.; Magdalou, J.; Wang, H. Nicotine-induced over-exposure to maternal glucocorticoid and activated glucocorticoid metabolism causes hypothalamic–pituitary–adrenal axis-associated neuroendocrine metabolic alterations in fetal rats. Toxicol. Lett. 2012, 209, 282–290. [Google Scholar] [CrossRef]
- Morgan, T.M.; Crawford, L.; Stoller, A.; Toth, D.; Yeo, K.-T.J.; Baron, J.A. Acute effects of nicotine on serum glucose insulin growth hormone and cortisol in healthy smokers. Metabolism 2004, 53, 578–582. [Google Scholar] [CrossRef]
- Luo, Z.-C.; Nuyt, A.-M.; Delvin, E.; Audibert, F.; Girard, I.; Shatenstein, B.; Cloutier, A.; Cousineau, J.; Djemli, A.; Deal, C.; et al. Maternal and Fetal IGF-I and IGF-II Levels, Fetal Growth, and Gestational Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 1720–1728. [Google Scholar] [CrossRef]
- Li, C.; Levitz, M.; Hubbard, G.; Jenkins, S.; Han, V.; Ferry, R.; McDonald, T.; Nathanielsz, P.; Schlabritz-Loutsevitch, N. The IGF Axis in Baboon Pregnancy: Placental and Systemic Responses to Feeding 70% Global Ad Libitum Diet. Placenta 2007, 28, 1200–1210. [Google Scholar] [CrossRef][Green Version]
- Olausson, H.; Sohlström, A. Effects of food restriction and pregnancy on the expression of insulin-like growth factors-I and -II in tIssues from guinea pigs. J. Endocrinol. 2003, 179, 437–445. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coan, P.M.; Vaughan, O.R.; Sekita, Y.; Finn-Sell, S.; Burton, G.J.; Constancia, M.; Fowden, A.L. Adaptations in placental phenotype support fetal growth during undernutrition of pregnant mice. J. Physiol. 2010, 588, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Sferruzzi-Perri, A.N. Regulating needs: Exploring the role of insulin-like growth factor-2 signalling in materno-fetal resource allocation. Placenta 2018, 64, S16–S22. [Google Scholar] [CrossRef]
- Cassidy, F.C.; Charalambous, M. Genomic imprinting, growth and maternal-fetal interactions. J. Exp. Biol. 2018, 221 (Suppl. S1), jeb164517. [Google Scholar] [CrossRef] [PubMed]
- Lopes, G.A.D.; Ribeiro, V.L.B.; Barbisan, L.F.; Rodrigues, M.A.M. Fetal developmental programing: Insights from human studies and experimental models. J. Matern. Neonatal Med. 2016, 30, 722–728. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, B.P.; Dicker, P.; Regan, C.L. Electronic cigarettes and obstetric outcomes: A prospective observational study. BJOG Int. J. Obstet. Gynaecol. 2020, 127, 750–756. [Google Scholar] [CrossRef]
- Furukawa, S.; Hayashi, S.; Usuda, K.; Abe, M.; Hagio, S.; Ogawa, I. Toxicological Pathology in the Rat Placenta. J. Toxicol. Pathol. 2011, 24, 95–111. [Google Scholar] [CrossRef]
- Ernst, M.; Moolchan, E.T.; Robinson, M.L. Behavioral and Neural Consequences of Prenatal Exposure to Nicotine. J. Am. Acad. Child Adolesc. Psychiatry 2001, 40, 630–641. [Google Scholar] [CrossRef]
- Feng, J.-H.; Yan, Y.-E.; Liang, G.; Liu, Y.-S.; Li, X.-J.; Zhang, B.-J.; Chen, L.-B.; Yu, H.; He, X.-H.; Wang, H. Maternal and fetal metabonomic alterations in prenatal nicotine exposure-induced rat intrauterine growth retardation. Mol. Cell. Endocrinol. 2014, 394, 59–69. [Google Scholar] [CrossRef]
- Gálvez-Prieto, B.; Bolbrinker, J.; Stucchi, P.; de Las Heras, A.I.; Merino, B.; Arribas, S.; Ruiz-Gayo, M.; Huber, M.; Wehland, M.; Kreutz, R.; et al. Comparative expression analysis of the renin-angiotensin system components between white and brown perivascular adipose tissue. J. Endocrinol. 2008, 197, 55–64. [Google Scholar] [CrossRef]
- Kamilic, J.; Hamming, I.; Kreutz, R.; Bolbrinker, J.; Siems, W.E.; Nassar, I.; Sluimer, J.C.; Walther, T.; Navis, G.J.; van Goor, H. Renal ACE2 expression and activity is unaltered during established hypertension in adult SHRSP and TGR (mREN2)27. Hypertens. Res. 2010, 33, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Aldubayan, M.A.; Ahmed, A.S.; Emara, A.M.; Ahmed, A.A.; Elgharabawy, R.M. Sinapic Acid Attenuates Cardiovascular Disorders in Rats by Modulating Reactive Oxygen Species and Angiotensin Receptor Expression. Oxidative Med. Cell. Longev. 2020, 2020, 1436858. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Xia, S.; Ragolia, L. Upregulation of AT2 receptor and iNOS impairs angiotensin II-induced contraction without endothelium influence in young normotensive diabetic rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R144–R154. [Google Scholar] [CrossRef] [PubMed]
- Briski, K.P.; Kale, A.Y.; Vavaiya, K.V. Impact of recurring intermediate insulin-induced hypoglycemia on hypothalamic paraventricular corticotropin-releasing hormone, oxytocin, vasopressin and glucokinase gene profiles: Role of type II glucocorticoid receptors. Exp. Brain Res. 2009, 195, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.A.; Fahmy, M.I.; Sayed, R.H.; El-Yamany, M.F.; El-Naggar, R.; Hegazy, A.A.; Al-Shorbagy, M. Eprosartan: A closer insight into its neuroprotective activity in rats with focal cerebral ischemia-reperfusion injury. J. Biochem. Mol. Toxicol. 2021, 35, e22796. [Google Scholar] [CrossRef]
- Ishikawa, K.; Tsunekawa, S.; Ikeniwa, M.; Izumoto, T.; Iida, A.; Ogata, H.; Uenishi, E.; Seino, Y.; Ozaki, N.; Sugimura, Y.; et al. Long-term pancreatic beta cell exposure to high levels of glucose but not palmitate induces DNA methylation within the insulin gene promoter and represses transcriptional activity. PLoS ONE 2015, 10, e0115350. [Google Scholar] [CrossRef]
- Dong, N.; Xu, B.; Xu, J. EGF-Mediated Overexpression of Myc Attenuates miR-26b by Recruiting HDAC3 to Induce Epithelial-Mesenchymal Transition of Lens Epithelial Cells. BioMed Res. Int. 2018, 2018, 7148023. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Chen, X.X.; Lin, H.; Fei, A.L.; Ge, R.S. 11beta-hydroxysteroid dehydrogenase types 1 and 2 in postnatal development of rat testis: Gene expression, localization and regulation by luteinizing hormone and androgens. Asian J. Androl. 2014, 16, 811–816. [Google Scholar] [CrossRef]
- Ye, X.; Kohtz, A.; Pollonini, G.; Riccio, A.; Alberini, C.M. Insulin Like Growth Factor 2 Expression in the Rat Brain Both in Basal Condition and following Learning Predominantly Derives from the Maternal Allele. PLoS ONE 2015, 10, e0141078. [Google Scholar] [CrossRef]
- Bae, G.D.; Park, E.Y.; Kim, K.; Jang, S.E.; Jun, H.S.; Oh, Y.S. Upregulation of caveolin-1 and its colocalization with cytokine receptors contributes to beta cell apoptosis. Sci. Rep. 2019, 9, 16785. [Google Scholar] [CrossRef]
- Hou, B.; Zhao, Y.; Qiang, G.; Yang, X.; Xu, C.; Chen, X.; Liu, C.; Wang, X.; Zhang, L.; Du, G. Puerarin Mitigates Diabetic Hepatic Steatosis and Fibrosis by Inhibiting TGF-beta Signaling Pathway Activation in Type 2 Diabetic Rats. Oxidative Med. Cell. Longev. 2018, 2018, 4545321. [Google Scholar] [CrossRef] [PubMed]
- Äijälä, M.; Malo, E.; Ukkola, O.; Bloigu, R.; Lehenkari, P.; Autio-Harmainen, H.; Santaniemi, M.; Kesäniemi, Y.A. Long-term fructose feeding changes the expression of leptin receptors and autophagy genes in the adipose tissue and liver of male rats: A possible link to elevated triglycerides. Genes Nutr. 2013, 8, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Merriman-Smith, R.; Donaldson, P.; Kistler, J. Differential expression of facilitative glucose transporters GLUT1 and GLUT3 in the lens. Investig. Ophthalmol. Vis. Sci. 1999, 40, 3224–3230. [Google Scholar]
- Jiang, M.; Li, C.; Liu, Q.; Wang, A.; Lei, M. Inhibiting Ceramide Synthesis Attenuates Hepatic Steatosis and Fibrosis in Rats with Non-alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Toral, M.; Robles-Vera, I.; De la Visitacion, N.; Romero, M.; Yang, T.; Sánchez, M.; Gómez-Guzmán, M.; Jiménez, R.; Raizada, M.K.; Duarte, J. Critical Role of the Interaction Gut Microbiota—Sympathetic Nervous System in the Regulation of Blood Pressure. Front. Physiol. 2019, 10, 231. [Google Scholar] [CrossRef] [PubMed]
- Han, K.S.; Balan, P.; Hong, H.D.; Choi, W.I.; Cho, C.W.; Lee, Y.C.; Moughan, P.J.; Singh, H. Korean ginseng modulates the ileal microbiota and mucin gene expression in the growing rat. Food Funct. 2014, 5, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; Humblot, C.; Noordine, M.L.; Wrzosek, L.; Tomas, J.; Mayeur, C.; Cherbuy, C.; Guyot, J.P.; Thomas, M. Behavior of lactobacilli isolated from fermented slurry (ben-saalga) in gnotobiotic rats. PLoS ONE 2013, 8, e57711. [Google Scholar] [CrossRef]
- Erjavec, I.; Bordukalo-Niksic, T.; Brkljacic, J.; Grcevic, D.; Mokrovic, G.; Kesic, M.; Rogic, D.; Zavadoski, W.; Paralkar, V.M.; Grgurevic, L.; et al. Constitutively Elevated Blood Serotonin Is Associated with Bone Loss and Type 2 Diabetes in Rats. PLoS ONE 2016, 11, e0150102. [Google Scholar] [CrossRef]
- López-Barradas, A.; González-Cid, T.; Vázquez, N.; Gavi-Maza, M.; Reyes-Camacho, A.; Velázquez-Villegas, L.A.; Ramírez, V.; Zandi-Nejad, K.; Mount, D.B.; Torres, N.; et al. Insulin and SGK1 reduce the function of Na+/monocarboxylate transporter 1 (SMCT1/SLC5A8). Am. J. Physiol. Cell Physiol. 2016, 311, C720–C734. [Google Scholar] [CrossRef]
- Katsurada, K.; Nandi, S.S.; Zheng, H.; Liu, X.; Sharma, N.M.; Patel, K.P. GLP-1 mediated diuresis and natriuresis are blunted in heart failure and restored by selective afferent renal denervation. Cardiovasc. Diabetol. 2020, 19, 57. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CON | NIC | |
|---|---|---|
| Virgin body weight | 216 ± 10.6 (n = 8) | 227 ± 18.1 (n = 8) |
| Pregnant body weight | 308 ± 11.5 (n = 6) | 328 ± 10.6 (n = 7) |
| Placental dry weight | 0.091 ± 0.008 (n = 16) | 0.086 ± 0.004 (n = 16) |
| Fetus dry weight | 0.41 ± 0.05 (n = 14) | 0.34 ± 0.06 (n = 15) |
| Dry fetus/placental weight | 4.36 ± 0.36 (n = 13) | 3.82 ± 0.20 (n = 15) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubcevic, J.; Watkins, J.; Lin, C.; Bautista, B.; Hatch, H.M.; Tevosian, S.G.; Hayward, L.F. Nicotine Exposure during Rodent Pregnancy Alters the Composition of Maternal Gut Microbiota and Abundance of Maternal and Amniotic Short Chain Fatty Acids. Metabolites 2022, 12, 735. https://doi.org/10.3390/metabo12080735
Zubcevic J, Watkins J, Lin C, Bautista B, Hatch HM, Tevosian SG, Hayward LF. Nicotine Exposure during Rodent Pregnancy Alters the Composition of Maternal Gut Microbiota and Abundance of Maternal and Amniotic Short Chain Fatty Acids. Metabolites. 2022; 12(8):735. https://doi.org/10.3390/metabo12080735
Chicago/Turabian StyleZubcevic, Jasenka, Jacqueline Watkins, Cindy Lin, Byrell Bautista, Heather M. Hatch, Sergei G. Tevosian, and Linda F. Hayward. 2022. "Nicotine Exposure during Rodent Pregnancy Alters the Composition of Maternal Gut Microbiota and Abundance of Maternal and Amniotic Short Chain Fatty Acids" Metabolites 12, no. 8: 735. https://doi.org/10.3390/metabo12080735
APA StyleZubcevic, J., Watkins, J., Lin, C., Bautista, B., Hatch, H. M., Tevosian, S. G., & Hayward, L. F. (2022). Nicotine Exposure during Rodent Pregnancy Alters the Composition of Maternal Gut Microbiota and Abundance of Maternal and Amniotic Short Chain Fatty Acids. Metabolites, 12(8), 735. https://doi.org/10.3390/metabo12080735