Abstract
Reproductive failure remains a significant challenge to the beef industry. The omics technologies have provided opportunities to improve reproductive efficiency. We used a multistaged analysis from blood profiles to integrate metabolome (plasma) and transcriptome (peripheral white blood cells) in beef heifers. We used untargeted metabolomics and RNA-Seq paired data from six AI-pregnant (AI-P) and six nonpregnant (NP) Angus-Simmental crossbred heifers at artificial insemination (AI). Based on network co-expression analysis, we identified 17 and 37 hub genes in the AI-P and NP groups, respectively. Further, we identified TGM2, TMEM51, TAC3, NDRG4, and PDGFB as more connected in the NP heifers’ network. The NP gene network showed a connectivity gain due to the rewiring of major regulators. The metabolomic analysis identified 18 and 15 hub metabolites in the AI-P and NP networks. Tryptophan and allantoic acid exhibited a connectivity gain in the NP and AI-P networks, respectively. The gene–metabolite integration identified tocopherol-a as positively correlated with ENSBTAG00000009943 in the AI-P group. Conversely, tocopherol-a was negatively correlated in the NP group with EXOSC2, TRNAUIAP, and SNX12. In the NP group, α-ketoglutarate-SMG8 and putrescine-HSD17B13 were positively correlated, whereas a-ketoglutarate-ALAS2 and tryptophan-MTMR1 were negatively correlated. These multiple interactions identified novel targets and pathways underlying fertility in bovines.
1. Introduction
Infertility has a negative impact on beef cattle production and the profit of the livestock industry. Despite technological advancements and improvements in management practices [1], reproductive failure remains a concern for cow-calf production due to the amount of capital invested [2]. Additionally, reproductive failure is one of the main reasons for culling, which reduces the longevity of the herd and affects the total profit of the producers [3]. Reproductive success relies on many factors, including breeding strategy, nutritional and health management, and genetic background [4]. Therefore, best management practices in heifer development often increase reproductive success [2].
Predominantly, producers have used traits, such as body condition score [5,6,7], reproductive tract score [8,9], and pelvic measurements [10,11], as tools to identify heifers with a high potential for fertility [5,6,7,8,9,10,11]. However, many heifers deemed reproductively mature even after the phenotypic assessments, fail to conceive [12]. Thus, new methods or tools are still required to identify and select animals with high fertility potential. Among the options, omics technologies have provided opportunities to understand the genetic mechanisms underlying fertility-related traits in cattle. Furthermore, the implementation of genomic evaluation and selection has increased the rate of genetic improvement [13]. Likewise, transcriptomics and metabolomics have provided promising results to assess fertility and identify potential biomarkers and gene signatures.
A plethora of transcriptomic studies from embryo [14,15], endometrium [16,17,18,19,20], uterine tissue [21], and peripheral white blood cells [22,23] have identified candidate genes for bovine fertility. Moorey et al. identified RPL39, SMIM26, N6AMT1, LONRF3, and GATA3 as differentially expressed in peripheral white blood cells associated with pregnancy outcomes in beef heifers [22]. Mazzoni et al. identified regulators, such as IGF1, AGT, TNF, PI3K, FGF2, TGF-β1, and related biological mechanisms important for pregnancy establishment in endometrial transcriptome [18]. Similarly, Geary et al. identified IGJ, IGLL1-2, CLCA1, PLA2G2F, and SHISA6 as differentially expressed in the endometrial biopsies of subfertile and fertile heifers [24]. Likewise, metabolomic profiling of blood [25,26,27] and follicular fluid [28,29] shed light on fertility-related metabolites. Gomez et al. identified pregnancy-specific metabolomic biomarkers, such as 2-oxoglutaric acid, 2-hydroxybutyric acid, dimethylamine, and ketoleucine, in the blood plasma of recipient beef cows [30]. Despite the insights into the novel genes or metabolites reported in these studies, these approaches rely solely on data from a single biological layer. In addition, differential expression analysis disregards the multiple interactions between genes or metabolites and their coordinated expression pattern across samples. Thus, a holistic approach that exploits the genomic layers and their interactions is still required to improve our understanding of the genetic architecture of fertility-related traits.
While the abovementioned studies provide insights into the molecular basis of fertility, to the best of our knowledge, there is a paucity of information about the integrated relationship between fertility, gene expression, and metabolite abundance. We hypothesized that fertility potential is partially determined by the co-expression of genes and metabolites and their coordinated interaction. Thus, our main objective was to unravel the transcriptomic and metabolomic architecture and the biological pathways underlying heifer fertility. To accomplish this, we integrated peripheral white blood cells transcriptome and plasma metabolome profiles using a network approach to unveil functional candidate genes and metabolites involved with heifer fertility. Additionally, we shed light on fertility-related pathways derived from the gene–metabolite pairs.
2. Results
2.1. Transcriptome and Metabolome Profiles
We used the RNA-Seq expression profile from peripheral white blood cells and the plasma metabolome to ascertain the putative mechanisms underlying fertility differences between AI-P and NP; six animals were in each group of beef heifers. A schematic representation of the study design and analysis steps is given in Figure 1.
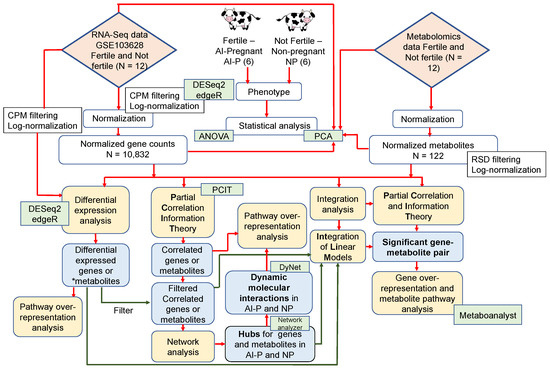
Figure 1.
Schematic representation of the study design and analysis steps. The yellow boxes represent the analysis steps; the green boxes represent the software/packages used; and the blue boxes represent the output obtained. The red connectors point to the main analysis steps, while the green connectors are the analysis after filtering. * Differentially expressed metabolites retrieved from Phillips et al. [25].
After quality control (see Section 4.2), we maintained 10,832 genes out of 24,616 from 12 heifers for differential expression analysis. Based on a combined approach using edgeR and DESeq2 (see Section 4), we identified 38 differentially expressed genes (DEGs, p-value ≤ 0.05 and absolute (log2 fold change ≥ 0.5)) (Figure 2, Supplementary Table S1). Among the DEGs, 29 genes were upregulated and 9 were downregulated in the NP group. Upregulated genes (top five in the list) included TFF2, TAC3, CNKSR3, ALAS2 and MYH10, while the downregulated genes (top five in the list) included SAXO2, ENSBTAG00000039555, ENSBTAG00000034871, ART3, and ENSBTAG00000048169.
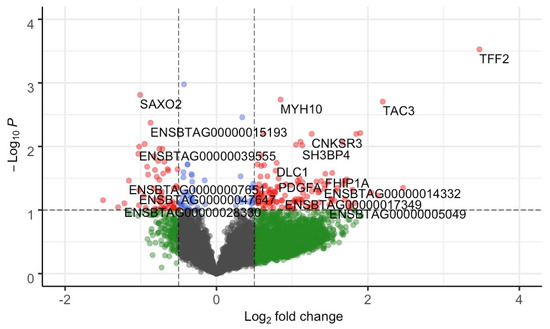
Figure 2.
Volcano plot of gene-based differential expression analysis for AI-P (n = 6) and NP (n = 6) crossbred heifers. Each dot corresponds to a gene. The x-axis represents log2 fold change for the difference in expression in the AI-P and NP groups. The y-axis corresponds to the negative log (base 10) of the p-value. Grey dots represent nonsignificant genes that did not cross the threshold of p-value or fold-change; green dots represent the genes with absolute (log2 fold change ≥ 0.5); blue dots represent the genes with significant p-value; and red dots represent 38 DEGs (p-value ≤ 0.05 and absolute (log2 fold change ≥ 0.5)). The genes on the left panel (0 to −2 of log2 fold change) were downregulated, while the genes on the right panel (0 to 4 of log2 fold change) were upregulated.
2.2. Gene Network and Functional Over-Representation Analyses
To identify genes or metabolites with coordinated expression patterns within the AI-P and NP groups, we used a co-expression network approach based on the partial correlation and information theory (PCIT) algorithm [31]. For each group, two gene networks and two metabolite networks were created.
For the gene network, the correlation of 10,832 genes identified 3,263,960 significantly correlated gene pairs in the AI-P group, while 5,058,326 significant correlations were identified for the NP group. To reduce data dimensionality and retrieve biologically meaningful information, we maintained the significantly correlated pairs with an absolute correlation greater than 0.99 (p-value ≤ 0.05) and co-expressed with the DEGs (Supplementary Table S1). After filtering, we maintained 320 and 844 significantly correlated gene pairs for the AI-P and NP groups, respectively (corresponding to 340 and 802 unique genes) (Supplementary Table S2).
To identify hub genes, we used the degree and betweenness centrality parameters from the Network Analyzer (Supplementary Table S3). After filtering, 17 hub genes were identified in the AI-P and 37 hub genes in the NP networks (Supplementary Table S3). Among them, CYTH3, SPN, TMEM51, GRB10, FHIP1A, TLCD3A, TGM2 and PDGFB were identified as shared hubs in both the AI-P and NP networks. The ENSBTAG00000047816, ENSBTAG00000015193, ENSBTAG00000009943 and ABI2 genes were unique to the AI-P group and over-represented biological processes, including innate immune response, regulation of actin dynamics for phagocytic cup formation, and signaling by VEGF and RHO GTPases activating WASPs and WAVEs signaling pathways. The SH3BP4, PDGFA, ART3, TAC3, PLEKHF1, NDRG4, SMG8, PCBP1, F2RL2, DIP2C, EXOSC2, ZNF16, TRNAU1AP, TTK, ALAS2, HSD17B13, SNX12, WWOX, MTMR1 and TFF2 genes were unique to the NP group. However, no significant biological processes were over-represented for these genes.
To visualize differences in connectivity between the AI-P and NP gene networks, we built a central reference network using DyNet. This network comprised 1095 nodes (genes) and 1164 edges (interactions) (Figure 3a). The nodes were filtered for betweenness centrality to highlight the hub genes from the AI-P and NP networks (Figure 3b).
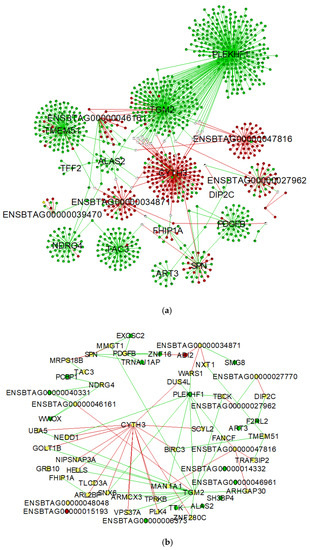
Figure 3.
Network comparison based on rewired nodes (genes) of AI-P and NP networks. Central reference network constructed using DyNet in Cytoscape. (a) The network comprises 1095 nodes and 1164 edges. The hub targets connected to the network are represented in this network. (b) The network was filtered for betweenness centrality to display the hub genes from both groups. The unique nodes are red (AI-P) or green (NP). Shared nodes between AI-P and NP are yellow.
The differential connectivity was calculated between the groups based on the frequency of the gene degree, followed by a z-score transformation for the 1095 genes. For the z-scores, 97.5 percentile points for standard normal distribution were considered to retrieve the genes with a z-score ± 1.96 (p-value ≤ 0.05). The NP group was used as the reference to determine the gain or loss in gene connectivity (Supplementary Table S4). Among the 1095 genes, PLEKHF1, TGM2, TMEM51, TAC3, NDRG4 and PDGFB exhibited a gain in connectivity in the NP group. Conversely, CYTH3, ENSBTAG00000027962, ENSBTAG00000034871 and ENSBTAG00000047816 genes were more connected in the AI-P group.
The co-expressed genes, including DEGs, hubs, and differentially connected genes, were used for pathway over-representation analysis separately for the AI-P and NP groups. For the AI-P, over-represented pathways were oocyte meiosis, Wnt signaling pathway, glucagon signaling pathway, propanoate metabolism, N-glycan biosynthesis and protein processing in the endoplasmic reticulum. Likewise, in the NP group, over-represented pathways included MAPK signaling pathway, ubiquitin-mediated proteolysis and mitophagy (Supplementary Table S5).
2.3. Metabolite Network Analyses
Similar to the gene network analysis, we used PCIT to identify co-expressed metabolite pairs. To this end, after quality control, 112 and 116 metabolites were maintained for the AI-P and NP groups, respectively. The PCIT analysis found 6216 significantly correlated metabolite pairs in the AI-P group (p-value ≤ 0.05). Similarly, 6670 metabolite pairs were identified in the NP group. Previously, Phillips et al. reported fifteen differentially expressed metabolites (DEMs) [25]. We then filtered these networks based on the 15 DEMs and significantly correlated with an absolute correlation greater than 0.90 (p-value ≤ 0.05). Considering this approach, 18 and 15 metabolite pairs for the AI-P and NP groups (corresponding to 23 unique metabolites in each) were maintained for further analysis (Supplementary Table S6).
Based on the degree and betweenness centrality topological parameters calculated by NetworkAnalyzer, we found eight and four hub metabolites for the AI-P and NP networks, respectively (Supplementary Table S7). Ornithine and cysteine were identified as hubs of both the AI-P and NP networks. Allantoic acid, methionine, putrescine, phenylethylamine, kynurenine and xylitol were unique to the AI-P group. Likewise, tryptophan and glutamine were unique to the NP network. The central reference metabolite network was constructed with DyNet for the AI-P and NP and comprised 37 nodes and 33 edges (Figure 4).
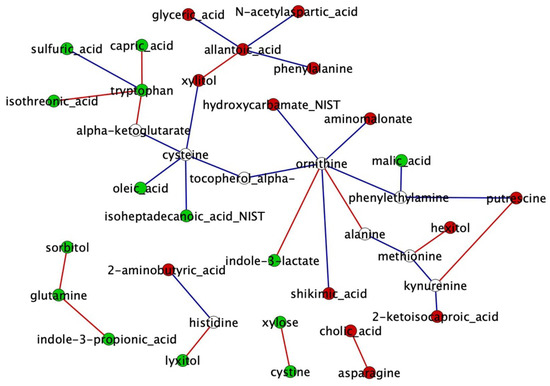
Figure 4.
Metabolite network comparison based on rewired nodes (metabolites) from the AI-P and NP groups. A central reference network was constructed using DyNet in Cytoscape. The unique nodes are red (AI-P) or green (NP). Shared nodes between AI-P and NP are white. The edges are red (negative correlation) and blue (positive correlation).
Next, we identified differentially connected metabolites between the AI-P and the NP groups. The differential connectivity was calculated between the groups based on the frequency of the metabolite degree for the 37 metabolites. Metabolites with z-scores ± 1.96 were significantly differentially connected (p-value ≤ 0.05). The gain or loss connectivity was calculated based on the NP group (Supplementary Table S8). Among 37 metabolites, tryptophan and allantoic acid were more connected in the NP and AI-P networks, respectively.
2.4. Gene–Metabolite Interaction Network
A combined PCIT analysis for 122 metabolites and 10,832 genes resulted in 3,280,267 and 5,075,367 significant co-expressed pairs, including gene–gene, metabolite–metabolite and gene–metabolites pairs, which were retrieved in the AI-P and NP groups, respectively. Only the significant gene–metabolite correlations were filtered based on the unique targets (see Section 4) from the AI-P and NP groups. To identify those targets, we considered the hub genes and metabolites (Supplementary Tables S3 and S7) and the significantly differentially connected genes and metabolites (Supplementary Tables S4 and S8) for each group. Thus, we retrieved 29 and 43 unique genes and metabolites from the AI-P and NP groups, respectively. Among these, 21 genes and metabolites (targets) were shared between both groups, eight were exclusive to the AI-P group and 22 to the NP group (Figure 5). The unique targets were then used as features for selecting gene–metabolite pairs with a correlation greater than 0.75 (p-value ≤ 0.05). Based on that, we identified 1161 gene–metabolite pairs in the AI-P network and 155 in the NP networks (Figure 6, Supplementary Table S9). These pairs were then used for functional over-representation analysis in MetaboAnalyst (Supplementary Table S10). In the AI-P group, the gene–metabolite pairs were over-represented by oocyte meiosis, progesterone-mediated oocyte maturation, and the PI3-Akt signaling pathway. In contrast, glyoxylate and dicarboxylate metabolism, alanine, aspartate and glutamate metabolism, and glucagon metabolism were over-represented in the NP group.
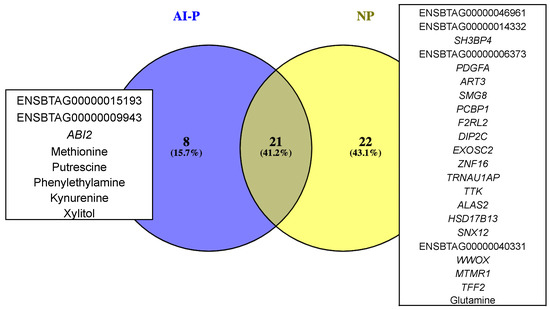
Figure 5.
Gene and metabolite targets exclusive for the AI-P and NP groups.
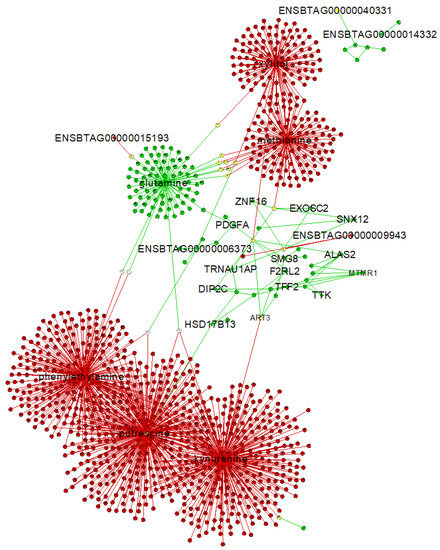
Figure 6.
Gene–metabolite interaction network from the AI-P and the NP groups. The unique exclusive targets are highlighted. A central reference network was constructed using DyNet in Cytoscape. The unique nodes are red (AI-P) or green (NP). Shared nodes between AI-P and NP are yellow.
To retrieve the gene–metabolite pairs based on the Integration of Linear model (IntLIM) approach, we utilized 1098 genes and 40 metabolites that were differentially expressed (DEGs and DEMs) or targets (co-expressed, hub genes, and differentially connected) for the AI-P and NP groups. After filtering for 5% variation, 1043 genes and 40 metabolites were maintained for data integration. This analysis resulted in 3177 gene–metabolite pairs (p-value ≤ 0.05) (Supplementary Table S11—Panel A). Next, we maintained 714 pairs (Supplementary Table S11—Panel B) by filtering with the unique exclusive targets as given in Figure 5.
Lastly, we overlapped the results from the PCIT and IntLIM to explore those gene–metabolite pairs that were identified in both approaches. These pairs included tocopherol-α, which was positively correlated in the AI-P group with ENSBTAG00000009943 and negatively correlated in the NP group with EXOSC2, TRNAUIAP and SNX12. In the NP group, the α-ketoglutarate-SMG8 and putrescine-HSD17B13 pairs were positively correlated, whereas α-ketoglutarate-ALAS2 and tryptophan-MTMR1 were negatively correlated.
3. Discussion
Currently, beef heifers are selected based on phenotypic assessments. However, a proportion of heifers that pass phenotypically remain NP after the breeding season. Understanding the genetic basis involved in complex traits, such as fertility, will be advantageous in efficiently selecting beef heifers and benefit cow-calf production. The phenotypic outcome of genomic and transcriptomic influences on the molecular state of the cell is reflected by the metabolome [32]. Therefore, in the context of reproductive biology, we focused on a multilayered approach to how regulation of upstream gene expression patterns exert changes on downstream metabolic indicators in the AI-P and NP beef heifers. In our study, we used the RNA-Seq approach to measure the expression of genes and untargeted metabolomics to analyze the metabolite concentration in the blood. Although fertility involves the regulation of multiple tissue organs, we focused on transcriptome and metabolome profiles in the blood because of two reasons: (i) The measurement of a specific product in the blood would be a noninvasive and valuable biomarker for predicting fertility in beef heifers, and (ii) circulating free-RNA and metabolites in the blood provides a potential window into health, phenotype, and developmental status of the organs in mammals [33,34,35]. Studies have reported blood biomarkers that predict infertility or pregnancy outcomes in humans [36,37] and cattle [30]. Furthermore, changes in metabolism affect the transport of nutrients from the blood to the uterine fluid altering fertility [30]. Considering all these factors, we used peripheral white blood cells and plasma to identify novel targets for fertility in beef heifers.
To systematically identify all molecules and their interactions exhibited in the blood for fertility, we used the gene and metabolite co-expression network approach. This approach identified the correlations and recognized the genes and metabolites playing a role in fertility. To accomplish this, we first identified the DEGs and retrieved the DEMs from our previous study [25]. Next, we built co-expression networks and used the DEGs and DEMs as features to prioritize candidate genes and metabolites. We identified the hubs and differentially connected genes and metabolites with significant biological roles, followed by an integrated analysis of the gene–metabolite pairs underlying fertility. Additionally, we identified a gain in gene connectivity in the network from the NP group compared to the AI-P group. This suggests network rewiring of major regulators modulating target gene expression with response to fertility status.
We identified 38 DEGs and 15 DEMs from blood plasma between the AI-P and NP beef heifers. Among the DEGs, TAC3 (tachykinin precursor 3) was identified as upregulated and a hub with a network connectivity gain in the NP group. The TAC3 gene encodes a neuropeptide essential to regulating human reproductive function. Supporting human reproductive biology, TAC3 has been identified in the bloodstream of patients with central pubertal disorders and hypogonadotropic hypogonadism [38,39]. The protein encoded by TAC3 modulates gonadotropin-releasing hormone (GnRH), which is responsible for the secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) [40]. However, TAC3 requires the presence of sex steroids to increase FSH and LH secretion [41]. Thus, the upregulation of TAC3 identified in blood, and yet the heifers not becoming pregnant may suggest an alteration in the hormonal and sex-steroid levels. FSH and LH signaling also regulate several pathways in ovarian granulosa cells, such as the mitogen-activated protein kinase (MAPK) pathway [42]. The MAPK pathway consists of protein kinases that regulate cell proliferation, inflammatory response, development, differentiation, and apoptosis [42]. In our study, the MAPK pathway was significantly over-represented in the NP group. Among the associated genes acting on the MAPK pathway, we identified PDGFB (platelet-derived growth factor) as significantly upregulated and differentially connected with a connectivity gain in the NP group. PDGFB, identified as a constituent of blood serum and platelets [43], modulates the primordial to primary follicle transition [44], which is essential for female fertility [45,46]. Studies have reported that PDGFB signaling promotes epithelial–mesenchymal transition (EMT), which is essential for embryogenesis [47]. EMT regulates critical processes during embryo development, and in the absence of EMT, development cannot proceed past the blastula stage [48]. The MAPK pathway allows cells to interpret external signals, specifically during EMT [49]. Furthermore, the MAPK pathway regulates extracellular signal-regulated kinase (ERK) proteins involved in the primary regulation of GnRH to stimulate the secretion of estradiol and progesterone [42]. In our study, PDGFB is negatively and differentially correlated with the hydrocinnamic acid in the NP group. Hydrocinnamic acid is an important class of phenolic acid, having antioxidant and anti-inflammatory properties [50]. Phenolic acids, also called polyphenols, have been associated with fertility, development, fetal health, and the bioavailability of nutrients [51]. A previous study reported that polyphenols inhibited the MAPK signaling pathway and mediated oxidative stress and inflammation [52]. Therefore, these findings suggest that the over-expression of the PDGFB gene and the downregulation of hydrocinnamic acid in the NP group triggered the MAPK signaling pathway, likely regulating the inflammatory response. Controlled inflammation is necessary for host defense, while an uncontrolled inflammatory response may affect fertility [53]. However, further validation of PDGFB and hydrocinnamic acid in reproductive cells or tissue types could highlight precise downstream mechanisms underlying fertility.
We identified the over-represented oocyte meiosis and Wnt signaling pathways in the AI-P group. The genes included ANAPC13, CAMK2G, CCNE2, PPP2R1B, PPP2R5A and PPP3CA. The CAMK2G gene encodes a calcium/calmodulin-dependent protein kinase involved with GnRH release [54]. CAMK2G was positively correlated with TGM2 (Transglutaminase 2) as given through PCIT in our study. TGM2 was upregulated and identified as a hub in the NP group. This result was further supported as TGM2 was significantly upregulated in uterine fluid collected from infertile women [55]. Furthermore, TGM2 levels were higher in the blood of patients with endometrial cancer [56]. TGM2 activates the β-catenin (Wnt) pathway, which impairs ovulation [57]. Balanced Wnt signaling is essential for the proper development of reproductive tract organs and fertility [58]. A previous study suggested overactivation of Wnt signaling has no effect on folliculogenesis but causes fertility failure due to abnormal fetal development [58]. Although the Wnt signaling pathway was over-represented in the AI-P group, differential regulation of co-expressed genes may regulate infertility in the NP group.
The PI3-Akt signaling pathway was over-represented by significant gene–metabolite pairs in the AI-P group. PI3-Akt signaling pathway plays an essential role in cell proliferation, apoptosis, metabolism, regulation of dormancy, and activation of mammalian primordial follicles [59]. Decreased PI3-Akt promotes Bax (a pro-apoptosis protein) translocation to the mitochondria and subsequent release of cytochrome c triggering apoptosis through the caspase pathway [59]. Higher apoptosis of granulosa cells has been associated with empty follicles and fewer or poor-quality oocytes and embryos in humans [60]. Furthermore, emerging evidence indicates the interrelationship of the PI3-Akt signaling pathway with pregnancy outcomes [61]. Akt deficient mice exhibited fewer mature follicles, reducing fertility [62]. This indicates the potential role of PI3-Akt in the AI-P group. This was further supported by one of the genes, NDRG4 (N-Myc downstream-regulated gene 4), downregulated in the AI-P group. NDRG4 has a crucial role in transferring its repression effect on PI3-Akt activity [63]. Studies in humans reported a negative feedback loop between NDRG4 activation and consequent PI3-Akt suppression [63], which could explain the role of NDRG4 in infertility via PI3-Akt suppression. Apart from being upregulated, NDRG4 was identified as a hub with a network connectivity gain in the NP group. Previous studies reported the NDRG4 expression in other reproductive tissue types [64], but its role in bovine fertility has not been reported yet. These findings suggest that NDRG4 has a potential role in regulating fertility and could be further explored.
Insulin-like growth factors modulate pathways, such as MAPK and PI3-Akt, by regulating gamete development, quality, and implantation [65]. Our study identified GRB10 (growth factor receptor bound protein 10) that was upregulated and differentially connected (loss in connectivity) in the NP group. GRB10 interacts negatively with insulin and insulin-like growth factor receptors [66,67,68]. Paulo et al. reported the involvement of the GRB10 gene during oocyte maturation in the bovine cumulus–oocyte complex [69]. The association of single nucleotide polymorphism in GRB10 with superovulation traits in cattle suggested the recovery of many embryos from heterozygous GRB10 cattle [70]. These studies suggested the role of GRB10 in regulating fertility, but a comprehensive understanding of bovine fertility requires further investigation.
The network analysis identified ART3, MTMR1, ALAS2, TFF2, SMG8, PLEKHF1 and EXOSC2 as unique hub genes in the NP group. ART3 (ADP-Ribosyltransferase-3) was downregulated and positively correlated with ornithine, putrescine and alpha-ketoglutarate in the NP group. As a representative arginine-specific ART-ribosyltransferase, ART3 has been reported to play a critical role in cell division, DNA repair, and inflammatory response [71]. Arginine and ornithine are precursors of nitric oxide and polyamines, respectively. In mammals, the ovaries produce ornithine decarboxylase (ODC) and putrescine during ovulation [72]. In previous studies, aged mice with reduced fertility exhibited low levels of ODC in ovaries; however, that did not improve after diet supplementation [72]. Interestingly, putrescine supplementation during in vitro maturation of aged mouse oocytes improved the quality of blastocysts [73]. These findings leave open questions about ART3 and its correlated metabolites (ornithine and putrescine) in the NP group.
Tryptophan was identified as differentially connected with a connectivity gain in the NP group. Tryptophan was negatively correlated with MTMR1 (myotubularin-related protein 1), a hub gene in the NP network. Tryptophan levels are influenced by dietary and hormonal factors [74]. Free tryptophan (not bound to albumin) is the protein carrier in plasma that is available to be taken by tissues and organs [75]. The previous literature shows that, besides fulfilling the demands of maternal proteins, tryptophan is required for fetal growth and development [76]. The need for tryptophan varies as the pregnancy progresses. In early and mid-pregnancy, increased maternal tryptophan availability helps meet the demand for protein synthesis and fetal development in humans [76]. Furthermore, tryptophan is reported as an antioxidant with a suppressive activity against cytochrome P-450-dependent lipid peroxidation in oxidative stress [77]. Additionally, levels of tryptophan were lower in women suffering from preeclampsia [77]. This evidence suggests that high levels of tryptophan are required during the initial phases of pregnancy. Downregulation of tryptophan could be one of the possible reasons for the infertility of beef heifers in our study. Evidence regarding the antioxidant effects of tryptophan has been reported, but the mechanism of action regulating fertility in bovines has not been reported yet [77].
The genes, metabolites, and their interactions identified in the present study over-represented some pathways playing a role in fertility. Some of the genes and metabolites were supported by the previous literature; however, some other targets identified were novel and warrant further detailed studies to evaluate the repeatability of our results in a larger cohort. As the heifers undergo variations during the estrus cycle and pregnancy, it is obvious to expect alterations in genes and metabolite profiles at a particular time point. This means that strategies for applying this information in real-time farming appear to be more complex and need a better understanding of the changes in genes, metabolites, and their interactions at different time points. Validation of these results in more samples and different time points would help to establish a framework for future fertility prediction using gene and metabolite biomarker profiles that could be practical for on-farm use and improve reproductive efficiency. Furthermore, validation of these genes and metabolites in the reproductive tissues and cells would help to identify the therapeutic targets for fertility.
4. Materials and Methods
4.1. Data Collection, Gene Expression, and Metabolite Profile
The RNA-Seq and metabolomic datasets previously generated by our group and publicly available were used in the current study. The experimental design, sample collection, and data generation were published elsewhere [23,25]. Briefly, blood samples from Angus-Simmental crossbred heifers were collected at AI and used for transcriptomic profiling of peripheral white blood cells and plasma metabolites.
Raw gene counts of 12 samples classified as AI-P or NP with six animals in each group from “station B” were downloaded from the GEO database (accession number GSE103628) [23]. RNA-Seq quality control and mapping were reported by Dickinson et al. [23]. From the same samples, blood plasma untargeted metabolomic profile was performed on gas chromatography (GC) coupled to time-of-flight mass spectrometry (TOF-MS) (GC-TOF-MS—Agilent 6890 GC). Analytical details were described by Phillips et al. [25].
4.2. Gene and Metabolite Differential Expression Analyses
Prior to statistical analysis, the read count data were transformed to counts per million (CPM) using edgeR v3.28.1 [78]. For the quality control, raw counts were filtered out based on the following criteria: (i) transcripts with zero counts (unexpressed); (ii) transcripts with less than 1 CPM on average (very lowly expressed); and (iii) transcripts that were not present in at least 50% of the samples (rarely expressed) [79].
We used the following two methods for the differential expression analysis: edgeR and DESeq2. This was performed to increase the robustness of our analysis and capture the biological variation. Thus, DEGs identified by both tools with a p-value ≤ 0.05 and absolute (log2 fold change) ≥ 0.5 were considered significant. The NP heifers were used as the reference group, and DEGs were classified as up- or downregulated based on the log2 fold change direction. The upregulated and downregulated genes were visualized using a volcano plot constructed using the R-package EnhancedVolcano v1.8.0 [80]. The fold change (FC) estimates by both the packages were similar. Thus, the FC output from DESeq2 was used.
For metabolomics, raw data files were preprocessed and quantification was reported as peak height using unique ion as default [25]. For each group of metabolites, only those with a relative standard deviation ≤0.15 were used based on the raw counts. Since this study focuses on the interactions between metabolites, the data were log normalized to unfold the differences in the AI-P and NP groups [35,81,82]. Based on this dataset, we retrieved 15 DEMs [25]. The DEMs and DEGs were then used as features for network filtering, as detailed below.
4.3. Gene and Metabolite Co-Expression Networks
The partial correlation and information theory (PCIT) approach was used to identify expression patterns and relationships within each regulatory layer (transcriptome and metabolome). PCIT reports the significantly correlated pairs after comparing all possible trios of genes or metabolites [31]. Gene co-expression networks were created considering 10,832 genes maintained after quality control (see methods in Section 4.2). Based on the normTransform function from DESeq2, read counts were normalized and used as input for PCIT analysis. For the metabolite networks, 122 metabolites were used after data normalization (see methods in Section 4.2).
Gene and metabolite networks were created separately for the AI-P and NP groups. This approach allowed us to identify changes in the network topology between conditions by comparing the connectivity of genes and metabolites. To reduce data dimensionality, a more stringent correlation value was used to select the gene pairs. Significantly correlated pairs (p-value ≤ 0.05) were determined based on partial correlation values greater than absolute (r ≥ 0.99) and absolute (r ≥ 0.9) for genes and metabolites, respectively. Additionally, significant pairs were maintained when a DEG or DEM was co-expressed.
Network topological parameters, including degree and betweenness centrality, were computed by the Cytoscape v3.8.2 [83] plugin Network Analyzer tool [84] and used for identifying hubs. The highly connected hubs were determined using the degree ≥ 1 and betweenness centrality ≥ 0 in each group for genes and metabolites and were referred to as “hub targets”. Based on network assumptions, highly connected nodes (genes or metabolites) are central to the network architecture and may be the primary regulators of biologically meaningful pathways.
To visualize changes in nodes and edge rewiring from the AI-P and NP groups, we used the Cytoscape plug-in—DyNet [85]. DyNet provides a central reference network constructed from the co-expressed pairs and highlights the most rewired nodes between the groups. Further, we identified the differentially connected genes or metabolites within each network. To this end, we calculated the differential connectivity (DK) index, which is the ratio of the node (gene or metabolite) connectivity to the maximum connectivity for each group [86]. The index is given by the following equation: . To determine the significance level, the index was transformed into a z-score (, where is the mean DKi and is the standard deviation of DKi. Significantly differentially connected genes or metabolites (referred to as differentially connected targets) were retrieved when z-score values were between from the standard deviation (p-value ≤ 0.05). The networks were constructed and visualized using Cytoscape v3.8.2.
4.4. Transcriptomic and Metabolomic Data Integration
Two approaches were used to unravel the complex relationship between genes and metabolites within the AI-P and NP groups. First, we created a single co-expression network combining the normalized genes (10,832) and metabolites (122) on PCIT to identify significantly correlated gene–metabolite pairs [87]. Only gene–metabolite pairs with absolute correlation values ≥ 0.75 and co-expressed with a hub or differentially connected targets (see methods in Section 4.3) were considered for further analysis.
To get an insight into the specific networks involved with fertility status, the correlated pairs were filtered with the “unique” targets. Herein, unique targets refer to the overlapping hub and differentially connected targets between the AI-P and NP groups. The networks with significant gene–metabolite pairs for the AI-P and NP groups were visualized in Cytoscape. Likewise, nodes and edge rewiring within the interaction networks were performed using DyNet (Cytoscape v3.8.2).
The second approach was based on a linear model framework using the IntLIM (Integration of Linear model) R-package [88]. IntLIM integrates the metabolomic and transcriptomic data considering a case-control design. For the present study, the differentially expressed (DEGs and DEMs), co-expressed hubs, and differentially connected targets previously identified from genes and metabolites for the AI-P and NP groups were used for the integration. The genes with the lowest 5% variation were excluded from the study as a quality control step. The linear model for data integration is given below:
where:
m = b1 + b2 g + b3 p+ b4 (g:p) + e
- m: is the log-normalized metabolite abundance;
- b1: is the intercept;
- b2g: is the normalized and adjusted gene-expression level;
- b3p: is the phenotype (AI-P and NP);
- b4 (g:p): is the interaction between gene expression and phenotype; and
- e: is the residual effect associated with each observation.
The absolute difference in the Spearman correlation was identified between the groups to identify significant gene–metabolite pairs (p-value ≤ 0.05). The absolute difference between the groups was estimated as (rPreg_cor—rNP_cor), where rPreg_cor and rNP_cor are the correlation values for the AI-P and NP groups.
4.5. Pathway Over-Representation Analysis
The pathway over-representation for DEGs, co-expressed, hub, and differentially connected genes were performed using ClueGO v2.5.4 and ShinyGO webtool. These approaches allowed us to identify the KEGG pathways and significant biological processes underlying differences within each group [89,90]. The redundant terms were clustered with a kappa score of 0.4, and the Bos taurus annotation was used as the background in ClueGO. Furthermore, the pathways were selected based on the p-value corrected for Bonferroni step down ≤ 0.05.
For the significant gene–metabolite pairs, joint-pathway analysis was performed using MetaboAnalyst 5.0 specific to Bos taurus. We used a hypergeometric test for enrichment, betweenness centrality as a topological measure and combined the p-values at the pathway level for integration. Pathways with p-value ≤ 0.05 (FDR ≤ 0.1) were considered significant.
5. Conclusions
The current study focused on identifying putative major regulators associated with fertility. To this end, we investigated the differences in the gene or metabolite expression and co-expression in beef heifers. Additionally, an integrated gene–metabolite analysis approach using correlation and linear model unveiled the potential gene–metabolite pairs affecting biological processes related to fertility in beef heifers. This research’s originality lies in multiple approaches used in determining gene–gene, metabolite–metabolite, and gene–metabolite interactions at AI. A detailed understanding of these targets and their underlying pathways in a bigger cohort at AI and at different time points could establish a framework for fertility prediction. More data and validation of these results would help model the complex relation of gene–metabolite profile over time.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/metabo12080708/s1. Table S1: Differentially expressed genes identified among AI-pregnant and nonpregnant groups. The genes up/downregulated are reported for nonpregnant heifers. Table S2: The genes correlated with 38 DEGs identified with the PCIT approach, with correlation ≥0.99. Table S3: Network analysis of genes co-expressed with DEGs in AI-pregnant and nonpregnant groups. The hub genes for each group are highlighted in bold. Table S4: Central reference network, including AI-pregnant and nonpregnant groups constructed using 1095 nodes. The genes highlighted in bold have a z-score of ± 1.96; the connectivity is given as gain or loss in the nonpregnant group. Table S5: Pathway over-representation analysis with significant genes from AI-pregnant and nonpregnant. Table S6: The metabolites correlated with 15 DEMs identified with the PCIT approach, with correlation ≥0.90. Table S7: Network analysis of metabolites co-expressed with DEMs in AI-pregnant and nonpregnant groups. The hub metabolites for each group are highlighted in bold. Table S8: Central reference metabolite network, including AI-pregnant and nonpregnant groups constructed using 37 nodes. The genes highlighted in bold have a z-score of ± 1.96; the connectivity is given as gain or loss in the nonpregnant group. Table S9: The gene–metabolite pairs correlated with significant targets (8 in AI-P and 22 in NP) identified with the PCIT approach, with correlation ≥0.75. Table S10: Significant pathways enriched by gene–metabolite pair identified from PCIT. Table S11: Gene–metabolite pairs by IntLIM approach.
Author Contributions
Conceptualization, P.B. and P.W.D.; methodology, P.B., S.P.R., W.J.S.D. and P.W.D.; software, P.B.; writing—original draft preparation, P.B.; writing—review and editing, P.B., W.J.S.D., P.W.D. and S.P.R.; funding acquisition, P.W.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Alabama Cattlemen’s Association, grant no. FF-NIA19-0000000048 from the Foundation for Food and Agriculture Research and by the Alabama Agricultural Experiment Station. The research was also supported by USDA—Agricultural Research Service—USDA-58-6010-1-005.
Institutional Review Board Statement
All procedures involving animals were approved by the Auburn University Institutional Animal Care and Use Committee (IACUC) 2015-2786 and conform with Planning Research and Experimental Procedures on Animals: Recommendations for Excellence (PREPARE) and Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
Informed Consent Statement
Not applicable.
Data Availability Statement
The transcriptomic data is available in the GEO database (accession number GSE103628). The metabolites data analyzed are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moorey, S.E.; Biase, F.H. Beef Heifer Fertility: Importance of Management Practices and Technological Advancements. J. Anim. Sci. Biotechnol. 2020, 11, 97. [Google Scholar] [CrossRef]
- Hindman, M.S.; Engelken, T.J. Beef Heifer Development. In Bovine Reproduction; Wiley Online library: Hoboken, NJ, USA, 2021; pp. 359–365. [Google Scholar] [CrossRef]
- Wathes, D.C.; Brickell, J.S.; Bourne, N.E.; Swali, A.; Cheng, Z. Factors Influencing Heifer Survival and Fertility on Commercial Dairy Farms. Animal 2008, 2, 1135–1143. [Google Scholar] [CrossRef]
- Bach, À. Effects of Nutrition and Genetics on Fertility in Dairy Cows. Fertil. Dev. 2019, 31, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Herd, D.B.; Sprott, L.R. Body Condition, Nutrition and Reproduction of Beef Cows. AgriLife Ext. Tex. AM Syst. 1998, B-1526, 1–12. [Google Scholar]
- Pryce, J.E.; Coffey, M.P.; Simm, G. The Relationship Between Body Condition Score and Reproductive Performance. J. Dairy Sci. 2001, 84, 1508–1515. [Google Scholar] [CrossRef]
- Markusfeld, O.; Galon, N.; Ezra, E. Body Condition Score, Health, Yield and Fertility in Dairy Cows. Vet. Rec. 1997, 141, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Pence, M.; Ensley, D.; Berghaus, R.; Rossi, J.; Wilson, T.; Cannon, P.T. Improving Reproductive Efficiency Through the Use of Reproductive Tract Scoring in a Group of Beef Replacelllent Heifers. Bov. Pract. 2007, 41, 35–40. [Google Scholar]
- Anderson, K.J.D.; LeFever, D.G.; Brinks, J.S.; Odde, K.G. The Use of Reproductive Tract Scoring in Beef Heifers. Agric. Pract. 1991, 12, 19–26. [Google Scholar]
- Larson, R.L.; White, B.J.; Laflin, S. Beef Heifer Development. Vet. Clin. N. Am. Food Anim. Pract. 2016, 32, 285–302. [Google Scholar] [CrossRef]
- Neville, W.E.; Mullinix, B.G.; Smith, J.B.; McCormick, W.C. Growth Patterns for Pelvic Dimensions and Other Body Measurements of Beef Females. J. Anim. Sci. 1978, 47, 1080–1088. [Google Scholar] [CrossRef]
- Dickinson, S.E.; Elmore, M.F.; Kriese-Anderson, L.; Elmore, J.B.; Walker, B.N.; Dyce, P.W.; Rodning, S.P.; Biase, F.H. Evaluation of Age, Weaning Weight, Body Condition Score, and Reproductive Tract Score in Pre-Selected Beef Heifers Relative to Reproductive Potential. J. Anim. Sci. Biotechnol. 2019, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Wiggans, G.R.; VanRaden, P.M.; Cooper, T.A. The Genomic Evaluation System in the United States: Past, Present, Future. J. Dairy Sci. 2011, 94, 3202–3211. [Google Scholar] [CrossRef] [PubMed]
- Mamo, S.; Mehta, J.P.; McGettigan, P.; Fair, T.; Spencer, T.E.; Bazer, F.W.; Lonergan, P. RNA Sequencing Reveals Novel Gene Clusters in Bovine Conceptuses Associated with Maternal Recognition of Pregnancy and Implantation1. Biol. Reprod. 2011, 85, 1143–1151. [Google Scholar] [CrossRef]
- Graf, A.; Krebs, S.; Zakhartchenko, V.; Schwalb, B.; Blum, H.; Wolf, E. Fine Mapping of Genome Activation in Bovine Embryos by RNA Sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 4139–4144. [Google Scholar] [CrossRef] [PubMed]
- Forde, N.; Carter, F.; Fair, T.; Crowe, M.A.; Evans, A.C.O.; Spencer, T.E.; Bazer, F.W.; McBride, R.; Boland, M.P.; O’Gaora, P.; et al. Progesterone-Regulated Changes in Endometrial Gene Expression Contribute to Advanced Conceptus Development in Cattle. Biol. Reprod. 2009, 81, 784–794. [Google Scholar] [CrossRef]
- Bauersachs, S.; Ulbrich, S.E.; Gross, K.; Schmidt, S.E.M.; Meyer, H.H.D.; Wenigerkind, H.; Vermehren, M.; Sinowatz, F.; Blum, H.; Wolf, E. Embryo-Induced Transcriptome Changes in Bovine Endometrium Reveal Species-Specific and Common Molecular Markers of Uterine Receptivity. Reproduction 2006, 132, 319–331. [Google Scholar] [CrossRef]
- Mazzoni, G.; Pedersen, H.S.; Rabaglino, M.B.; Hyttel, P.; Callesen, H.; Kadarmideen, H.N. Characterization of the Endometrial Transcriptome in Early Diestrus Influencing Pregnancy Status in Dairy Cattle after Transfer of in Vitro-Produced Embryos. Physiol. Genom. 2020, 52, 269–279. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Ramos, R.S.; Pugliesi, G.; Andrade, S.C.S.; Van Hoeck, V.; Langbeen, A.; Oliveira, M.L.; Gonella-Diaza, A.M.; Gasparin, G.; Fukumasu, H.; et al. Endometrial Transcriptional Profiling of a Bovine Fertility Model by Next-Generation Sequencing. Genom. Data 2016, 7, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Chankeaw, W.; Lignier, S.; Richard, C.; Ntallaris, T.; Raliou, M.; Guo, Y.; Plassard, D.; Bevilacqua, C.; Sandra, O.; Andersson, G.; et al. Analysis of the Transcriptome of Bovine Endometrial Cells Isolated by Laser Micro-Dissection (1): Specific Signatures of Stromal, Glandular and Luminal Epithelial Cells. BMC Genom. 2021, 22, 451. [Google Scholar] [CrossRef]
- Binelli, M.; Scolari, S.C.; Pugliesi, G.; Van Hoeck, V.; Gonella-Diaza, A.M.; Andrade, S.C.S.; Gasparin, G.R.; Coutinho, L.L. The Transcriptome Signature of the Receptive Bovine Uterus Determined at Early Gestation. PLoS ONE 2015, 10, e0122874. [Google Scholar] [CrossRef]
- Moorey, S.E.; Walker, B.N.; Elmore, M.F.; Elmore, J.B.; Rodning, S.P.; Biase, F.H. Rewiring of Gene Expression in Circulating White Blood Cells Is Associated with Pregnancy Outcome in Heifers (Bos Taurus). Sci. Rep. 2020, 10, 16786. [Google Scholar] [CrossRef]
- Dickinson, S.E.; Griffin, B.A.; Elmore, M.F.; Kriese-Anderson, L.; Elmore, J.B.; Dyce, P.W.; Rodning, S.P.; Biase, F.H. Transcriptome Profiles in Peripheral White Blood Cells at the Time of Artificial Insemination Discriminate Beef Heifers with Different Fertility Potential. BMC Genom. 2018, 19, 129. [Google Scholar] [CrossRef] [PubMed]
- Geary, T.W.; Burns, G.W.; Moraes, J.G.N.; Moss, J.I.; Denicol, A.C.; Dobbs, K.B.; Ortega, M.S.; Hansen, P.J.; Wehrman, M.E.; Neibergs, H.; et al. Identification of Beef Heifers with Superior Uterine Capacity for Pregnancy. Biol. Reprod. 2016, 95, 47. [Google Scholar] [CrossRef]
- Phillips, K.M.; Read, C.C.; Kriese-Anderson, L.A.; Rodning, S.P.; Brandebourg, T.D.; Biase, F.H.; Marks, M.L.; Elmore, J.B.; Stanford, M.K.; Dyce, P.W. Plasma Metabolomic Profiles Differ at the Time of Artificial Insemination Based on Pregnancy Outcome, in Bos Taurus Beef Heifers. Sci. Rep. 2018, 8, 13196. [Google Scholar] [CrossRef]
- Kusama, K.; Bai, R.; Matsuno, Y.; Ideta, A.; Sakurai, T.; Nagaoka, K.; Hori, M.; Imakawa, K. Characterization of Serum Metabolome and Proteome Profiles Identifies SNX5 Specific for Pregnancy Failure in Holstein Heifers. Life 2022, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Gómez, E.; Salvetti, P.; Gatien, J.; Carrocera, S.; Martín-González, D.; Muñoz, M. Blood Plasma Metabolomics Predicts Pregnancy in Holstein Cattle Transferred with Fresh and Vitrified/Warmed Embryos Produced in Vitro. J. Proteome Res. 2020, 19, 1169–1182. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, T.M.; Gonçalves, R.F.; Melo, C.F.O.R.; de Oliveira, D.N.; Lima, E.d.O.; Visintin, J.A.; de Achilles, M.A.; Catharino, R.R. A Metabolomic Overview of Follicular Fluid in Cows. Front. Vet. Sci. 2018, 5, 10. [Google Scholar] [CrossRef]
- Aranciaga, N.; Morton, J.D.; Berg, D.K.; Gathercole, J.L. Proteomics and Metabolomics in Cow Fertility: A Systematic Review. Reproduction 2020, 160, 639–658. [Google Scholar] [CrossRef]
- Gómez, E.; Muñoz, M.; Gatien, J.; Carrocera, S.; Martín-González, D.; Salvetti, P. Metabolomic Identification of Pregnancy-Specific Biomarkers in Blood Plasma of BOS TAURUS Beef Cattle after Transfer of in Vitro Produced Embryos. J. Proteom. 2020, 225, 103883. [Google Scholar] [CrossRef]
- Reverter, A.; Chan, E.K.F. Combining Partial Correlation and an Information Theory Approach to the Reversed Engineering of Gene Co-Expression Networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef]
- Choueiry, F.; Singh, S.; Sircar, A.; Laliotis, G.; Sun, X.; Chavdoula, E.; Zhang, S.; Helmig-Mason, J.; Hart, A.; Epperla, N.; et al. Integration of Metabolomics and Gene Expression Profiling Elucidates IL4I1 as Modulator of Ibrutinib Resistance in ABC-Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 2146. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.; Pan, W.; Gawad, C.; Fan, H.C.; Kerchner, G.A.; Wyss-Coray, T.; Blumenfeld, Y.J.; El-Sayed, Y.Y.; Quake, S.R. Noninvasive in Vivo Monitoring of Tissue-Specific Global Gene Expression in Humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7361–7366. [Google Scholar] [CrossRef]
- Basu, M.; Wang, K.; Ruppin, E.; Hannenhalli, S. Predicting Tissue-Specific Gene Expression from Whole Blood Transcriptome. Sci. Adv. 2021, 7, eabd6991. [Google Scholar] [CrossRef]
- Carmelo, V.A.O.; Banerjee, P.; da Silva Diniz, W.J.; Kadarmideen, H.N. Metabolomic Networks and Pathways Associated with Feed Efficiency and Related-Traits in Duroc and Landrace Pigs. Sci. Rep. 2020, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Michou, V.I.; Kanavaros, P.; Athanassiou, V.; Chronis, G.B.; Stabamas, S.; Tsilivakos, V. Fraction of the Peripheral Blood Concentration of CD56+/CD16−/CD3− Cells in Total Natural Killer Cells as an Indication of Fertility and Infertility. Fertil. Steril. 2003, 80, 691–697. [Google Scholar] [CrossRef]
- Thum, M.Y.; Bhaskaran, S.; Abdalla, H.I.; Ford, B.; Sumar, N.; Shehata, H.; Bansal, A.S. An Increase in the Absolute Count of CD56dimCD16+CD69+ NK Cells in the Peripheral Blood Is Associated with a Poorer IVF Treatment and Pregnancy Outcome. Hum. Reprod. 2004, 19, 2395–2400. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, A.K.; Reimann, F.; Guclu, M.; Yalin, A.S.; Kotan, L.D.; Porter, K.M.; Serin, A.; Mungan, N.O.; Cook, J.R.; Ozbek, M.N.; et al. TAC3 and TACR3 Mutations in Familial Hypogonadotropic Hypogonadism Reveal a Key Role for Neurokinin B in the Central Control of Reproduction. Nat. Genet. 2009, 41, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Tusset, C.; Noel, S.D.; Trarbach, E.B.; Silveira, L.F.G.; Jorge, A.A.L.; Brito, V.N.; Cukier, P.; Seminara, S.B.; Mendonça, B.B.d.; Kaiser, U.B.; et al. Mutational Analysis of TAC3 and TACR3 Genes in Patients with Idiopathic Central Pubertal Disorders. Arq. Bras. Endocrinol. Metabol. 2012, 56, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Blasco, V.; Pinto, F.M.; Fernández-Atucha, A.; Prados, N.; Tena-Sempere, M.; Fernández-Sánchez, M.; Candenas, L. Altered Expression of the Kisspeptin/KISS1R and Neurokinin B/NK3R Systems in Mural Granulosa and Cumulus Cells of Patients with Polycystic Ovarian Syndrome. J. Assist. Reprod. Genet. 2019, 36, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.; Navarro, V.M. Novel Biology of Tachykinins in Gonadotropin-Releasing Hormone Secretion. Semin. Reprod. Med. 2019, 37, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Kahnamouyi, S.; Nouri, M.; Farzadi, L.; Darabi, M.; Hosseini, V.; Mehdizadeh, A. The Role of Mitogen-Activated Protein Kinase–Extracellular Receptor Kinase Pathway in Female Fertility Outcomes: A Focus on Pituitary Gonadotropins Regulation. Ther. Adv. Endocrinol. Metab. 2018, 9, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Liebanas, E.; Nahar, K.; Bertuzzi, G.; Keller, A.; Betsholtz, C.; Mäe, M.A. Adult-Induced Genetic Ablation Distinguishes PDGFB Roles in Blood-Brain Barrier Maintenance and Development. J. Cereb. Blood Flow Metab. 2022, 42, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.E.; Detzel, C.; Skinner, M.K. Platelet-Derived Growth Factor Modulates the Primordial to Primary Follicle Transition. Reproduction 2006, 131, 1007–1015. [Google Scholar] [CrossRef]
- Schmahl, J.; Rizzolo, K.; Soriano, P. The PDGF Signaling Pathway Controls Multiple Steroid-Producing Lineages. Genes Dev. 2008, 22, 3255–3267. [Google Scholar] [CrossRef] [PubMed]
- Lindblom, P.; Gerhardt, H.; Liebner, S.; Abramsson, A.; Enge, M.; Hellström, M.; Bäckström, G.; Fredriksson, S.; Landegren, U.; Nyström, H.C.; et al. Endothelial PDGF-B Retention Is Required for Proper Investment of Pericytes in the Microvessel Wall. Genes Dev. 2003, 17, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Hou, X.; Xia, J.; Qian, X.; Miele, L.; Sarkar, F.H.; Wang, Z. Emerging Roles of PDGF-D in EMT Progression during Tumorigenesis. Cancer Treat. Rev. 2013, 39, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex Networks Orchestrate Epithelial–Mesenchymal Transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Sun, Y.; Shimokado, A.; Muragaki, Y. The Roles of Mitogen-Activated Protein Kinase Pathways in TGF- β -Induced Epithelial-Mesenchymal Transition. J. Signal Transduct. 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Korkina, L.G. Phenylpropanoids as Naturally Occurring Antioxidants: From Plant Defense to Human Health. Cell. Mol. Biol. 2007, 53, 15–25. [Google Scholar] [PubMed]
- Ly, C.; Yockell-Lelièvre, J.; Ferraro, Z.M.; Arnason, J.T.; Ferrier, J.; Gruslin, A. The Effects of Dietary Polyphenols on Reproductive Health and Early Development. Hum. Reprod. Update 2015, 21, 228–248. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Rana, T.; Alotaibi, G.H.; Shamsuzzaman, M.; Naqvi, M.; Sehgal, A.; Singh, S.; Sharma, N.; Almoshari, Y.; Abdellatif, A.A.H.; et al. Polyphenols Inhibiting MAPK Signalling Pathway Mediated Oxidative Stress and Inflammation in Depression. Biomed. Pharmacother. 2022, 146, 112545. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, H.N.; Sales, K.J.; Catalano, R.D.; Norman, J.E. Inflammatory Pathways in Female Reproductive Health and Disease. Reproduction 2009, 138, 903–919. [Google Scholar] [CrossRef] [PubMed]
- Waters, W.W.; Chen, P.L.; McArthur, N.H.; Moreno, P.A.; Harms, P.G. Calcium/Calmodulin-Dependent Protein Kinase II Involvement in Release of Gonadotropin-Releasing Hormone. Neuroendocrinology 1998, 67, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, H.C.; Evans, J.; Johnson, N.; Infusini, G.; Webb, A.; Rombauts, L.J.R.; Vollenhoven, B.J.; Salamonsen, L.A.; Edgell, T.A. Idiopathic Infertility in Women Is Associated with Distinct Changes in Proliferative Phase Uterine Fluid Proteins. Biol. Reprod. 2018, 98, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Mu, C.; Wang, Z.; Wang, Y.; Li, Y.; Mai, Y.; Li, S.; Xu, H.; Gu, B.; Luo, L.; et al. Diagnostic and Prognostic Values of Serum EpCAM, TGM2, and HE4 Levels in Endometrial Cancer. Front. Oncol. 2020, 10, 1697. [Google Scholar] [CrossRef] [PubMed]
- Beazley, K.E.; Nurminskaya, M. Effects of Dietary Quercetin on Female Fertility in Mice: Implication of Transglutaminase 2. Reprod. Fertil. Dev. 2016, 28, 974. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Camlin, N.J.; Holt, J.E.; Teixeira, J.M.; McLaughlin, E.A.; Tanwar, P.S. Germ Cell Specific Overactivation of WNT/Βcatenin Signalling Has No Effect on Folliculogenesis but Causes Fertility Defects Due to Abnormal Foetal Development. Sci. Rep. 2016, 6, 27273. [Google Scholar] [CrossRef]
- Li, H.; Shi, L.; Liang, S.; Fang, C.; Xu, Q.; Lu, G.; Wang, Q.; Cheng, J.; Shen, J.; Shen, M. Moxibustion Alleviates Decreased Ovarian Reserve in Rats by Restoring the PI3K/AKT Signaling Pathway. J. Integr. Med. 2022, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Chang, Y.; Wei, L.; Chen, J.; Li, J.; Goldsmith, S.; Silber, S.; Liang, X. Apoptosis of Mural Granulosa Cells Is Increased in Women with Diminished Ovarian Reserve. J. Assist. Reprod. Genet. 2019, 36, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.L.; Bridgham, J.T.; Swenson, J.A. Activation of the Akt/Protein Kinase B Signaling Pathway Is Associated with Granulosa Cell Survival1. Biol. Reprod. 2001, 64, 1566–1574. [Google Scholar] [CrossRef]
- Brown, C.; LaRocca, J.; Pietruska, J.; Ota, M.; Anderson, L.; Duncan Smith, S.; Weston, P.; Rasoulpour, T.; Hixon, M.L. Subfertility Caused by Altered Follicular Development and Oocyte Growth in Female Mice Lacking PKBalpha/Akt11. Biol. Reprod. 2010, 82, 246–256. [Google Scholar] [CrossRef]
- Chu, D.; Zhang, Z.; Zhou, Y.; Li, Y.; Zhu, S.; Zhang, J.; Zhao, Q.; Ji, G.; Wang, W.; Zheng, J. NDRG4, a Novel Candidate Tumor Suppressor, Is a Predictor of Overall Survival of Colorectal Cancer Patients. Oncotarget 2015, 6, 7584–7596. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Gu, Y.; Zhang, X.; Wang, J.-M.; He, Y.-P.; Shi, Y.; Sun, Z.-G.; Shi, H.-J.; Wang, J. Uterine Expression of NDRG4 Is Induced by Estrogen and Up-Regulated during Embryo Implantation Process in Mice. PLoS ONE 2016, 11, e0155491. [Google Scholar] [CrossRef] [PubMed]
- Ipsa, E.; Cruzat, V.F.; Kagize, J.N.; Yovich, J.L.; Keane, K.N. Growth Hormone and Insulin-Like Growth Factor Action in Reproductive Tissues. Front. Endocrinol. 2019, 10, 777. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, M.; Cowley, M.; Geoghegan, F.; Smith, F.M.; Radford, E.J.; Marlow, B.P.; Graham, C.F.; Hurst, L.D.; Ward, A. Maternally-Inherited Grb10 Reduces Placental Size and Efficiency. Dev. Biol. 2010, 337, 1–8. [Google Scholar] [CrossRef]
- Ooi, J.; Yajnik, V.; Immanuel, D.; Gordon, M.; Moskow, J.J.; Buchberg, A.M.; Margolis, B. The Cloning of Grb10 Reveals a New Family of SH2 Domain Proteins. Oncogene 1995, 10, 1621–1630. [Google Scholar]
- Shiura, H.; Miyoshi, N.; Konishi, A.; Wakisaka-Saito, N.; Suzuki, R.; Muguruma, K.; Kohda, T.; Wakana, S.; Yokoyama, M.; Ishino, F.; et al. Meg1/Grb10 Overexpression Causes Postnatal Growth Retardation and Insulin Resistance via Negative Modulation of the IGF1R and IR Cascades. Biochem. Biophys. Res. Commun. 2005, 329, 909–916. [Google Scholar] [CrossRef]
- Rosa, P.R.A.d.; Bohrer, R.C.; Ludke, C.A.; Cesaro, M.P.D.; Pereira, G.R.; Mondadori, R.G.; Antoniazzi, A.Q.; Gonçalves, P.B.D. Grb10 Characterization in Bovine Cumulus Oocyte Complexes from Different Follicle Sizes. Ciência Rural. 2015, 45, 898–904. [Google Scholar] [CrossRef][Green Version]
- Wu, Y.; Zhang, Z.; Zhang, J.B.; Deng, Q. Association of Growth Factor Receptor-Bound Protein 10 Gene Polymorphism with Superovulation Traits in Changbaishan Black Cattle. Genet. Mol. Res. 2016, 15, 1–9. [Google Scholar] [CrossRef]
- He, J.; Li, Y.; Wang, Y.; Zhang, H.; Ge, S.; Fan, X. Targeted Silencing of the ADP-Ribosyltransferase 3 Gene Inhibits the Migration Ability of Melanoma Cells. Oncol. Lett. 2018, 15, 7053–7059. [Google Scholar] [CrossRef]
- Lavergne, C.L.J.; Tao, Y.; Ren, Y.; Lafleur, N.; Liu, X.J. Systemic L-Ornithine Supplementation Specifically Increases Ovarian Putrescine Levels during Ovulation in Mice. Biol. Reprod. 2022, 106, 792–801. [Google Scholar] [CrossRef]
- Liu, D.; Mo, G.; Tao, Y.; Wang, H.; Liu, X.J. Putrescine Supplementation during in Vitro Maturation of Aged Mouse Oocytes Improves the Quality of Blastocysts. Reprod. Fertil. Dev. 2017, 29, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 117864691769193. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan Res. 2009, 2, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Tryptophan Metabolism, Disposition and Utilization in Pregnancy. Biosci. Rep. 2015, 35, e00261. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Liu, H.; Bai, M.; Gao, J.; Wu, X.; Yin, Y. Redox Properties of Tryptophan Metabolism and the Concept of Tryptophan Use in Pregnancy. Int. J. Mol. Sci. 2017, 18, 1595. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Cesar, A.S.M.; Regitano, L.C.A.; Koltes, J.E.; Fritz-Waters, E.R.; Lanna, D.P.D.; Gasparin, G.; Mourão, G.B.; Oliveira, P.S.N.; Reecy, J.M.; Coutinho, L.L. Putative Regulatory Factors Associated with Intramuscular Fat Content. PLoS ONE 2015, 10, e0128350. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2018. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 30 June 2022).
- van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, Scaling, and Transformations: Improving the Biological Information Content of Metabolomics Data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef]
- Jauhiainen, A.; Madhu, B.; Narita, M.; Narita, M.; Griffiths, J.; Tavaré, S. Normalization of Metabolomics Data with Applications to Correlation Maps. Bioinformatics 2014, 30, 2155–2161. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.-E.; Lengauer, T.; Albrecht, M. Computing Topological Parameters of Biological Networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Goenawan, I.H.; Bryan, K.; Lynn, D.J. DyNet: Visualization and Analysis of Dynamic Molecular Interaction Networks. Bioinformatics 2016, 32, 2713–2715. [Google Scholar] [CrossRef] [PubMed]
- Fuller, T.F.; Ghazalpour, A.; Aten, J.E.; Drake, T.A.; Lusis, A.J.; Horvath, S. Weighted Gene Coexpression Network Analysis Strategies Applied to Mouse Weight. Mamm. Genome 2007, 18, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.; Fortes, M.R.S.; Reverter, A.; Diniz, W.J.d.S.; Cesar, A.S.M.; Lima, A.O.d.; Petrini, J.; de Souza, M.M.; Coutinho, L.L.; Mourão, G.B.; et al. Genetic Regulators of Mineral Amount in Nelore Cattle Muscle Predicted by a New Co-Expression and Regulatory Impact Factor Approach. Sci. Rep. 2020, 10, 8436. [Google Scholar] [CrossRef]
- Siddiqui, J.K.; Baskin, E.; Liu, M.; Cantemir-Stone, C.Z.; Zhang, B.; Bonneville, R.; McElroy, J.P.; Coombes, K.R.; Mathé, E.A. IntLIM: Integration Using Linear Models of Metabolomics and Gene Expression Data. BMC Bioinform. 2018, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).