
Preanalytical Pitfalls in Untargeted Plasma Nuclear Magnetic Resonance Metabolomics of Endocrine Hypertension

 , , , , , ,
, , , , , ,  , , , , , , , , , , and
, , , , , , , , , , and
Abstract
:1. Introduction
2. Results
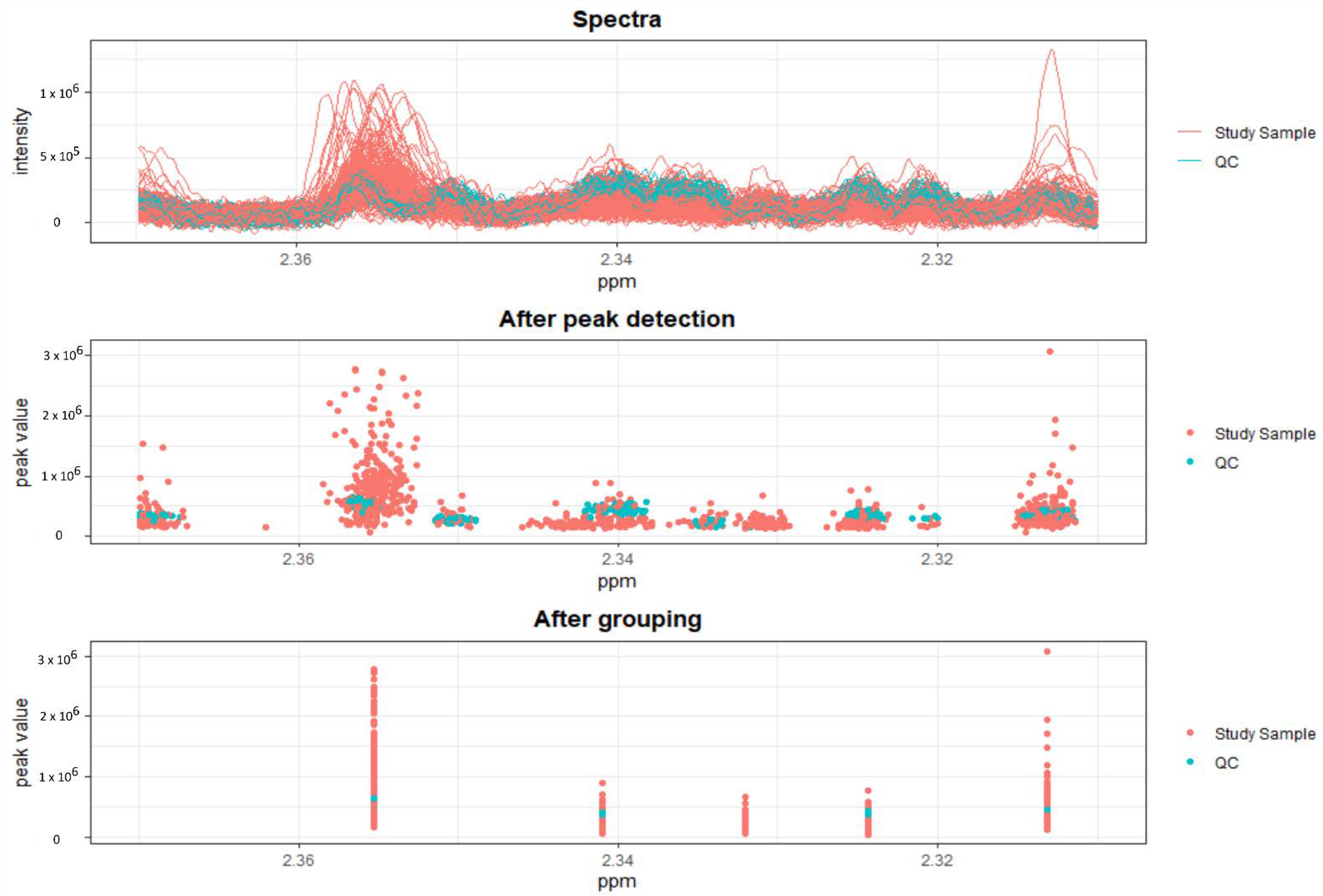
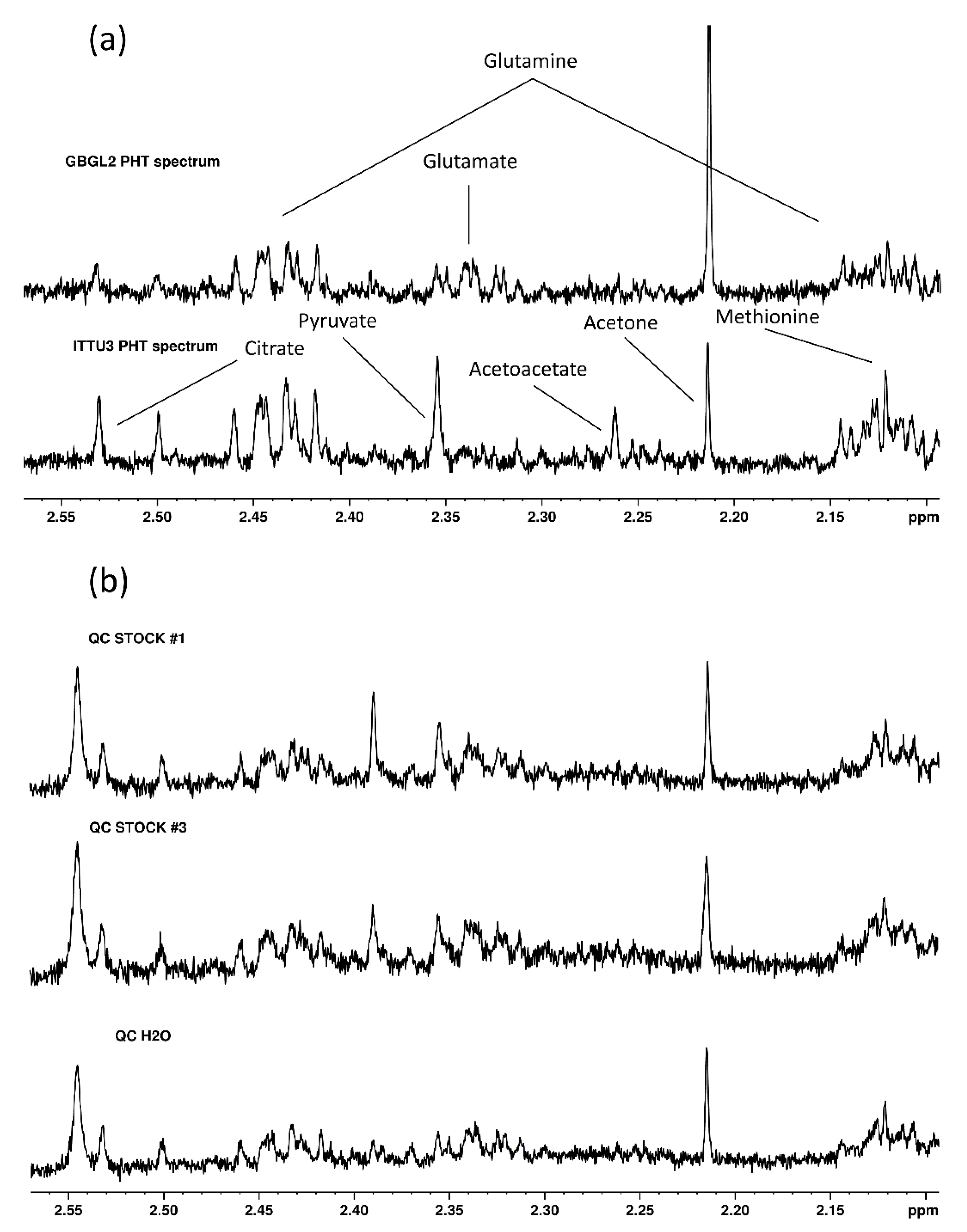
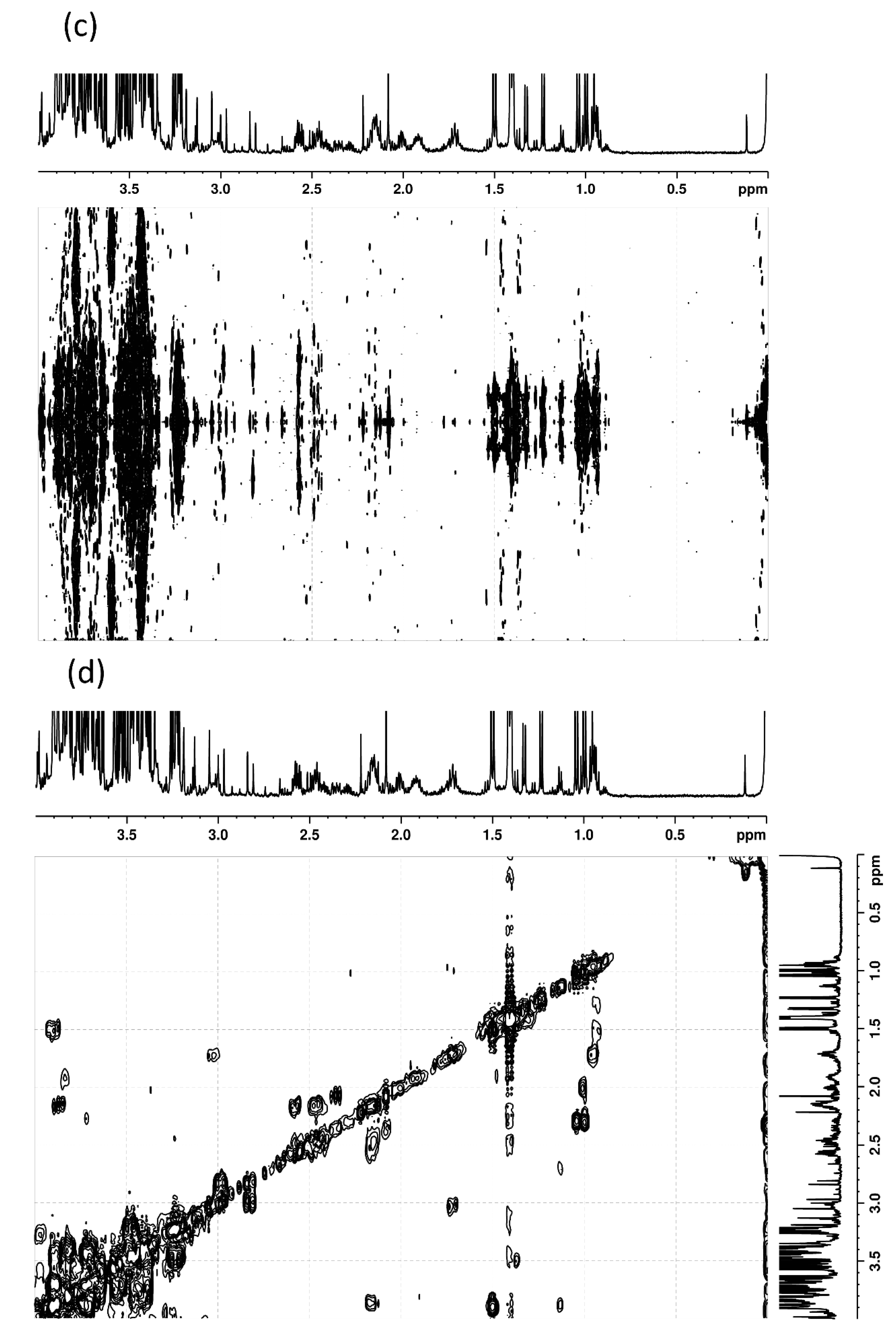
2.1. Initial Approach: Establishing Possible EHT–PHT Biomarkers
2.2. Correcting for Confounders: Approach A (ASCA Correction)
2.3. Correcting for Confounders: Approach B (Metabolite Exclusions)
2.4. Correcting for Confounders: Approach C (Whole Center Exclusions)
3. Discussion
4. Materials and Methods
4.1. Patient and Sample Characteristics
4.2. Untargeted 1H-NMR Metabolomics
4.3. Data Analysis and Statistics
4.4. Confounders
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Raised Blood Pressure (SBP>=140 OR DBP>=90) (Crude Estimate). 2017. Available online: https://www.who.int/data/gho/data/indicators/indicator-details/GHO/raised-blood-pressure-(sbp-=140-or-dbp-=90)-(crude-estimate) (accessed on 27 May 2022).
- GBD 2017 Risk Factor Collaborators, Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994.
- Reincke, M.; Fischer, E.; Gerum, S.; Merkle, K.; Schulz, S.; Pallauf, A.; Quinkler, M.; Hanslik, G.; Lang, K.; Hahner, S.; et al. Observational study mortality in treated primary aldosteronism: The German Conn’s registry. Hypertension 2012, 60, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Monticone, S.; D’Ascenzo, F.; Moretti, C.; Williams, T.A.; Veglio, F.; Gaita, F.; Mulatero, P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 41–50. [Google Scholar] [CrossRef]
- Stolk, R.F.; Bakx, C.; Mulder, J.; Timmers, H.J.; Lenders, J.W. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J. Clin. Endocrinol. Metab. 2013, 98, 1100–1106. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; El Kawkgi, O.M.; Henriquez, A.F.; Bancos, I. Cardiovascular risk and mortality in patients with active and treated hypercortisolism. Gland Surg. 2020, 9, 43–58. [Google Scholar] [CrossRef]
- Clayton, R.N.; Raskauskiene, D.; Reulen, R.C.; Jones, P.W. Mortality and morbidity in Cushing’s disease over 50 Years in Stoke-on-Trent, UK: Audit and meta-analysis of literature. J. Clin. Endocrinol. Metab. 2011, 96, 632–642. [Google Scholar] [CrossRef]
- Mulatero, P.; Monticone, S.; Deinum, J.; Amar, L.; Prejbisz, A.; Zennaro, M.C.; Beuschlein, F.; Rossi, G.P.; Nishikawa, T.; Morganti, A.; et al. Genetics, prevalence, screening and confirmation of primary aldosteronism: A position statement and consensus of the Working Group on Endocrine Hypertension of The European Society of Hypertension. J. Hypertens. 2020, 38, 1919–1928. [Google Scholar] [CrossRef]
- Young, W.F.; Calhoun, D.A.; Lenders, J.W.; Stowasser, M.; Textor, S.C. Screening for endocrine hypertension: An endocrine society scientific statement. Endocr. Rev. 2017, 38, 103–122. [Google Scholar] [CrossRef] [Green Version]
- Ceccato, F.; Boscaro, M. Cushing’s Syndrome: Screening and Diagnosis. High Blood Press. Cardiovasc. Prev. 2016, 23, 209–215. [Google Scholar] [CrossRef]
- Yorke, E.; Atiase, Y.; Akpalu, J.; Sarfo-Kantanka, O. Screening for Cushing Syndrome at the Primary Care Level: What Every General Practitioner Must Know. Int. J. Endocrinol. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Lenders, J.W.; Kerstens, M.N.; Amar, L.; Prejbisz, A.; Robledo, M.; Taieb, D.; Pacak, K.; Crona, J.; Zelinka, T.; Mannelli, M.; et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J. Hypertens. 2020, 38, 1443–1456. [Google Scholar] [CrossRef]
- Rossi, G.P.; Bisogni, V.; Rossitto, G.; Maiolino, G.; Cesari, M.; Zhu, R.; Seccia, T.M. Practice Recommendations for Diagnosis and Treatment of the Most Common Forms of Secondary Hypertension. High Blood Press. Cardiovasc. Prev. 2020, 27, 547–560. [Google Scholar] [CrossRef]
- Coene, K.; Kluijtmans, L.; Van der Heeft, E.; Engelke, U.; De Boer, S.; Hoegen, B.; Kwast, H.; Van de Vorst, M.; Huigen, M.; Keularts, I.; et al. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Hou, E.; Wang, L.; Wang, Y.; Yang, L.; Zheng, X.; Xie, G.; Sun, Q.; Liang, M.; Tian, Z. Reconstruction and analysis of correlation networks based on GC-MS metabolomics data for young hypertensive men. Anal. Chim. Acta 2015, 854, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Ameta, K.; Gupta, A.; Kumar, S.; Sethi, R.; Kumar, D.; Mahdi, A.A. Essential hypertension: A filtered serum based metabolomics study. Sci. Rep. 2017, 7, 1–11. [Google Scholar]
- Roberts, L.D.; Koulman, A.; Griffin, J.L. Towards metabolic biomarkers of insulin resistance and type 2 diabetes: Progress from the metabolome. Lancet Diabetes Endocrinol. 2014, 2, 65–75. [Google Scholar] [CrossRef]
- ens@t-ht. Available online: https://www.ensat-ht.eu/ (accessed on 27 May 2022).
- Erlic, Z.; Reel, P.; Reel, S.; Amar, L.; Pecori, A.; Larsen, C.K.; Tetti, M.; Pamporaki, C.; Prehn, C.; Adamski, J.; et al. Targeted metabolomics as a tool in discriminating endocrine from primary hypertension. J. Clin. Endocrinol. Metab. 2020, 106, 1111–1128. [Google Scholar] [CrossRef]
- Broadhurst, D.I.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef] [Green Version]
- Brindle, J.T.; Antti, H.; Holmes, E.; Tranter, G.; Nicholson, J.K.; Bethell, H.W.; Clarke, S.; Schofield, P.M.; McKilligin, E.; Mosedale, D.E.; et al. Rapid and noninvasive diagnosis of the presence and severity of coronary heart disease using 1H-NMR-based metabonomics. Nat. Med. 2002, 8, 1439–1444. [Google Scholar] [CrossRef]
- Kirschenlohr, H.L.; Griffin, J.L.; Clarke, S.C.; Rhydwen, R.; Grace, A.A.; Schofield, P.M.; Brindle, K.M.; Metcalfe, J.C. Proton NMR analysis of plasma is a weak predictor of coronary artery disease. Nat. Med. 2006, 12, 705–710. [Google Scholar] [CrossRef]
- Ransohoff, D.F. Bias as a threat to the validity of cancer molecular-marker research. Nat. Rev. Cancer 2005, 5, 142–149. [Google Scholar] [CrossRef]
- Smilde, A.K.; Jansen, J.J.; Hoefsloot, H.C.J.; Lamers, R.-j.A.N.; Greef, J.V.D.; Timmerman, M.E. ANOVA-simultaneous component analysis (ASCA): A new tool for analyzing designed metabolomics data. Bioinformatics 2005, 21, 3043–3048. [Google Scholar] [CrossRef]
- Posma, J.M.; Garcia-perez, I.; Ebbels, T.M.D.; Lindon, J.C.; Stamler, J.; Elliott, P.; Holmes, E.; Nicholson, J.K. Optimized Phenotypic Biomarker Discovery and Confounder Elimination via Covariate-Adjusted Projection to Latent Structures from Metabolic Spectroscopy Data. J. Proteome Res. 2018, 17, 1586–1595. [Google Scholar] [CrossRef] [Green Version]
- Engel, J.; Blanchet, L.; Bloemen, B.; Van den Heuvel, L.P.; Engelke, U.H.; Wevers, R.A.; Buydens, L.M. Regularized MANOVA (rMANOVA) in untargeted metabolomics. Anal. Chim. Acta 2015, 899, 1–12. [Google Scholar] [CrossRef]
- Munda, M.; Legrand, C. Adjusting for centre heterogeneity in multicentre clinical trials with a time-to-event outcome. Pharm. Stat. 2014, 13, 145–152. [Google Scholar] [CrossRef]
- Anisimov, V.V. Effects of unstratified and centre-stratified randomization in multi-centre clinical trials. Pharm. Stat. 2011, 10, 50–59. [Google Scholar] [CrossRef]
- Würtz, P.; Raiko, J.R.; Magnussen, C.G.; Soininen, P.; Kangas, A.J.; Tynkkynen, T.; Thomson, R.; Laatikainen, R.; Savolainen, M.J.; Laurikka, J.; et al. High-throughput quantification of circulating metabolites improves prediction of subclinical atherosclerosis. Eur. Heart J. 2012, 33, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.H.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Crosslin, D.R.; Haynes, C.; Dungan, J.; Newby, L.K.; Hauser, E.R.; Ginsburg, G.S.; et al. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ. Cardiovasc. Genet. 2010, 3, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Mels, C.M.; Delles, C.; Louw, R.; Schutte, A.E. Central systolic pressure and a nonessential amino acid metabolomics profile: The African Prospective study on the Early Detection and Identification of Cardiovascular disease and Hypertension. J. Hypertens. 2019, 37, 1157–1166. [Google Scholar] [CrossRef]
- Goïta, Y.; Chao de la Barca, J.M.; Keïta, A.; Diarra, M.B.; Dembélé, K.C.; Chabrun, F.; Dramé, B.S.I.; Kassogué, Y.; Diakité, M.; Mirebeau-Prunier, D.; et al. Sexual Dimorphism of Metabolomic Profile in Arterial Hypertension. Sci. Rep. 2020, 10, 7517. [Google Scholar] [CrossRef]
- Gonzalez-Calero, L.; Martin-Lorenzo, M.; Martínez, P.J.; Baldan-Martin, M.; Ruiz-Hurtado, G.; Segura, J.; De la Cuesta, F.; Barderas, M.G.; Ruilope, L.M.; Vivanco, F.; et al. Hypertensive patients exhibit an altered metabolism. A specific metabolite signature in urine is able to predict albuminuria progression. Transl. Res. 2016, 178, 25–37.e7. [Google Scholar] [CrossRef]
- Øvrehus, M.A.; Bruheim, P.; Ju, W.; Zelnick, L.R.; Langlo, K.A.; Sharma, K.; De Boer, I.H.; Hallan, S.I. Gene Expression Studies and Targeted Metabolomics Reveal Disturbed Serine, Methionine, and Tyrosine Metabolism in Early Hypertensive Nephrosclerosis. Kidney Int. Rep. 2019, 4, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Bliziotis, N.G.; Kluijtmans, L.A.J.; Soto, S.; Tinnevelt, G.H.; Langton, K.; Robledo, M.; Pamporaki, C.; Engelke, U.F.H.; Erlic, Z.; Engel, J.; et al. Pre-versus post-operative untargeted plasma nuclear magnetic resonance spectroscopy metabolomics of pheochromocytoma and paraganglioma. Endocrine 2022, 75, 254–265. [Google Scholar] [CrossRef]
- Teahan, O.; Gamble, S.; Holmes, E.; Waxman, J.; Nicholson, J.K.; Bevan, C.; Keun, H.C. Impact of analytical bias in metabonomic studies of human blood serum and plasma. Anal. Chem. 2006, 78, 4307–4318. [Google Scholar] [CrossRef]
- Nishiumi, S.; Suzuki, M.; Kobayashi, T.; Yoshida, M. Differences in metabolite profiles caused by pre-analytical blood processing procedures. J. Biosci. Bioeng. 2018, 125, 613–618. [Google Scholar] [CrossRef] [Green Version]
- Bervoets, L.; Louis, E.; Reekmans, G.; Mesotten, L.; Thomeer, M.; Adriaensens, P.; Linsen, L. Influence of preanalytical sampling conditions on the 1H NMR metabolic profile of human blood plasma and introduction of the Standard PREanalytical Code used in biobanking. Metabolomics 2015, 11, 1197–1207. [Google Scholar] [CrossRef]
- Brunius, C.; Pedersen, A.; Malmodin, D.; Karlsson, B.G.; Andersson, L.I.; Tybring, G.; Landberg, R. Prediction and modeling of pre-analytical sampling errors as a strategy to improve plasma NMR metabolomics data. Bioinformatics 2017, 33, 3567–3574. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Kennedy, A.D.; Elsea, S.H.; Miller, M.J. Analytes related to erythrocyte metabolism are reliable biomarkers for preanalytical error due to delayed plasma processing in metabolomics studies. Clin. Chim. Acta 2017, 466, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Jobard, E.; Trédan, O.; Postoly, D.; André, F.; Martin, A.L.; Elena-Herrmann, B.; Boyault, S. A systematic evaluation of blood serum and plasma pre-analytics for metabolomics cohort studies. Int. J. Mol. Sci. 2016, 17, 2035. [Google Scholar] [CrossRef] [Green Version]
- Kamlage, B.; Maldonado, S.G.; Bethan, B.; Peter, E.; Schmitz, O.; Liebenberg, V.; Schatz, P. Quality markers addressing preanalytical variations of blood and plasma processing identified by broad and targeted metabolite profiling. Clin. Chem. 2014, 60, 399–412. [Google Scholar] [CrossRef]
- Ferreira, D.L.; Maple, H.J.; Goodwin, M.; Brand, J.S.; Yip, V.; Min, J.L.; Groom, A.; Lawlor, D.A.; Ring, S. The effect of pre-analytical conditions on blood metabolomics in epidemiological studies. Metabolites 2019, 9. [Google Scholar]
- Bernini, P.; Bertini, I.; Luchinat, C.; Nincheri, P.; Staderini, S.; Turano, P. Standard operating procedures for pre-analytical handling of blood and urine for metabolomic studies and biobanks. J. Biomol. NMR 2011, 49, 231–243. [Google Scholar] [CrossRef]
- Yang, W.; Chen, Y.; Xi, C.; Zhang, R.; Song, Y.; Zhan, Q.; Bi, X.; Abliz, Z. Liquid chromatography-tandem mass spectrometry-based plasma metabonomics delineate the effect of metabolites’ stability on reliability of potential biomarkers. Anal. Chem. 2013, 85, 2606–2610. [Google Scholar] [CrossRef]
- Cao, Z.; Kamlage, B.; Wagner-Golbs, A.; Maisha, M.; Sun, J.; Schnackenberg, L.K.; Pence, L.; Schmitt, T.C.; Daniels, J.R.; Rogstad, S.; et al. An Integrated Analysis of Metabolites, Peptides, and Inflammation Biomarkers for Assessment of Preanalytical Variability of Human Plasma. J. Proteome Res. 2019, 18, 2411–2421. [Google Scholar] [CrossRef]
- Breier, M.; Wahl, S.; Prehn, C.; Fugmann, M.; Ferrari, U.; Weise, M.; Banning, F.; Seissler, J.; Grallert, H.; Adamski, J.; et al. Targeted metabolomics identifies reliable and stable metabolites in human serum and plasma samples. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Hirayama, A.; Sugimoto, M.; Suzuki, A.; Hatakeyama, Y.; Enomoto, A.; Harada, S.; Soga, T.; Tomita, M.; Takebayashi, T. Effects of processing and storage conditions on charged metabolomic profiles in blood. Electrophoresis 2015, 36, 2148–2155. [Google Scholar] [CrossRef]
- Pinto, J.; Domingues, M.R.M.; Galhano, E.; Pita, C.; Do Ceu Almeida, M.; Carreira, I.M.; Gil, A.M. Human plasma stability during handling and storage: Impact on NMR metabolomics. Analyst 2014, 139, 1168–1177. [Google Scholar] [CrossRef]
- Wagner-Golbs, A.; Neuber, S.; Kamlage, B.; Christiansen, N.; Bethan, B.; Rennefahrt, U.; Schatz, P.; Lind, L. Effects of long-term storage at −80 °C on the human plasma metabolome. Metabolites 2019, 9, 99. [Google Scholar] [CrossRef] [Green Version]
- Haid, M.; Muschet, C.; Wahl, S.; Römisch-Margl, W.; Prehn, C.; Möller, G.; Adamski, J. Long-Term Stability of Human Plasma Metabolites during Storage at −80 °C. J. Proteome Res. 2018, 17, 203–211. [Google Scholar] [CrossRef]
- Westerhuis, J.A.; Van Velzen, E.J.; Hoefsloot, H.C.; Smilde, A.K. Multivariate paired data analysis: Multilevel PLSDA versus OPLSDA. Metabolomics 2010, 6, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Bi, H.; Guo, Z.; Jia, X.; Liu, H.; Ma, L.; Xue, L. The key points in the pre-analytical procedures of blood and urine samples in metabolomics studies. Metabolomics 2020, 16. [Google Scholar] [CrossRef]
- Gowda, G.A.; Raftery, D.; Hong, N.N. Evaluation of fumaric acid and maleic acid as internal standards for nmr analysis of protein precipitated plasma, serum, and whole blood. Anal. Chem. 2021, 93, 3233–3240. [Google Scholar] [CrossRef]
- Bliziotis, N.G.; Engelke, U.F.H.; Aspers, R.L.E.G.; Engel, J.; Deinum, J.; Timmers, H.J.L.M.; Wevers, R.A.; Kluijtmans, L.A.J. A comparison of high-throughput plasma NMR protocols for comparative untargeted metabolomics. Metab. Off. J. Metab. Soc. 2020, 16, 64. [Google Scholar]
- Hubert, M.; Rousseeuw, P.; Verdonck, T. Robust PCA for skewed data and its outlier map. Comput. Stat. Data Anal. 2009, 53, 2264–2274. [Google Scholar] [CrossRef]
- Tom Reynkens. Rospca: Robust Sparse PCA Using the ROSPCA Algorithm. Software Version 1.0.4. 2018. Available online: https://rdrr.io/cran/rospca/ (accessed on 27 May 2022).
- R Studio Team. RStudio: Integrated Development for R; RStudio Inc.: Boston, MA, USA, Software versions 1.1.463 (2018) and 1.2.5033 (2019).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, Software versions 3.4.4 (2018) and 3.6.3 (2020).
- Hao, J.; Astle, W.; Iorio, M.D.; Ebbels, T.M.D. BATMAN—An R package for the automated quantification of metabolites from nuclear magnetic resonance spectra using a Bayesian model. Bioinformatics 2012, 28, 2088–2090. [Google Scholar] [CrossRef] [Green Version]
- Beirnaert, C.; Meysman, P.; Vu, T.N.; Hermans, N.; Apers, S.; Pieters, L.; Covaci, A.; Laukens, K. speaq 2.0: A complete workflow for high-throughput 1D NMR spectra processing and quantification. PLoS Comput. Biol. 2018, 14. [Google Scholar] [CrossRef] [Green Version]
- Southam, A.D.; Weber, R.J.; Engel, J.; Jones, M.R.; Viant, M.R. A complete workflow for high-resolution spectral-stitching nanoelectrospray direct-infusion mass-spectrometry-based metabolomics and lipidomics. Nat. Protoc. 2017, 12, 255–273. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application in 1H NMR Metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.; Tibshirani, R.; Narasimhan, B.; Chu, G. Impute: Imputation for Microarray Data. 2019. Available online: https://bioconductor.riken.jp/packages/3.9/bioc/html/impute.html (accessed on 27 May 2022).
- Parsons, H.M.; Ludwig, C.; Günther, U.L.; Viant, M.R. Improved classification accuracy in 1- and 2-dimensional NMR metabolomics data using the variance stabilising generalised logarithm transformation. BMC Bioinform. 2007, 8, 234. [Google Scholar] [CrossRef] [Green Version]
- Rocke, D.; Lee, G.C.; Tillinghast, J.; Durbin-Johnson, B.; Wu, S. LMGene: LMGene Software for Data Transformation and Identification of Differentially Expressed Genes in Gene Expression Arrays, R Package Version 2.43.0; 2019. Available online: https://rdrr.io/bioc/LMGene/ (accessed on 27 May 2022).
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted Profiling: Quantitative Analysis of H NMR Metabolomics Data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.; Feunang, Y.; Marcu, A.; AC, G.; Liang, K. HMDB 4.0—The Human Metabolome Database for 2018. Nucleic Acids Res. 2018, 4, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Lewis, I.A.; Hegeman, A.D.; Anderson, M.E.; Li, J.; Schulte, C.F.; Westler, W.M.; Eghbalnia, H.R.; Sussman, M.R.; Markley, J.L. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 2008, 26, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Wevers, R.A.; Engelke, U.; Heerschap, A. High-resolution 1H-NMR spectroscopy of blood plasma for metabolic studies. Clin. Chem. 1994, 40, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13. [Google Scholar] [CrossRef] [Green Version]
- Jolliffe, I. Principal Component Analysis. In Encyclopedia of Statistics in Behavioral Science; Everitt, B.S., Howell, D.C., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2005; Volume 3, pp. 1580–1584. [Google Scholar]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Mehmood, T.; Liland, K.H.; Snipen, L.; Sæbø, S. A review of variable selection methods in Partial Least Squares Regression. Chemom. Intell. Lab. Syst. 2012, 118, 62–69. [Google Scholar] [CrossRef]
- Lê Cao, K.-A.; Rossow, D.; Robert- Granié, C.; Besse, P. Sparse PLS: Variable Selection when Integrating Omics data. Stat. Appl. Genet. Mol. Biol. 2008, 7, 35. [Google Scholar] [CrossRef]
- Lê Cao, K.-A.; Boitard, S.; Besse, P. Sparse PLS discriminant analysis: Biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymanska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerhuis, J.A.; Hoefsloot, H.C.; Smit, S.; Vis, D.J.; Smilde, A.K.; Velzen, E.J.; Duijnhoven, J.P.; Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.F. Constructing confidence sets using rank statistics. J. Am. Stat. Assoc. 1972, 67, 687–690. [Google Scholar] [CrossRef]
- Best, D.J.; Roberts, D.E. The Upper Tail Probabilities of Spearman’s Rho. J. R. Stat. Soc. 1975, 24, 377–379. [Google Scholar]
- Gils, C.; Nybo, M. Quality Control of Preanalytical Handling of Blood Samples for Future Research: A National Survey. J. Appl. Lab. Med. 2020, 5, 83–90. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PHT (n = 106) | EHT (n = 231) | PA (n = 104) | PPGL (n = 94) | CS (n = 33) | |
|---|---|---|---|---|---|
| PATIENT CHARACTERISTICS | |||||
| PATIENT AGE | 55 [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79] * | 49 [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77] | 48 [26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74] | 50 [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77] | 47 [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76] |
| p-value ** | 0.003 | 0.002 | 0.1 | 0.03 | |
| PATIENT SEX (F/M) | 61/45 | 135/96 | 47/57 | 58/36 | 30/3 |
| p-value *** | 0.9 | 0.1 | 0.6 | 0.0003 | |
| PREANALYTICAL SAMPLE CHARACTERISTICS | |||||
| SAMPLE AGE (days) | 2393 [127–6418] * | 1125 [11–3442] * | 535 [52–2280] * | 1548 [11–3442] * | 162 [19–1186] * |
| p-value ** | 6 × 10−11 | 4 × 10−10 | 0.002 | 3 × 10−12 | |
| SAMPLE CENTER OF ORIGIN | |||||
| FRPA1 | 17 (16%) | 66 (29%) | 40 (38%) | 26 (28%) | 0 |
| FRPA2 | 0 | 11 (4.8%) | 0 | 0 | 11 (33%) |
| GBGL2 | 49 (46%) | 0 | 0 | 0 | 0 |
| GYDR | 20 (19%) | 28 (12%) | 8 (7.7%) | 19 (20%) | 1 (3.0%) |
| GYLU | 0 | 1 (0.4%) | 0 | 1 (1.1%) | 0 |
| GYMU | 0 | 4 (1.7%) | 0 | 4 (4.3%) | 0 |
| GYWU | 0 | 1 (0.4%) | 0 | 1 (1.1%) | 0 |
| IRGA | 0 | 3 (1.3%) | 0 | 0 | 3 (9.1%) |
| ITPD | 0 | 20 (8.7%) | 2 (1.9%) | 4 (4.3%) | 14 (42%) |
| ITPD3 | 0 | 9 (3.9%) | 8 (7.7%) | 0 | 1 (3.0%) |
| ITTU3 | 20 (16%) | 51 (22%) | 46 (44%) | 2 (2.1%) | 3 (9.1%) |
| NLNI | 0 | 6 (2.6%) | 0 | 6 (6.4%) | 0 |
| PLWW | 0 | 31 (13%) | 0 | 31 (33%) | 0 |
| p-value *** | 5 × 10−4 | 5 × 10−4 | 5 × 10−4 | 6 × 10−26 | |
| ANALYTICAL SAMPLE CHARACTERISTICS | |||||
| BATCH | 22 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42] * | 25 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48] * | 22 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44] | 31 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48] * | 24 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46] * |
| p-value ** | 0.1 | 0.8 | 0.006 | 0.9 | |
| RUN ORDER | 7 [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16] * | 7 [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17] * | 7 [2,3,4,5,6,7,8,9,10,11,12,13,14] * | 9 [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17] * | 7 [2,3,4,5,6,7,8,9,10,11,12,13,14,15] * |
| p-value ** | 0.6 | 0.7 | 0.5 | 0.9 | |
| Scenario | Metric | Initial Approach | Approach A | Approach B | Approach C |
|---|---|---|---|---|---|
| EHT-PHT | Balanced Accuracy | 79 (78–79) | 79 (79–79) | 67 (66–67) | 58 (57–59) |
| Sensitivity ** | 87 (87–87) | 84 (83–84) | 69 (69–70) | 62 (61–63) | |
| Specificity *** | 70 (70–71) | 74 (73–74) | 64 (63–65) | 53 (51–55) | |
| PA-PHT | Balanced Accuracy | 83 (83–84) | 83 (83–83) | 69 (69–70) | 69 (68–70) |
| Sensitivity ** | 90 (89–90) | 89 (89–90) | 70 (69–71) | 77 (76–79) | |
| Specificity *** | 77 (77–78) | 77 (77–77) | 69 (68–70) | 61 (60–62) | |
| PPGL-PHT | Balanced Accuracy | 79 (78–79) | 81 (80–81) | 68 (68–69) | 68 (67–69) |
| Sensitivity ** | 88 (87–88) | 86 (85–87) | 69 (68–70) | 69 (68–70) | |
| Specificity *** | 70 (69–70) | 75 (75–76) | 67 (66–68) | 66 (65–68) | |
| CS-PHT | Balanced Accuracy | 85 (84–85) | - | 82 (81–82) | - |
| Sensitivity ** | 71 (71–72) | - | 79 (78–80) | - | |
| Specificity *** | 98 (98–99) | - | 84 (84–85) | - | |
| ALL-ALL | Balanced Accuracy | 65 (64–65) | - | 53 (52–53) | 57 (57–58) |
| CS TP Rate | 73 (72–74) | - | 72 (71–73) | - | |
| PA TP * Rate | 65 (64–65) | - | 53 (52–54) | 69 (67–70) | |
| PHT TP * Rate | 72 (72–72) | - | 45 (45–46) | 35 (33–36) | |
| PPGL TP * Rate | 50 (49–51) | - | 42 (41–43) | 69 (68–70) |
| Metabolite | NMR Signal (ppm) | Initial Approach | Approach A | Approach B | Approach C |
|---|---|---|---|---|---|
| Alanine | 1.457 | −0.19975 | −0.07187 | 0 | 0 |
| Creatine | 3.917 | 0 | 0 | 0.019157 | |
| Creatinine | 4.041 | 0 | 0 | 0.177419 | 0 |
| Dimethyl sulfone | 3.137 | 0 | 0 | 0.109629 | |
| Dimethylamine | 2.695 | 0 | 0 | −0.03439 | 0 |
| Dimethylglycine | 2.91 | 0 | 0 | 0.04487 | 0.027703 |
| Formate | 8.441 | −0.01988 | −0.01755 | 0.023133 | 0 |
| Glutamine | 2.433 | 0.148614 | 0.135957 | 0 | |
| Glutamate | 2.325 | −0.12554 | −0.14981 | 0 | |
| Glucose | 5.22 | 0.039396 | 0.0097 | 0.147391 | |
| Glycine | 3.548 | −0.0108 | 0 | 0 | |
| Glycerol | 3.555 | 0 | 0 | 0.091654 | |
| Lactate | 4.108 | 0.025885 | 0 | −0.08734 | |
| Lysine | 2.997 | 0 | 0 | 0.03347 | 0 |
| Methionine | 2.122 | 0.052659 | 0.02404 | 0 | |
| Methanol | 3.346 | 0.062726 | 0.050343 | 0.04658 | |
| Proline | 1.996 | −0.02291 | −0.00628 | −0.13954 | −0.01636 |
| Pyruvate | 2.356 | 0.312859 | 0.32791 | 0.197295 | |
| Threonine | 4.24 | 0 | 0 | 0.040696 | 0 |
| Tyrosine | 7.168 | 0 | 0 | −0.0194 | 0 |
| Valine | 0.981 | 0 | 0 | 0 | −0.00058 |
| Unknown Metabolites | 3.162 | 0.009448 | 0.017788 | 0.236056 | 0 |
| 3.262 | 0 | 0 | −0.15528 | −0.05692 | |
| 3.284 | −0.03909 | −0.02878 | 0 | ||
| 3.612 | 0 | 0 | −0.11482 | 0 | |
| 3.67 | 0 | 0 | −0.12957 | 0 |
| Metabolite | NMR Peaks (ppm) | Dataset | Reason * | FRPA1 PHT/Cluster 2/High Sample Age |
|---|---|---|---|---|
| Acetylcarnitine | 3.177 | PA-PHT, PPGL-PHT | PLSDA CLUSTER, SAMPLE AGE | ↓ |
| Creatine | 3.021, 3.917 | PA-PHT, PPGL-PHT | PLSDA CLUSTER, SAMPLE AGE | ↑ |
| Dimethyl sulfone | 3.137 | PA-PHT, PPGL-PHT | PLSDA SAMPLE AGE | ↑ |
| Glucose | 5.220, 5.227 | PA-PHT, PPGL-PHT | PLSDA CLUSTER, SAMPLE AGE | ↓ |
| Glutamate | 2.047, 2.060, 2.075, 2.095, 2.103, 2.108, 2.113, 2.122, 2.132, 2.140, 2.145, 2.325, 2.332, 2.341, 2.356 | PA-PHT, PPGL-PHT | FRPA1 PHT, PLSDA CLUSTER, SAMPLE AGE | ↑ |
| Glutamine | 2.095, 2.103, 2.108, 2.113, 2.122, 2.132, 2.140, 2.145, 2.418, 2.428, 2.433, 2.444, 2.449, 2.460 | PA-PHT, PPGL-PHT | FRPA1 PHT, PLSDA CLUSTER, SAMPLE AGE | ↓ |
| Glycerol | 3.555, 3.567 | PA-PHT | PLSDA CLUSTER, SAMPLE AGE | ↑ |
| Glycine | 3.548 | PA-PHT, PPGL-PHT | PLSDA SAMPLE AGE | ↓ |
| Lactate | 1.321, 1.307, 4.080, 4.094, 4.108, 4.121 | PA-PHT, PPGL-PHT | PLSDA CLUSTER, SAMPLE AGE | ↑ |
| Methanol | 3.346 | PA-PHT, PPGL-PHT | PLSDA CLUSTER, SAMPLE AGE | ↓ |
| Methionine | 2.122 | PA-PHT, PPGL-PHT | FRPA1 PHT, PLSDA SAMPLE AGE | ↓ |
| Ornithine | 3.041, 3.057 | PA-PHT, PPGL-PHT | PLSDA CLUSTER, SAMPLE AGE | ↑ |
| Pyruvate | 2.356 | PA-PHT, PPGL-PHT | PLSDA CLUSTER, SAMPLE AGE | ↓ |
| Unknown metabolite | 3.284 | PA-PHT, PPGL-PHT | PLSDA CLUSTER, SAMPLE AGE | ↑ |
| PHT (n= 40) | EHT (n = 118) | PA (n = 54) | PPGL (n = 64) | |
|---|---|---|---|---|
| PATIENT CHARACTERISTICS | ||||
| PATIENT AGE | 44 [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70] | 49 [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74] | 48 [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74] | 50 [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74] |
| p-value ** | 0.03 | 0.05 | 0.04/0.6 | |
| PATIENT SEX (F/M) | 15/25 | 65/53 | 24/30 | 41/23 |
| p-value *** | 0.07 | 0.5 | 0.009/0.04 | |
| PREANALYTICAL SAMPLE CHARACTERISTICS | ||||
| SAMPLE AGE | 366 [127–1307] * | 748 [83–2841] * | 380 [83–1598] * | 1419 [121–2841] |
| p-value ** | 1 × 10−5 | 1 | 4 × 10−13/1 × 10−15 | |
| SAMPLE CENTER OF ORIGIN | ||||
| GYDR | 20 (50%) | 27 (23%) | 8 (15%) | 19 (30%) |
| GYLU | 0 | 1 (0.8%) | 0 | 1 (1.6%) |
| GYMU | 0 | 4 (3.4%) | 0 | 4 (6.3%) |
| GYWU | 0 | 1 (0.8%) | 0 | 1 (1.6%) |
| ITTU3 | 20 (50%) | 48 (41%) | 46 (85%) | 2 (3.1%) |
| NLNI | 0 | 6 (5.1%) | 0 | 6 (9.4%) |
| PLWW | 0 | 31 (26%) | 0 | 31 (48%) |
| p-value *** | 6 × 10−5 | 5 × 10−4 | 2 × 10−13/4 × 10−23 | |
| ANALYTICAL SAMPLE CHARACTERISTICS | ||||
| BATCH | 19 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42] | 27 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48] * | 22 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42] | 37 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48] * |
| p-value ** | 0.001 | 0.1 | 7 × 10−5/0.0001 | |
| RUN ORDER | 7 [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16] * | 9 [2,3,4,5,6,7,8,9,10,11,12,13,14] * | 8 [2,3,4,5,6,7,8,9,10,11,12,13,14] * | 9 [2,3,4,5,6,7,8,9,10,11,12,13,14] * |
| p-value ** | 0.3 | 0.4 | 0.4/1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bliziotis, N.G.; Kluijtmans, L.A.J.; Tinnevelt, G.H.; Reel, P.; Reel, S.; Langton, K.; Robledo, M.; Pamporaki, C.; Pecori, A.; Van Kralingen, J.; et al. Preanalytical Pitfalls in Untargeted Plasma Nuclear Magnetic Resonance Metabolomics of Endocrine Hypertension. Metabolites 2022, 12, 679. https://doi.org/10.3390/metabo12080679
Bliziotis NG, Kluijtmans LAJ, Tinnevelt GH, Reel P, Reel S, Langton K, Robledo M, Pamporaki C, Pecori A, Van Kralingen J, et al. Preanalytical Pitfalls in Untargeted Plasma Nuclear Magnetic Resonance Metabolomics of Endocrine Hypertension. Metabolites. 2022; 12(8):679. https://doi.org/10.3390/metabo12080679
Chicago/Turabian StyleBliziotis, Nikolaos G., Leo A. J. Kluijtmans, Gerjen H. Tinnevelt, Parminder Reel, Smarti Reel, Katharina Langton, Mercedes Robledo, Christina Pamporaki, Alessio Pecori, Josie Van Kralingen, and et al. 2022. "Preanalytical Pitfalls in Untargeted Plasma Nuclear Magnetic Resonance Metabolomics of Endocrine Hypertension" Metabolites 12, no. 8: 679. https://doi.org/10.3390/metabo12080679
APA StyleBliziotis, N. G., Kluijtmans, L. A. J., Tinnevelt, G. H., Reel, P., Reel, S., Langton, K., Robledo, M., Pamporaki, C., Pecori, A., Van Kralingen, J., Tetti, M., Engelke, U. F. H., Erlic, Z., Engel, J., Deutschbein, T., Nölting, S., Prejbisz, A., Richter, S., Adamski, J., ... Timmers, H. J. L. M. (2022). Preanalytical Pitfalls in Untargeted Plasma Nuclear Magnetic Resonance Metabolomics of Endocrine Hypertension. Metabolites, 12(8), 679. https://doi.org/10.3390/metabo12080679