
Metabolic Profiling of Rheumatoid Arthritis Neutrophils Reveals Altered Energy Metabolism That Is Not Affected by JAK Inhibition

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Demographics
2.2. Neutrophil Preparation
2.3. Intracellular Metabolite Extraction and NMR Processing
2.4. H-NMR Measurements
2.5. Intracellular ROS Production in Response to fMLP
2.6. Bacterial Killing Assay
2.7. ROS Production in Response to Live S. aureus
2.8. Visualisation of NET Production by Immuno-Histochemistry
2.9. Quantitative Measurement of DNA Released during NETosis
2.10. Statistical Analysis
3. Results
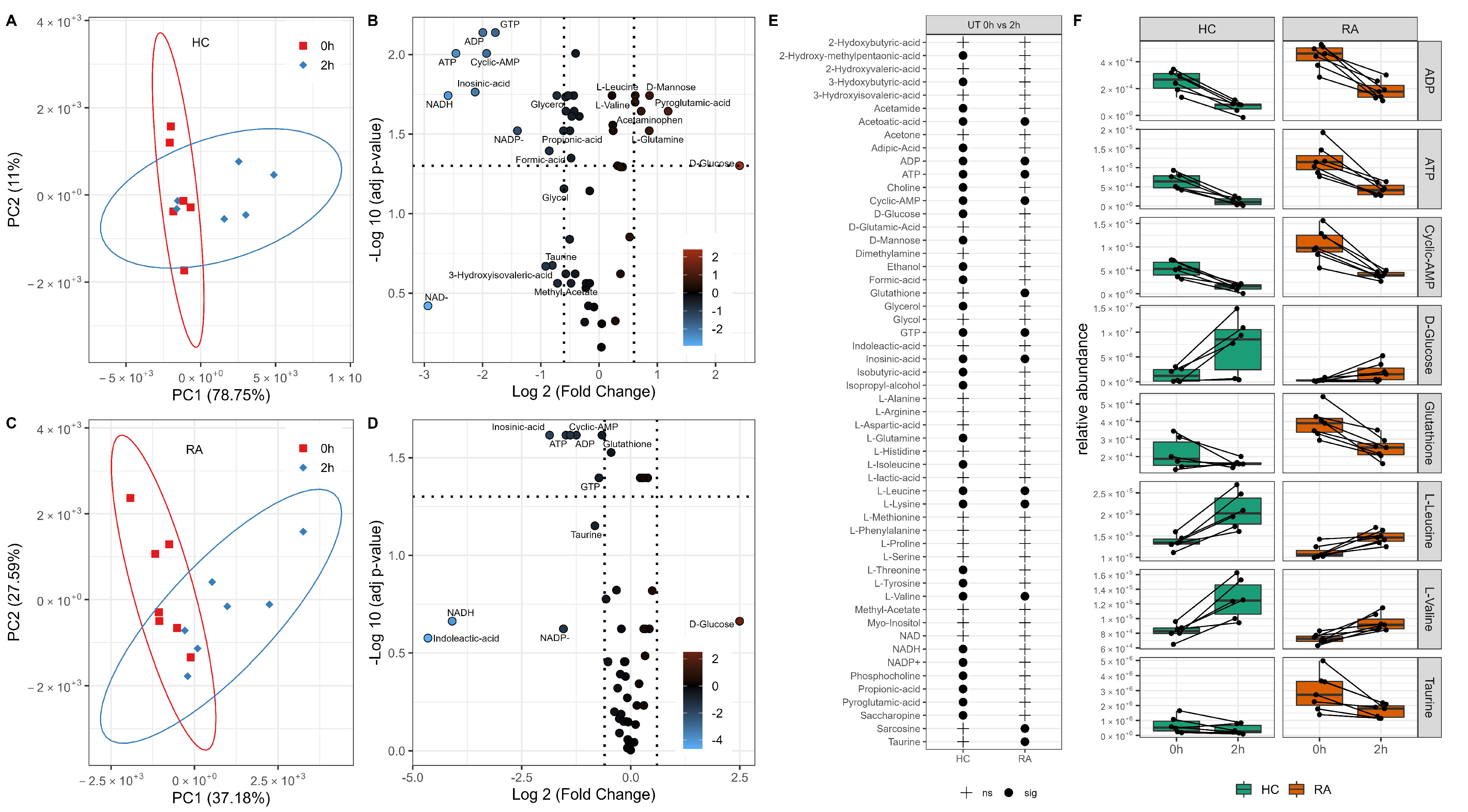
3.1. Changes in Neutrophil Metabolome Associated with RA
3.2. Changes in Neutrophil Metabolome after 2 h Incubation
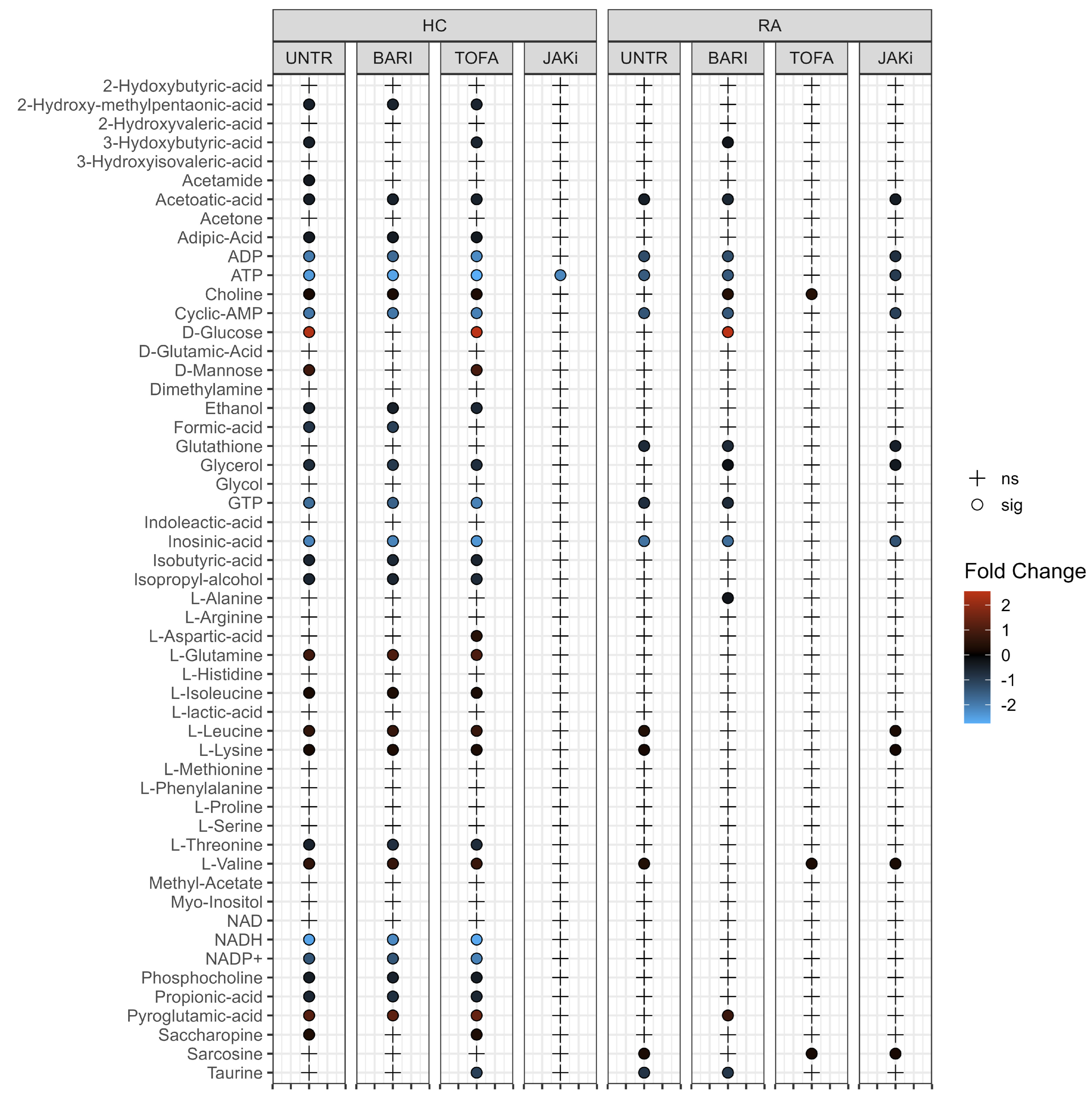
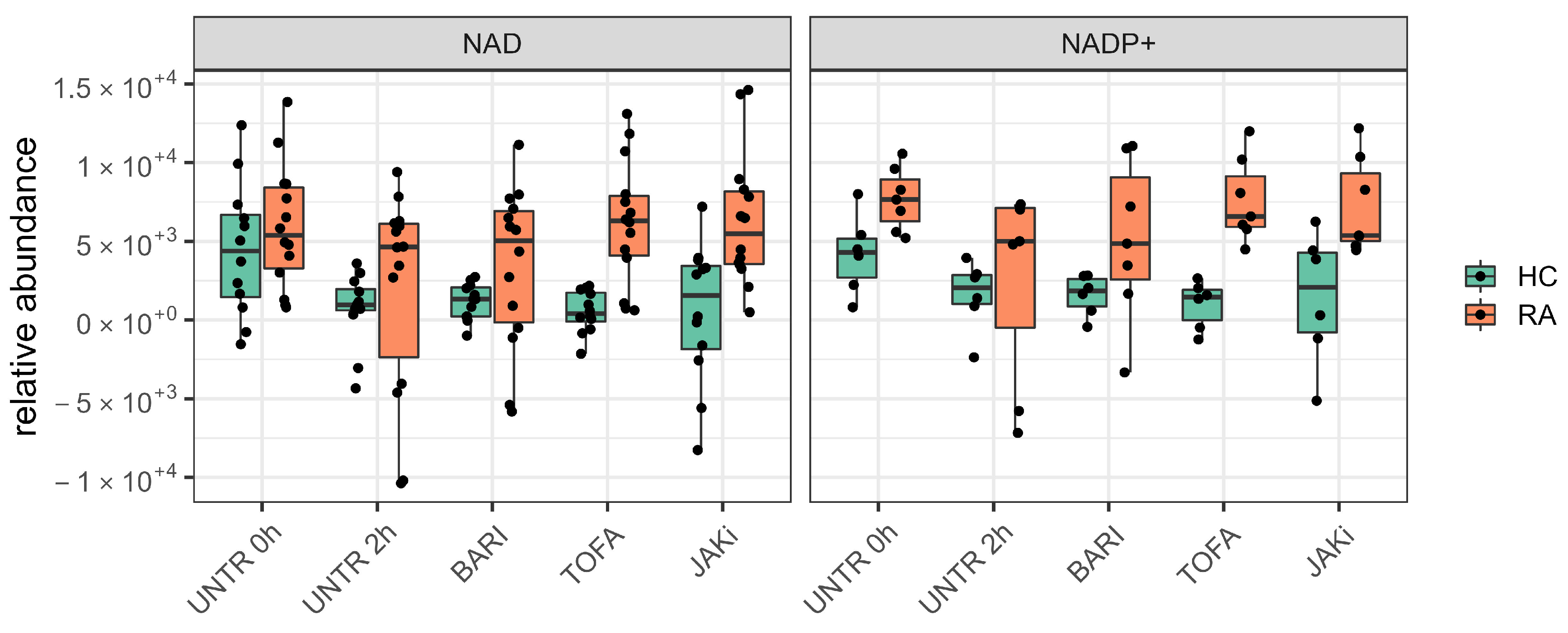
3.3. Changes in Neutrophil Metabolome Following Treatment with JAK Inhibitors
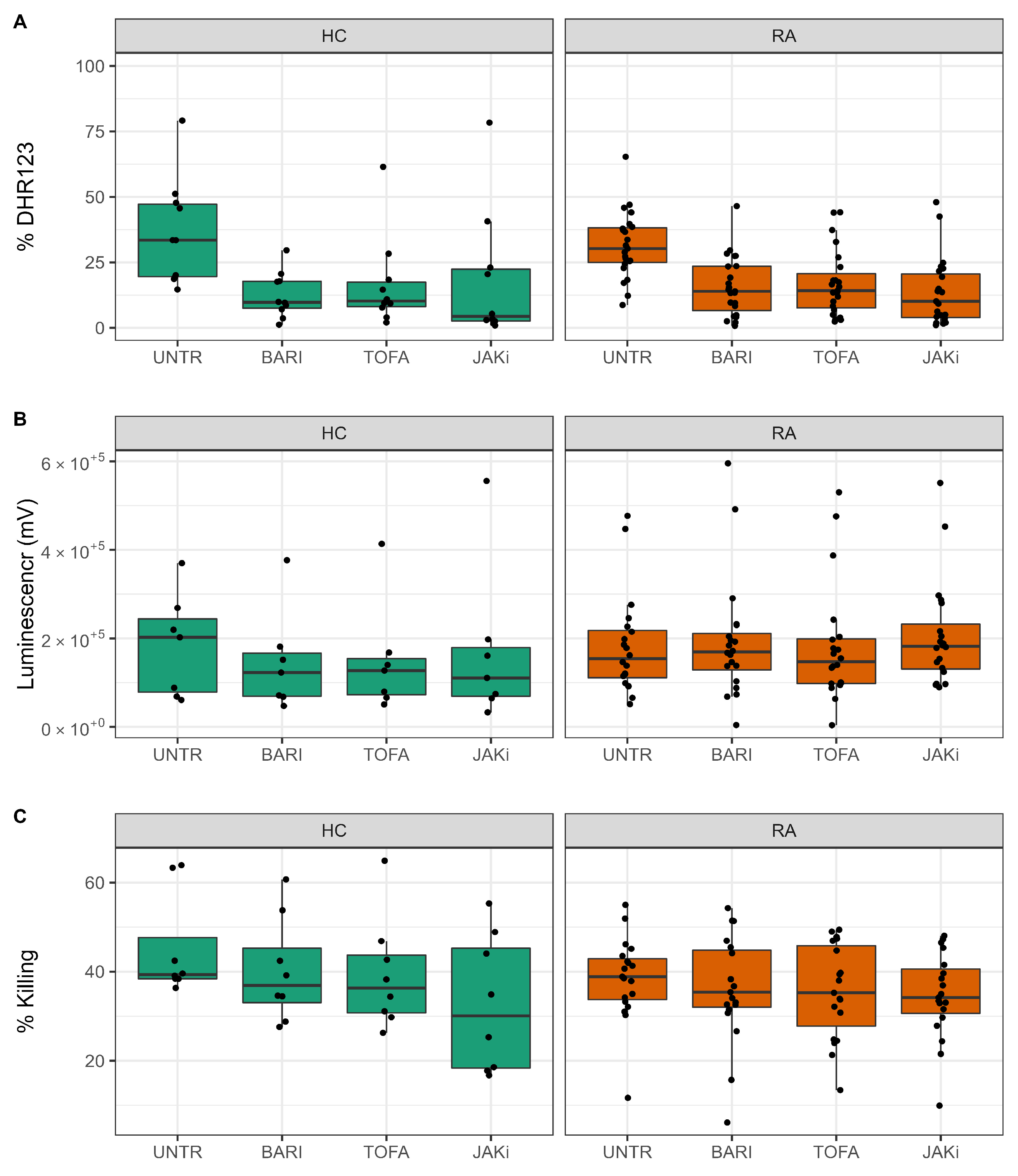
3.4. Effect of JAK Inhibitors on ROS and NET Production
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACR | American College of Rheumatology |
| DHR123 | Dihydrorhodamine-123 |
| DMARD | Disease-modifying anti-rheumatic drug |
| fMLP | f-Met-Leu-Phe |
| GM-CSF | Granulocyte/macrophage-colony stimulating factor |
| JAK | Janus Kinase |
| JAKi | Janus kinase inhibitor |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NET | Neutrophil extracellular trap |
| NMR | nuclear magnetic resonance |
| PMA | Phorbol 12-myristate 13-acetate |
| RA | Rheumatoid arthritis |
| ROS | Reactive Oxygen Species |
| STAT | Signal transducers and activators of transcription |
| TNF | Tumour necrosis factor alpha |
References
- Edwards, S.W. Biochemistry and Physiology of the Neutrophil; Cambridge University Press: Cambridge, UK, 1994. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Wright, H.L.; Moots, R.J.; Bucknall, R.C.; Edwards, S.W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 2010, 49, 1618–1631. [Google Scholar] [CrossRef] [Green Version]
- Wright, H.L.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, L.J.; Kaplan, M.J. Neutrophils in Rheumatoid Arthritis: Breaking Immune Tolerance and Fueling Disease. Trends Mol. Med. 2019, 25, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Tanaka, Y. Targeting the Janus kinases in rheumatoid arthritis: Focus on tofacitinib. Expert Opin. Pharmacother. 2014, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Blanco, R.; Charles-Schoeman, C.; Wollenhaupt, J.; Zerbini, C.; Benda, B.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; Zwillich, S.H.; et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: A randomised phase 3 trial. Lancet 2013, 381, 451–460. [Google Scholar] [CrossRef]
- Fleischmann, R.; Kremer, J.; Cush, J.; Schulze-Koops, H.; Connell, C.A.; Bradley, J.D.; Gruben, D.; Wallenstein, G.V.; Zwillich, S.H.; Kanik, K.S.; et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 495–507. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Yamaoka, K. JAK inhibitor tofacitinib for treating rheumatoid arthritis: From basic to clinical. Mod. Rheumatol. 2013, 23, 415–424. [Google Scholar] [CrossRef]
- Laurence, A.; Pesu, M.; Silvennoinen, O.; O’Shea, J. JAK Kinases in Health and Disease: An Update. Open Rheumatol. J. 2012, 6, 232–244. [Google Scholar] [CrossRef]
- Shi, J.G.; Chen, X.; Lee, F.; Emm, T.; Scherle, P.A.; Lo, Y.; Punwani, N.; Williams, W.V.; Yeleswaram, S. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J. Clin. Pharmacol. 2014, 54, 1354–1361. [Google Scholar] [CrossRef]
- Kaur, K.; Kalra, S.; Kaushal, S. Systematic review of tofacitinib: A new drug for the management of rheumatoid arthritis. Clin. Ther. 2014, 36, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Strand, V.; van Vollenhoven, R.F.; Lee, E.B.; Fleischmann, R.; Zwillich, S.H.; Gruben, D.; Koncz, T.; Wilkinson, B.; Wallenstein, G. Tofacitinib or adalimumab versus placebo: Patient-reported outcomes from a phase 3 study of active rheumatoid arthritis. Rheumatology 2016, 55, 1031–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Radominski, S.C.; Gomez-Reino, J.J.; Wang, L.; Krishnaswami, S.; Wood, S.P.; Soma, K.; Nduaka, C.I.; Kwok, K.; Valdez, H.; et al. Analysis of infections and all-cause mortality in phase II, phase III, and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 2924–2937. [Google Scholar] [CrossRef] [PubMed]
- Garufi, C.; Maclean, M.; Gadina, M.; Spinelli, F.R. Affecting the effectors: JAK inhibitors modulation of immune cell numbers and functions in patients with rheumatoid arthritis. Expert Rev. Clin. Immunol. 2022, 18, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.Y.; Asano, T.; Sumichika, Y.; Sato, S.; Kobayashi, H.; Watanabe, H.; Suzuki, E.; Kozuru, H.; Yatsuhashi, H.; Koga, T.; et al. Tofacitinib inhibits granulocyte–macrophage colony-stimulating factor-induced NLRP3 inflammasome activation in human neutrophils. Arthritis Res. Ther. 2018, 20, 196. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Immunometabolism in early and late stages of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 291–301. [Google Scholar] [CrossRef]
- Wishart, D.S. Metabolomics for Investigating Physiological and Pathophysiological Processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef]
- Emwas, A.H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef] [Green Version]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef]
- Thomas, H.B.; Moots, R.J.; Edwards, S.W.; Wright, H.L. Whose Gene Is It Anyway? The Effect of Preparation Purity on Neutrophil Transcriptome Studies. PLoS ONE 2015, 10, e0138982. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.S.; Moots, R.J.; Wright, H.L. Janus kinase inhibitors prevent migration of rheumatoid arthritis neutrophils towards interleukin-8, but do not inhibit priming of the respiratory burst or reactive oxygen species production. Clin. Exp. Immunol. 2017, 189, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Dowty, M.E.; Jesson, M.I.; Ghosh, S.; Lee, J.; Meyer, D.M.; Krishnaswami, S.; Kishore, N. Preclinical to clinical translation of tofacitinib, a Janus kinase inhibitor, in rheumatoid arthritis. J. Pharmacol. Exp. Ther. 2014, 348, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chokesuwattanaskul, S.; Phelan, M.M.; Edwards, S.W.; Wright, H.L. A robust intracellular metabolite extraction protocol for human neutrophil metabolic profiling. PLoS ONE 2018, 13, e0209270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.D.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolom. Off. J. Metabolom. Soc. 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted profiling: Quantitative analysis of 1H-NMR metabolomics data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef]
- Grosman, R. Nmr Metabolic Profiling of Mosquito Species to Understand Insecticide Resistance. Ph.D. Thesis, The University of Liverpool, Liverpool, UK, 2020. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [Green Version]
- Sommer, C.; Straehle, C.N.; K’othe, U.; Hamprecht, F.A. ilastik: Interactive Learning and Segmentation Toolkit. In Proceedings of the Eighth IEEE International Symposium on Biomedical Imaging (ISBI 2011), Chicago, IL, USA, 30 March–2 April 2011; Volume 1, pp. 230–233. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. mixOmics: An R package for ’omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- Kohl, S.M.; Klein, M.S.; Hochrein, J.; Oefner, P.J.; Spang, R.; Gronwald, W. State-of-the art data normalization methods improve NMR-based metabolomic analysis. Metabolomics 2012, 8, 146–160. [Google Scholar] [CrossRef] [Green Version]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H-NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Sokolova, M.; Lapalme, G. A systematic analysis of performance measures for classification tasks. Inf. Process. Manag. 2009, 45, 427–437. [Google Scholar] [CrossRef]
- Broadhurst, D.I.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef] [Green Version]
- Westerhuis, J.A.; Hoefsloot, H.C.J.; Smit, S.; Vis, D.J.; Smilde, A.K.; van Velzen, E.J.J.; van Duijnhoven, J.P.M.; van Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Alarcon, M.F.; McLaren, Z.; Wright, H.L. Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O. Front. Immunol. 2021, 12, 649693. [Google Scholar] [CrossRef]
- Edwards, S.W. The O-2Generating NADPH Oxidase of Phagocytes: Structure and Methods of Detection. Methods 1996, 9, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs Are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, H.L.; Makki, F.A.; Moots, R.J.; Edwards, S.W. Low-density granulocytes: Functionally distinct, immature neutrophils in rheumatoid arthritis with altered properties and defective TNF signalling. J. Leukoc. Biol. 2016, 101, 599–611. [Google Scholar] [CrossRef]
- Chapman, E.A.; Lyon, M.; Simpson, D.; Mason, D.; Beynon, R.J.; Moots, R.J.; Wright, H.L. Caught in a Trap? Proteomic Analysis of Neutrophil Extracellular Traps in Rheumatoid Arthritis and Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 423. [Google Scholar] [CrossRef]
- Clish, C.B. Metabolomics: An emerging but powerful tool for precision medicine. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [Green Version]
- Hur, B.; Gupta, V.K.; Huang, H.; Wright, K.A.; Warrington, K.J.; Taneja, V.; Davis, J.M.; Sung, J. Plasma metabolomic profiling in patients with rheumatoid arthritis identifies biochemical features predictive of quantitative disease activity. Arthritis Res. Ther. 2021, 23, 164. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.; Hiraishi, T.; Oku, T.; Okuma, K.; Suzumura, K.; Hashimoto, M.; Ito, H.; Aramori, I.; Hirayama, Y. Metabolomic approach to the exploration of biomarkers associated with disease activity in rheumatoid arthritis. PLoS ONE 2019, 14, e0219400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Dikshit, M. Metabolic Insight of Neutrophils in Health and Disease. Front. Immunol. 2019, 10, 2099. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Ghosh, P.; Datta, S.; Ghosh, A.; Chattopadhyay, S.; Chatterjee, M. Oxidative stress as a potential biomarker for determining disease activity in patients with Rheumatoid Arthritis. Free Radic. Res. 2012, 46, 1482–1489. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Kontny, E. Taurine and inflammatory diseases. Amino Acids 2014, 46, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.G.; Kwon, Y.M.; Kang, I.S.; Kim, C. Taurine chloramine selectively regulates neutrophil degranulation through the inhibition of myeloperoxidase and upregulation of lactoferrin. Amino Acids 2020, 52, 1191–1199. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Grabowska, A.; Bereta, J.; Stelmaszynska, T. Taurine chloramine, a product of activated neutrophils, inhibits in vitro the generation of nitric oxide and other macrophage inflammatory mediators. J. Leukoc. Biol. 1995, 58, 667–674. [Google Scholar] [CrossRef]
- Richer, B.C.; Salei, N.; Laskay, T.; Seeger, K. Changes in Neutrophil Metabolism upon Activation and Aging. Inflammation 2018, 41, 710–721. [Google Scholar] [CrossRef]
- Bilzer, M.; Lauterburg, B.H. Glutathione metabolism in activated human neutrophils: Stimulation of glutathione synthesis and consumption of glutathione by reactive oxygen species. Eur. J. Clin. Investig. 1991, 21, 316–322. [Google Scholar] [CrossRef]
- Yuyun, X.; Fan, Y.; Weiping, W.; Qing, Y.; Bingwei, S. Metabolomic analysis of spontaneous neutrophil apoptosis reveals the potential involvement of glutathione depletion. Innate Immun. 2020, 27, 31–40. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Jammal, T.E.; Vuitton, L.; Gerfaud-Valentin, M.; Kerever, S.; Sève, P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun. Rev. 2019, 18, 102390. [Google Scholar] [CrossRef]
- Maeshima, K.; Yamaoka, K.; Kubo, S.; Nakano, K.; Iwata, S.; Saito, K.; Ohishi, M.; Miyahara, H.; Tanaka, S.; Ishii, K.; et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon-γ and interleukin-17 production by human CD4+ T cells. Arthritis Rheum. 2012, 64, 1790–1798. [Google Scholar] [CrossRef]
- Kubo, S.; Yamaoka, K.; Kondo, M.; Yamagata, K.; Zhao, J.; Iwata, S.; Tanaka, Y. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann. Rheum. Dis. 2013, 73, 2192–2198. [Google Scholar] [CrossRef]
- Kubo, S.; Nakayamada, S.; Sakata, K.; Kitanaga, Y.; Ma, X.; Lee, S.; Ishii, A.; Yamagata, K.; Nakano, K.; Tanaka, Y. Janus Kinase Inhibitor Baricitinib Modulates Human Innate and Adaptive Immune System. Front. Immunol. 2018, 9, 1510. [Google Scholar] [CrossRef]
- Isailovic, N.; Ceribelli, A.; Cincinelli, G.; Vecellio, M.; Guidelli, G.; Caprioli, M.; Luciano, N.; Motta, F.; Selmi, C.; Santis, M.D. Lymphocyte modulation by tofacitinib in patients with rheumatoid arthritis. Clin. Exp. Immunol. 2021, 205, 142–149. [Google Scholar] [CrossRef]
- Sun, L.; Guo, R.F.; Newstead, M.W.; Standiford, T.J.; Macariola, D.R.; Shanley, T.P. Effect of IL-10 on Neutrophil Recruitment and Survival after Pseudomonas aeruginosa Challenge. Am. J. Respir. Cell Mol. Biol. 2009, 41, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, K.; Kumar, U.; Das, S.; Chaudhuri, J.; Kumar, P.; Kanjilal, M.; Ghosh, P.; Sircar, G.; Basyal, R.K.; Kanga, U.; et al. Synovial IL-9 facilitates neutrophil survival, function and differentiation of Th17 cells in rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Xiao, X.; Ghobrial, R.M.; Li, X.C. IL-9 and Th9 cells: Progress and challenges. Int. Immunol. 2013, 25, 547–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chokesuwattanaskul, S.; Fresneda Alarcon, M.; Mangalakumaran, S.; Grosman, R.; Cross, A.L.; Chapman, E.A.; Mason, D.; Moots, R.J.; Phelan, M.M.; Wright, H.L. Metabolic Profiling of Rheumatoid Arthritis Neutrophils Reveals Altered Energy Metabolism That Is Not Affected by JAK Inhibition. Metabolites 2022, 12, 650. https://doi.org/10.3390/metabo12070650
Chokesuwattanaskul S, Fresneda Alarcon M, Mangalakumaran S, Grosman R, Cross AL, Chapman EA, Mason D, Moots RJ, Phelan MM, Wright HL. Metabolic Profiling of Rheumatoid Arthritis Neutrophils Reveals Altered Energy Metabolism That Is Not Affected by JAK Inhibition. Metabolites. 2022; 12(7):650. https://doi.org/10.3390/metabo12070650
Chicago/Turabian StyleChokesuwattanaskul, Susama, Michele Fresneda Alarcon, Sangeetha Mangalakumaran, Rudi Grosman, Andrew L. Cross, Elinor A. Chapman, David Mason, Robert J. Moots, Marie M. Phelan, and Helen L. Wright. 2022. "Metabolic Profiling of Rheumatoid Arthritis Neutrophils Reveals Altered Energy Metabolism That Is Not Affected by JAK Inhibition" Metabolites 12, no. 7: 650. https://doi.org/10.3390/metabo12070650
APA StyleChokesuwattanaskul, S., Fresneda Alarcon, M., Mangalakumaran, S., Grosman, R., Cross, A. L., Chapman, E. A., Mason, D., Moots, R. J., Phelan, M. M., & Wright, H. L. (2022). Metabolic Profiling of Rheumatoid Arthritis Neutrophils Reveals Altered Energy Metabolism That Is Not Affected by JAK Inhibition. Metabolites, 12(7), 650. https://doi.org/10.3390/metabo12070650