
Effects of Metabolic Disorders in Immune Cells and Synoviocytes on the Development of Rheumatoid Arthritis

 ,
, 
Abstract
:1. Introduction
2. Metabolic Disorders in Rheumatoid Arthritis Connected with Mitochondrial Dysfunction
2.1. Hypoxia
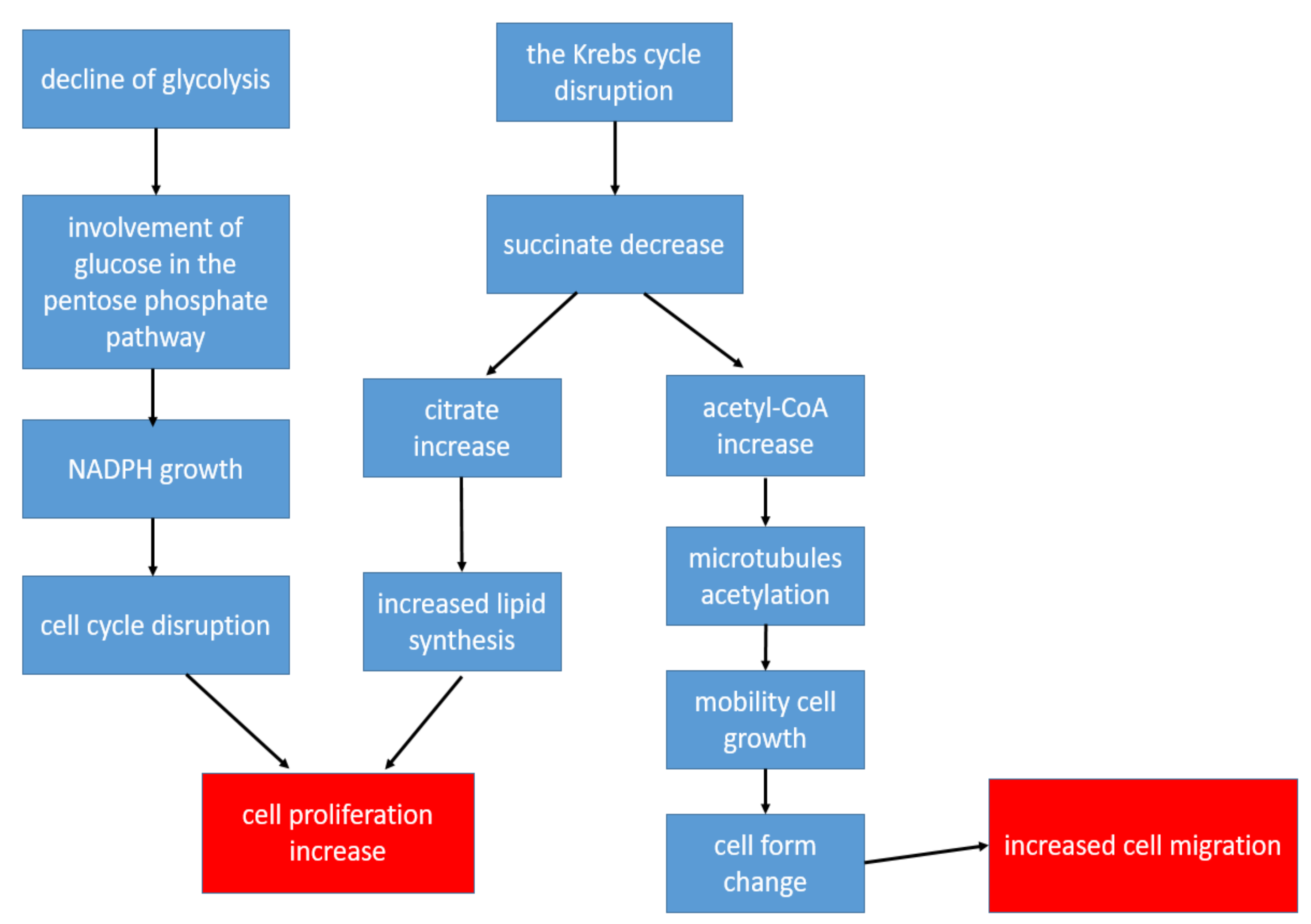
2.2. Glycolysis
2.3. The Krebs Cycle
2.4. Oxidative Phosphorylation
2.5. Fatty Acid β-Oxidation
2.6. Glutaminolysis
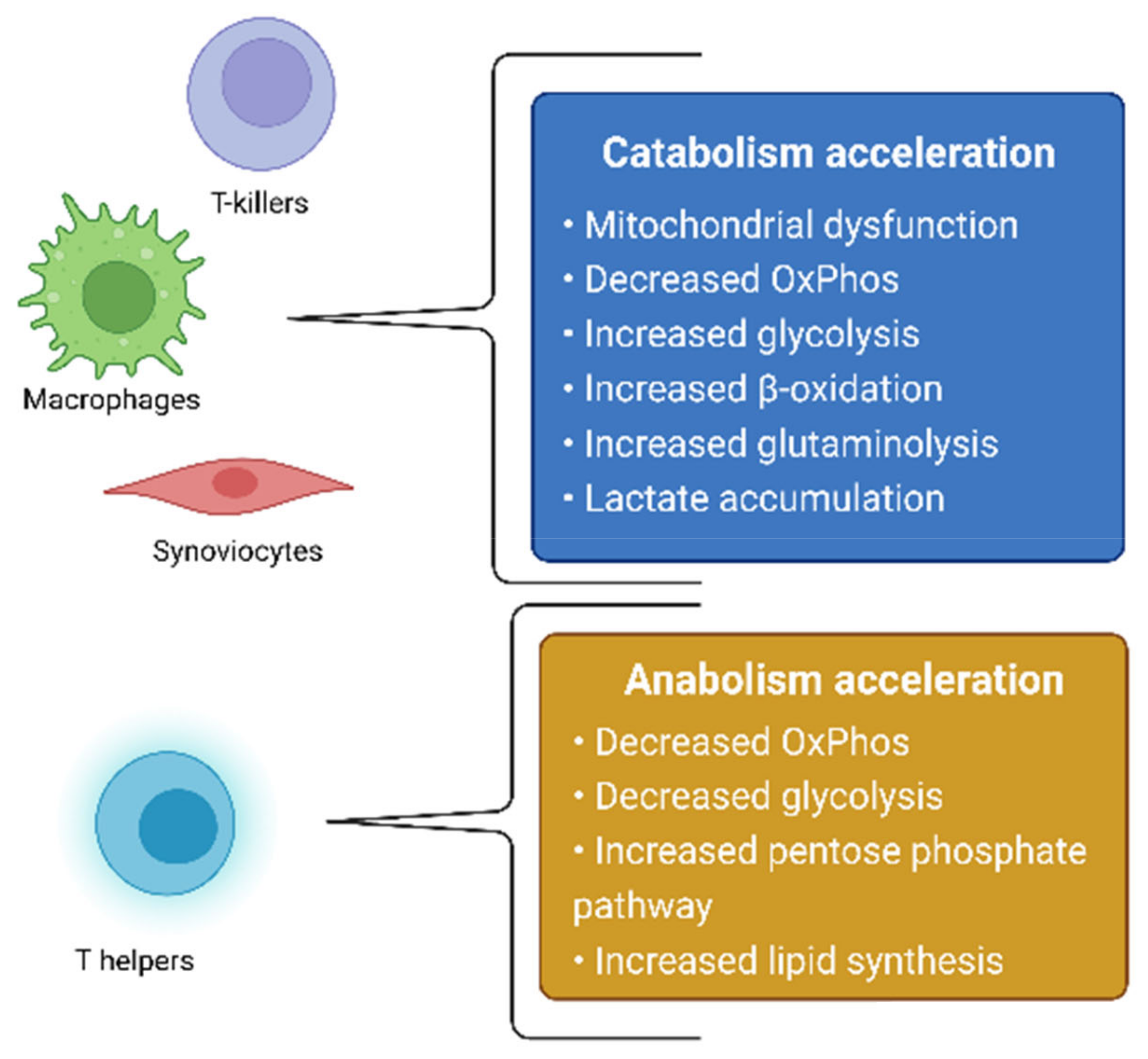
3. Models of Rheumatoid Arthritis Pathogenesis Based on Metabolic Changes in Different Cell Types
3.1. Synoviocytes
3.2. Macrophages
3.3. CD8+ T-Lymphocytes
3.4. CD4+ T-Lymphocytes
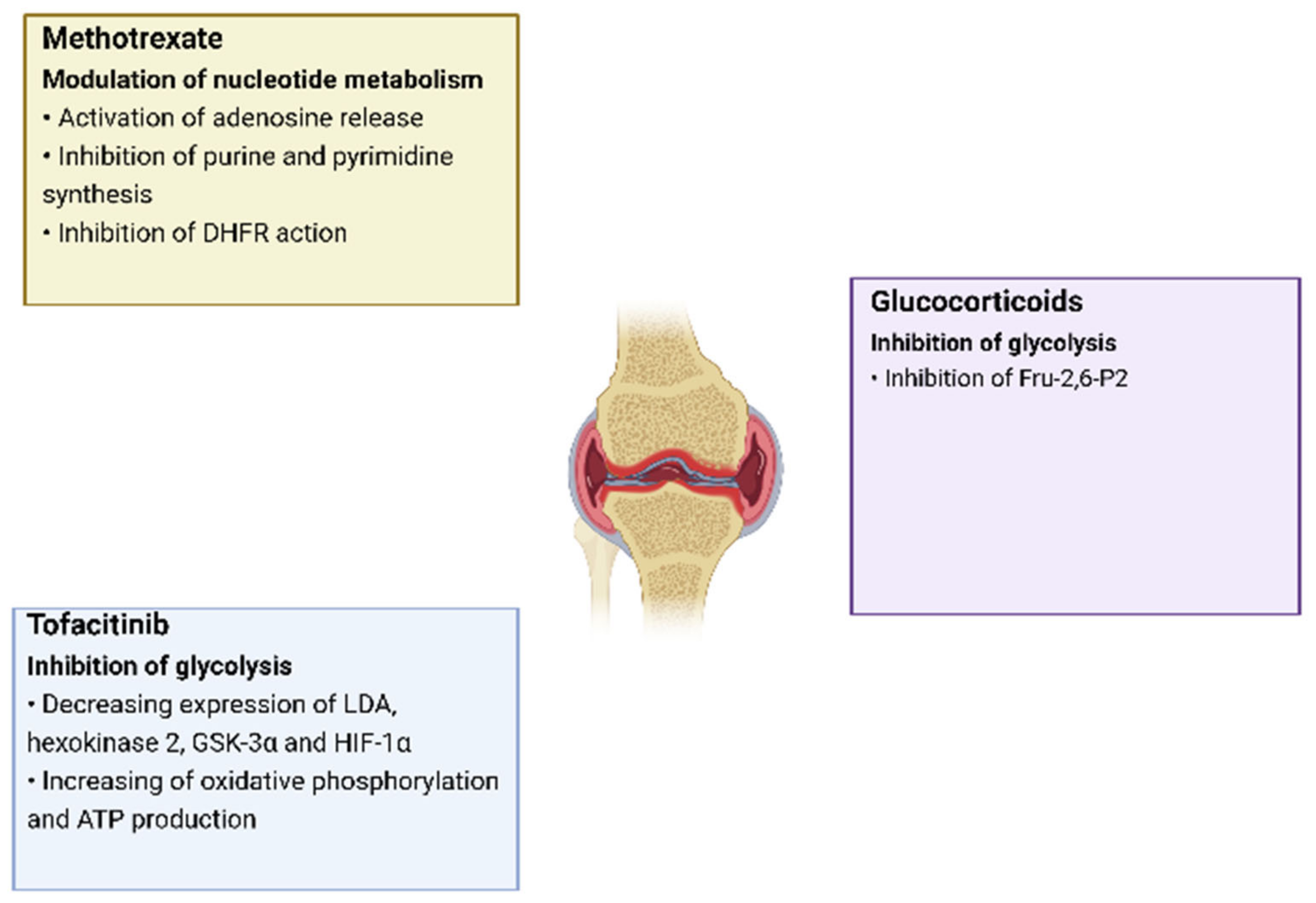
4. Therapy of Rheumatoid Arthritis Aimed at Regulating Metabolic Pathways
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gibofsky, A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: A Synopsis. Am. J. Manag. Care 2014, 20 (Suppl. S7), S128–S135. [Google Scholar]
- Rheumatoid Arthritis. Symptoms & Causes. Available online: https://www.mayoclinic.org/diseases-conditions/rheumatoid-arthritis/symptoms-causes/syc-20353648 (accessed on 28 May 2021).
- Köhler, B.M.; Günther, J.; Kaudewitz, D.; Lorenz, H.-M. Current Therapeutic Options in the Treatment of Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Rheumatoid Arthritis. Diagnosis & Treatment. Available online: https://www.mayoclinic.org/diseases-conditions/rheumatoid-arthritis/diagnosis-treatment/drc-20353653 (accessed on 28 May 2021).
- Clayton, S.A.; MacDonald, L.; Kurowska-Stolarska, M.; Clark, A.R. Mitochondria as Key Players in the Pathogenesis and Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 673916. [Google Scholar] [CrossRef] [PubMed]
- Mitochondrion. Available online: https://www.britannica.com/science/mitochondrion (accessed on 4 June 2021).
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; Oxidative Phosphorylation; W H Freeman: New York, NY, USA, 2002; Chapter 18. Available online: https://www.ncbi.nlm.nih.gov/books/NBK21208/ (accessed on 1 June 2021).
- Mitochondrial Diseases. Available online: https://my.clevelandclinic.org/health/diseases/15612-mitochondrial-diseases (accessed on 4 June 2021).
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Filippin, L.I.; Vercelino, R.; Marroni, N.P.; Xavier, R.M. Redox signalling and the inflammatory response in rheumatoid arthritis. Clin. Exp. Immunol. 2008, 152, 415–422. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Moodley, D.; Mody, G.; Patel, N.; Chuturgoon, A.A. Mitochondrial depolarisation and oxidative stress in rheumatoid arthritis patients. Clin. Biochem. 2008, 41, 1396–1401. [Google Scholar] [CrossRef]
- Da Sylva, T.R.; Connor, A.; Mburu, Y.; Keystone, E.; E Wu, G. Somatic mutations in the mitochondria of rheumatoid arthritis synoviocytes. Arthritis Res. Ther. 2005, 7, R844–R851. [Google Scholar] [CrossRef] [Green Version]
- Biniecka, M.; Kennedy, A.; Ng, C.T.; Chang, T.C.; Balogh, E.; Fox, E.; Veale, D.J.; Fearon, U.; O’Sullivan, J.N. Successful tumour necrosis factor (TNF) blocking therapy suppresses oxidative stress and hypoxia-induced mitochondrial mutagenesis in inflammatory arthritis. Arthritis Res. Ther. 2011, 13, R121. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; DeOliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-Secreted Lactic Acid Promotes IL-23/IL-17 Proinflammatory Pathway. J. Immunol. 2008, 180, 7175–7183. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef] [PubMed]
- Lindy, S.; Uitto, J.; Turto, H.; Rokkanen, P.; Vainio, K. Lactate dehydrogenase in the synovial tissue in rheumatoid arthritis: Total activity and isoenzyme composition. Clin. Chim. Acta 1971, 31, 19–23. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Wu, B.; Goodman, S.B.; Berry, G.J.; Goronzy, J.J.; Weyand, C.M. Metabolic Control of Autoimmunity and Tissue Inflammation in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 652771. [Google Scholar] [CrossRef]
- Saraiva, A.L.; Veras, F.P.; Peres, R.S.; Talbot, J.; Lima, K.A.; Luiz, J.P.; Carballido, J.M.; Cunha, T.M.; Cunha, F.Q.; Ryffel, B.; et al. Succinate receptor deficiency attenuates arthritis by reducing dendritic cell traffic and expansion of Th 17 cells in the lymph nodes. FASEB J. 2018, 32, 6550–6558. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Qiu, J.; Zhao, T.V.; Wang, Y.; Maeda, T.; Goronzy, I.N.; Akiyama, M.; Ohtsuki, S.; Jin, K.; Tian, L.; et al. Succinyl-CoA Ligase Deficiency in Pro-inflammatory and Tissue-Invasive T Cells. Cell Metab. 2020, 32, 967–980. [Google Scholar] [CrossRef]
- Yang, X.Y.; Di Zheng, K.; Lin, K.; Zheng, G.; Zou, H.; Wang, J.M.; Lin, Y.Y.; Chuka, C.M.; Ge, R.S.; Zhai, W.; et al. Energy Metabolism Disorder as a Contributing Factor of Rheumatoid Arthritis: A Comparative Proteomic and Metabolomic Study. PLoS ONE 2015, 10, e0132695. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J. 2007, 405, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poderoso, J.J.; Helfenberger, K.; Poderoso, C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 2019, 88, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230. [Google Scholar] [CrossRef] [Green Version]
- St Clair, E.W.; Wilkinson, W.E.; Lang, T.; Sanders, L.; Misukonis, M.A.; Gilkeson, G.S.; Pisetsky, D.S.; Granger, D.I.; Weinberg, J.B. Increased expression of blood mononuclear cell nitric oxide synthase type 2 in rheumatoid arthritis patients. J. Exp. Med. 1996, 184, 1173–1178. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013, 227, 283–288. [Google Scholar] [CrossRef]
- Soldatov, V.O.; Malorodova, T.N.; Balamutova, T.I.; Ksenofontov, A.O.; Dovgan, A.P.; Urozhevskaya, Z.S. Endothelial dys-function: Comparative evaluation of ultrasound dopplerography, laser dopplerflowmetry and direct monitoring of arterial pressure for conducting pharmacological tests in rats. Res. Results Pharmacol. 2018, 4, 73–80. [Google Scholar] [CrossRef]
- Sobenin, I.; Sazonova, M.; Postnov, A.; Bobryshev, Y.V.; Orekhov, A. Mitochondrial Mutations are Associated with Atherosclerotic Lesions in the Human Aorta. Clin. Dev. Immunol. 2012, 2012, 832464. [Google Scholar] [CrossRef]
- Duvvuri, B.; Duvvuri, V.R.; Wang, C.; Chen, L.; Wagar, L.E.; Jamnik, V.; Wu, J.; Yeung, R.S.M.; Grigull, J.; Watts, T.H.; et al. The Human Immune System Recognizes Neopeptides Derived from Mitochondrial DNA Deletions. J. Immunol. 2014, 192, 4581–4591. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Myeloid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Immunobiology 2015, 220, 833–844. [Google Scholar] [CrossRef]
- Harty, L.C.; Biniecka, M.; O’Sullivan, J.; Fox, E.; Mulhall, K.; Veale, D.; Fearon, U. Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann. Rheum. Dis. 2011, 71, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yu, S.; Wang, D.; Chen, S.; Chen, S.; Zheng, Y.; Wang, N.; Chen, S.; Li, J.; Shen, B. Germline and somatic mtDNA mutation spectrum of rheumatoid arthritis patients in the Taizhou area, China. Rheumatology 2020, 59, 2982–2991. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.; Mitrofanov, K.Y.; Zhelankin, A.V.; Sazonova, M.; Postnov, A.; Revin, V.V.; Bobryshev, Y.V.; Orekhov, A.N. Quantitative Assessment of Heteroplasmy of Mitochondrial Genome: Perspectives in Diagnostics and Methodological Pitfalls. BioMed Res. Int. 2014, 2014, 292017. [Google Scholar] [CrossRef]
- Rodgers, L.C.; Cole, J.; Rattigan, K.M.; Barrett, M.P.; Kurian, N.; McInnes, I.B.; Goodyear, C.S. The rheumatoid synovial environment alters fatty acid metabolism in human monocytes and enhances CCL20 secretion. Rheumatology 2019, 59, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Sobenin, I.; Bobryshev, Y.V. Plasmacytoid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Front. Physiol. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curi, R.; Newsholme, P.; Marzuca-Nassr, G.N.; Takahashi, H.K.; Hirabara, S.M.; Cruzat, V.; Krause, M.; de Bittencourt, P.I.H. Regulatory principles in metabolism–then and now. Biochem. J. 2016, 473, 1845–1857. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Saegusa, J.; Sendo, S.; Okano, T.; Akashi, K.; Irino, Y.; Morinobu, A. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 76. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795.e19. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.-F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Carbonell, R.; Divakaruni, A.S.; Lodi, A.; Vicente-Suarez, I.; Saha, A.; Cheroutre, H.; Boss, G.R.; Tiziani, S.; Murphy, A.N.; Guma, M. Critical Role of Glucose Metabolism in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2016, 68, 1614–1626. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Kwon, J.-E.; Lee, S.-Y.; Lee, E.-J.; Kim, D.S.; Moon, S.-J.; Lee, J.; Kwok, S.-K.; Park, S.-H.; Cho, M.-L. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. 2017, 8, e2565. [Google Scholar] [CrossRef] [PubMed]
- Valcarcel-Ares, M.N.; Riveiro-Naveira, R.R.; Garcia, F.J.B.; Loureiro, J.; Hermida-Carballo, L.; Blanco, F.J.; López-Armada, M.J. Mitochondrial dysfunction promotes and aggravates the inflammatory response in normal human synoviocytes. Rheumatology 2014, 53, 1332–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyand, C.M.; Zeisbrich, M.; Goronzy, J.J. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr. Opin. Immunol. 2017, 46, 112–120. [Google Scholar] [CrossRef]
- Zeisbrich, M.; Yanes, R.E.; Zhang, H.; Watanabe, R.; Li, Y.; Brosig, L.; Hong, J.; Wallis, B.B.; Giacomini, J.C.; Assimes, T.L.; et al. Hypermetabolic macrophages in rheumatoid arthritis and coronary artery disease due to glycogen synthase kinase 3b inactivation. Ann. Rheum. Dis. 2018, 77, 1053–1062. [Google Scholar] [CrossRef]
- Yamashita, T.; Hagino, H.; Hayashi, I.; Hayashibara, M.; Tanida, A.; Nagira, K.; Fukui, R.; Nagashima, H. Effect of a cathepsin K inhibitor on arthritis and bone mineral density in ovariectomized rats with collagen-induced arthritis. Bone Rep. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Souto-Carneiro, M.M.; Klika, K.D.; Abreu, M.T.; Meyer, A.P.; Saffrich, R.; Sandhoff, R.; Jennemann, R.; Kraus, F.V.; Tykocinski, L.-O.; Eckstein, V.; et al. Effect of Increased Lactate Dehydrogenase An Activity and Aerobic Glycolysis on the Proinflammatory Profile of Autoimmune CD8+ T Cells in Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 2050–2064. [Google Scholar] [CrossRef]
- Yang, Z.; Shen, Y.; Oishi, H.; Matteson, E.L.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 331ra38. [Google Scholar] [CrossRef] [Green Version]
- McGarry, T.; Orr, C.; Wade, S.; Biniecka, M.; Wade, S.; Gallagher, L.; Low, C.; Veale, D.; Fearon, U. JAK/STATBlockade Alters Synovial Bioenergetics, Mitochondrial Function, and Proinflammatory Mediators in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1959–1970. [Google Scholar] [CrossRef] [Green Version]
- Falconer, J.; Murphy, A.N.; Young, S.P.; Clark, A.R.; Tiziani, S.; Guma, M.; Buckley, C.D. Review: Synovial Cell Metabolism and Chronic Inflammation in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.; Hasseli, R.; Ohl, S.; Lange, U.; Frommer, K.; Müller-Ladner, U. Adipokines and Autoimmunity in Inflammatory Arthritis. Cells 2021, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Pucino, V.; Certo, M.; Varricchi, G.; Marone, G.; Ursini, F.; Rossi, F.W.; de Paulis, A.; Mauro, C.; Raza, K.; Buckley, C.D. Metabolic Checkpoints in Rheumatoid Arthritis. Front. Physiol. 2020, 11, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.C.; O’Donnell, A.F. ‘Sugarcoating’ 2-deoxyglucose: Mechanisms that suppress its toxic effects. Curr. Genet. 2020, 67, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.; Varma, M.; Bryant, C.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2017, 167, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guagnano, M.T.; D’Angelo, C.; Caniglia, D.; Di Giovanni, P.; Celletti, E.; Sabatini, E.; Speranza, L.; Bucci, M.; Cipollone, F.; Paganelli, R. Improvement of Inflammation and Pain after Three Months’ Exclusion Diet in Rheumatoid Arthritis Patients. Nutrients 2021, 13, 3535. [Google Scholar] [CrossRef] [PubMed]
- Jutley, G.S.; Sahota, K.; Sahbudin, I.; Filer, A.; Arayssi, T.; Young, S.P.; Raza, K. Relationship between Inflammation and Metabolism in Patients with Newly Presenting Rheumatoid Arthritis. Front. Immunol. 2021, 12, 3359. [Google Scholar] [CrossRef]
- Mašić, D.; Stengaard-Pedersen, K.; Løgstrup, B.B.; Hørslev-Petersen, K.; Hetland, M.L.; Junker, P.; Østergaard, M.; Ammitzbøll, C.; Möller, S.; Christensen, R.; et al. Similar lipid level changes in early rheumatoid arthritis patients following 1-year treat-to-target strategy with adalimumab plus methotrexate versus placebo plus methotrexate: Secondary analyses from the randomised controlled OPERA trial. Rheumatol. Int. 2021, 41, 543–549. [Google Scholar] [CrossRef]
- Mei, L.; Yang, Z.; Zhang, X.; Liu, Z.; Wang, M.; Wu, X.; Chen, X.; Huang, Q.; Huang, R. Sustained Drug Treatment Alters the Gut Microbiota in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 4238. [Google Scholar] [CrossRef]
- Nogueira-Recalde, U.; Lorenzo-Gómez, I.; Blanco, F.J.; Loza, M.I.; Grassi, D.; Shirinsky, V.; Shirinsky, I.; Lotz, M.; Robbins, P.D.; Domínguez, E.; et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. eBioMedicine 2019, 45, 588–605. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Metabolic Agent | Role in Inflammation |
|---|---|
| Lactate | growth of mitochondrial mutations, release of pro-inflammatory cytokines |
| Succinate | activation of innate immunity through binding to GPR91 |
| Citrate | activation of the synthesis of lipids to create the membranes of new T-helper cells |
| Acetyl-CoA | increased migration of T cells to the synovial membrane |
| LDHA | activation of Th1 cell maturation, increase in IFN-γ production |
| (HIF) -1α | development of hypoxia in the cells of an inflamed joint |
| NO-synthase | synthesis of NO, which causes mitochondrial dysfunction |
| Carnitine | increased production of CCL20, which attracts lymphocytes |
| GLS1 | important for the proliferation of synoviocytes and Th17 lymphocytes |
| SLC1A5 | participation in the differentiation of naive CD4+ T-lymphocytes into Th1 and Th17 lymphocytes |
| Pyruvate kinase | production of proinflammatory cytokines IL-1β and IL-6 by macrophages |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blagov, A.V.; Grechko, A.V.; Nikiforov, N.G.; Zhuravlev, A.D.; Sadykhov, N.K.; Orekhov, A.N. Effects of Metabolic Disorders in Immune Cells and Synoviocytes on the Development of Rheumatoid Arthritis. Metabolites 2022, 12, 634. https://doi.org/10.3390/metabo12070634
Blagov AV, Grechko AV, Nikiforov NG, Zhuravlev AD, Sadykhov NK, Orekhov AN. Effects of Metabolic Disorders in Immune Cells and Synoviocytes on the Development of Rheumatoid Arthritis. Metabolites. 2022; 12(7):634. https://doi.org/10.3390/metabo12070634
Chicago/Turabian StyleBlagov, Alexander V., Andrey V. Grechko, Nikita G. Nikiforov, Alexander D. Zhuravlev, Nikolay K. Sadykhov, and Alexander N. Orekhov. 2022. "Effects of Metabolic Disorders in Immune Cells and Synoviocytes on the Development of Rheumatoid Arthritis" Metabolites 12, no. 7: 634. https://doi.org/10.3390/metabo12070634
APA StyleBlagov, A. V., Grechko, A. V., Nikiforov, N. G., Zhuravlev, A. D., Sadykhov, N. K., & Orekhov, A. N. (2022). Effects of Metabolic Disorders in Immune Cells and Synoviocytes on the Development of Rheumatoid Arthritis. Metabolites, 12(7), 634. https://doi.org/10.3390/metabo12070634