
TraVis Pies: A Guide for Stable Isotope Metabolomics Interpretation Using an Intuitive Visualization



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Visualization Rationale
2.2. Travis Pies, a Software to Generate the Proposed Visualizations
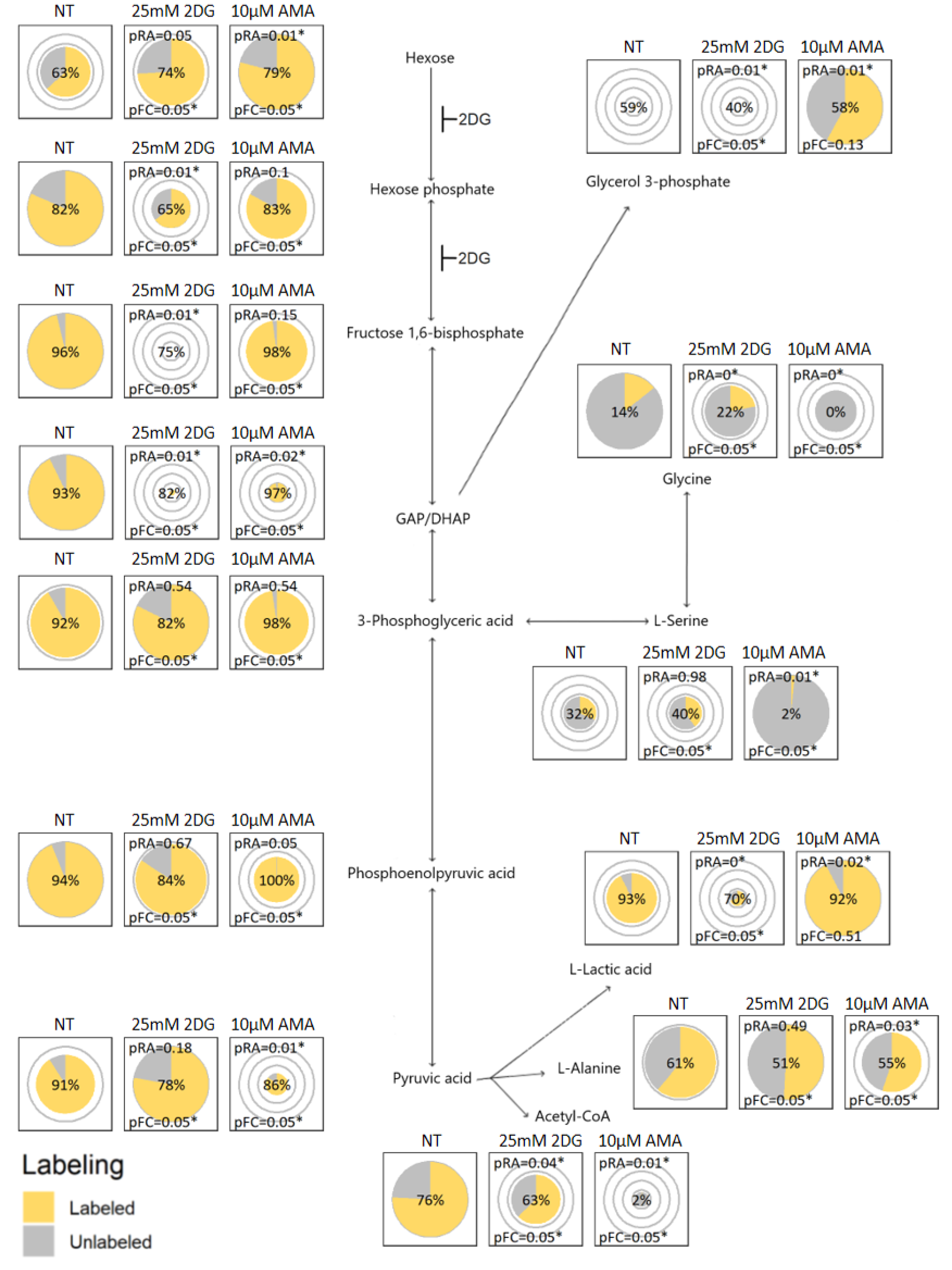
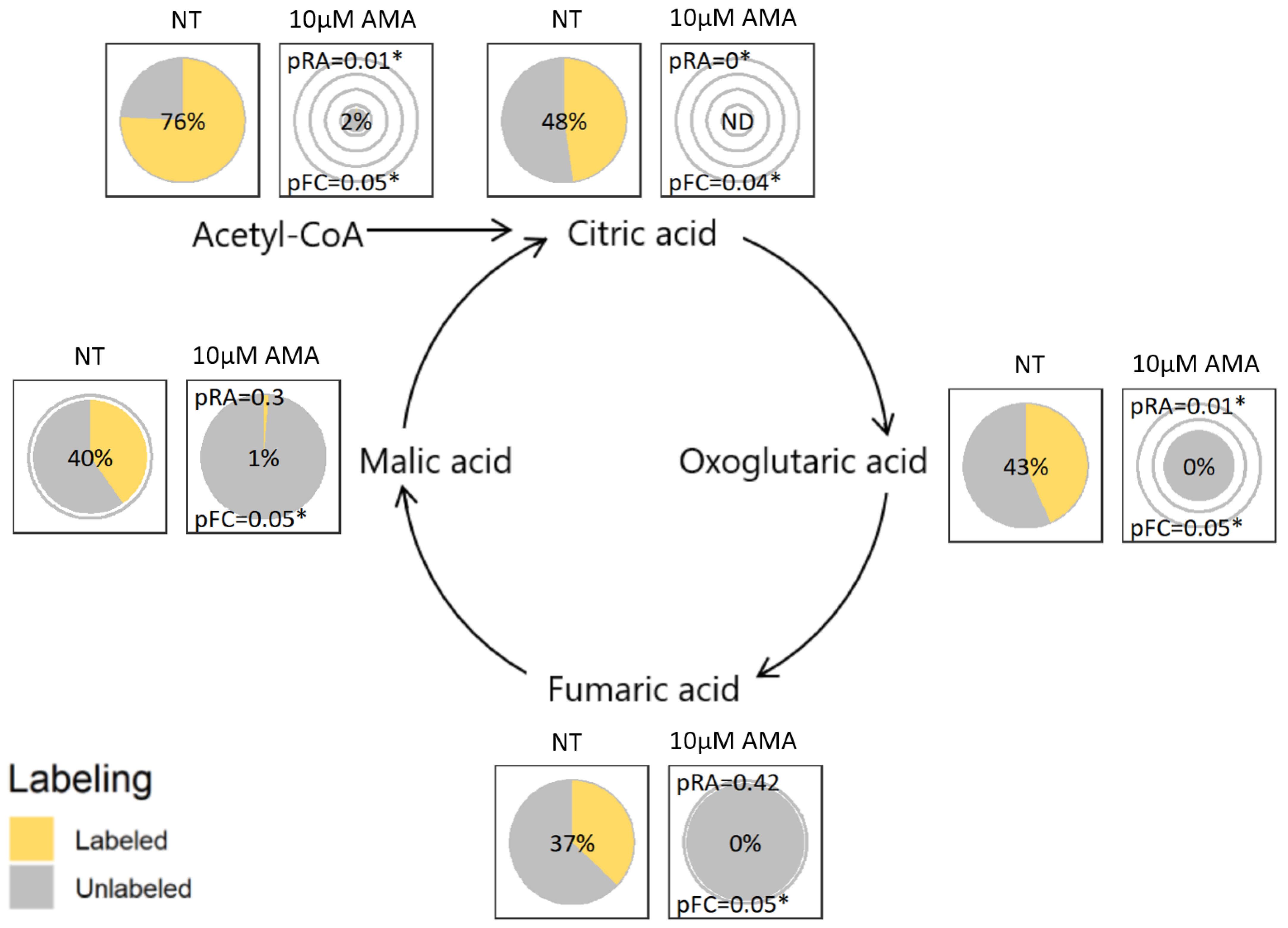
2.3. Visualization Application and Interpretation
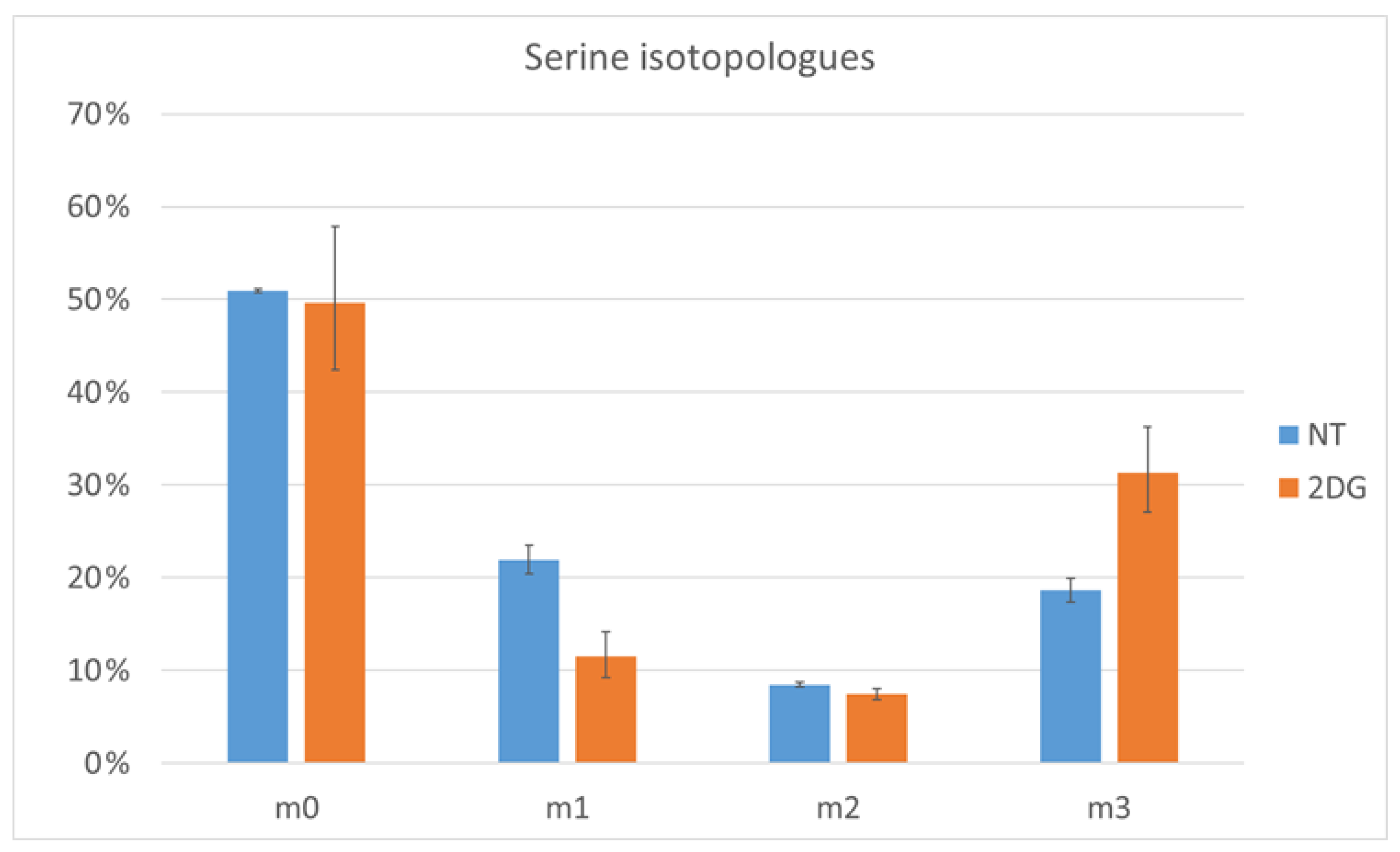
2.4. Verification by Labeling Patterns
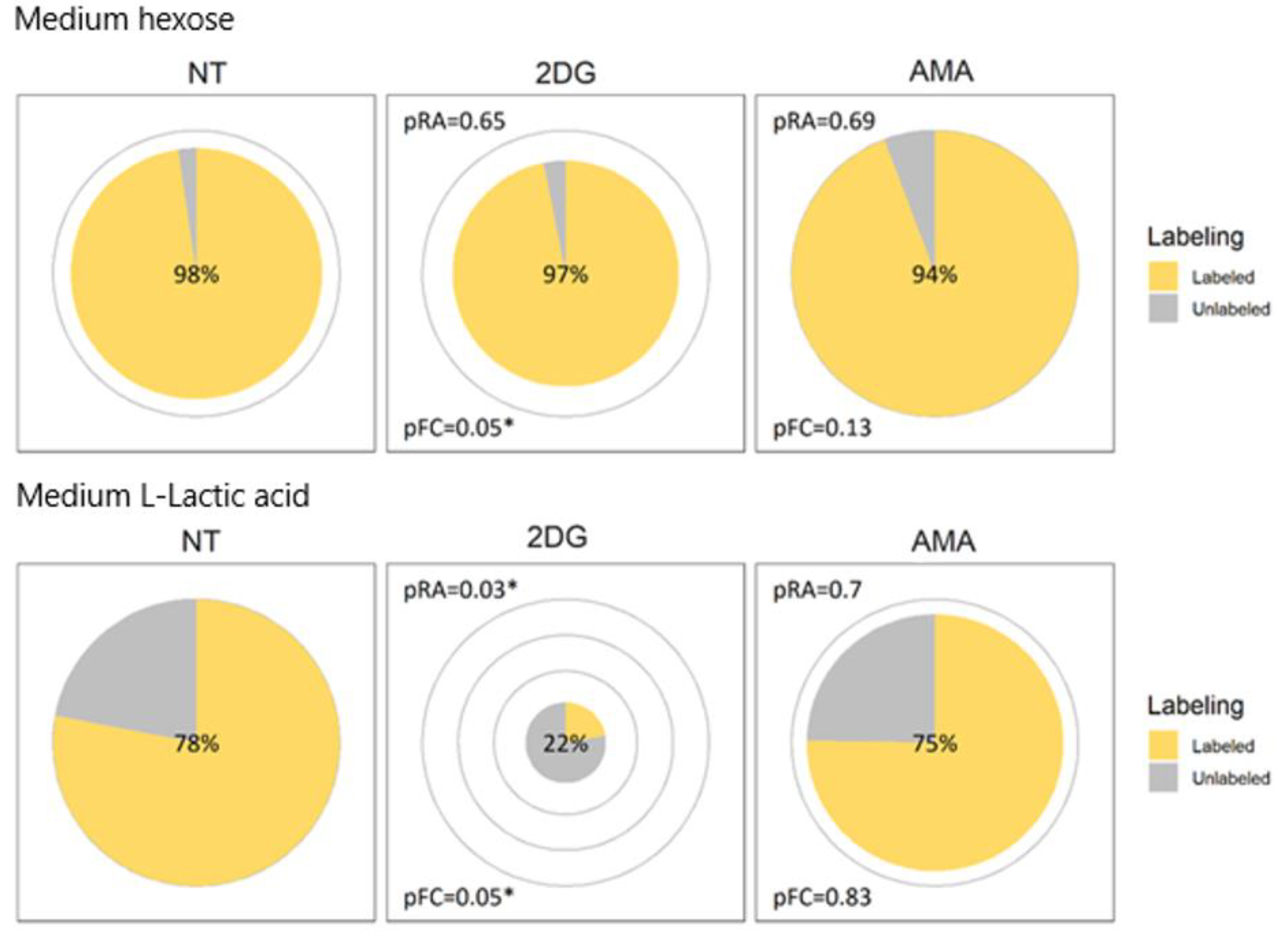
2.5. Support for Application on Other Experiments
3. Materials and Methods
3.1. Cell Culture Experiments
3.2. LC-MS Method
3.3. Data Acquisition and Preliminary Analysis
3.4. Calculating the Fractional Contribution of Acetyl-CoA
3.5. TraVis Pies Data Visualization
3.6. Statistics and Reproducibility
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mitchener, J.; King, Z.A.; Metallo, C.M. Escher-Trace: A web application for pathway-based visualization of stable isotope tracing data. BMC Bioinform. 2020, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, S.; Bird, M.J.; Emmerzaal, T.L.; Wong, S.Y.; Felgueira, C.; Stiers, K.M.; Sabbagh, L.; Himmelreich, N.; Poschet, G.; Windmolders, P.; et al. The Metabolic Map into the Pathomechanism and Treatment of PGM1-CDG. Am. J. Hum. Genet. 2019, 104, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, Y.; Katsumata, Y.; Sano, M.; Honda, K.; Kajimura, M.; Fukuda, K.; Suematsu, M. Visualization of in vivo metabolic flows reveals accelerated utilization of glucose and lactate in penumbra of ischemic heart. Sci. Rep. 2016, 6, 32361. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.C.; O’Donnell, A.F. ‘Sugarcoating’ 2-deoxyglucose: Mechanisms that suppress its toxic effects. Curr. Genet. 2021, 67, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Potter, V.R.; Reif, A.E. Inhibition of an electron transport component by antimycin A. J. Biol. Chem. 1952, 194, 287–297. [Google Scholar] [CrossRef]
- Gaude, E.; Schmidt, C.; Gammage, P.A.; Dugourd, A.; Blacker, T.; Chew, S.P.; Saez-Rodriguez, J.; O’Neill, J.S.; Szabadkai, G.; Minczuk, M.; et al. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol. Cell 2018, 69, 581–593.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polat, I.H.; Tarrado-Castellarnau, M.; Bharat, R.; Perarnau, J.; Benito, A.; Cortés, R.; Sabatier, P.; Cascante, M. Oxidative Pentose Phosphate Pathway Enzyme 6-Phosphogluconate Dehydrogenase Plays a Key Role in Breast Cancer Metabolism. Biology 2021, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Chokkathukalam, A.; Kim, D.H.; Barrett, M.P.; Breitling, R.; Creek, D.J. Stable isotope-labeling studies in metabolomics: New insights into structure and dynamics of metabolic networks. Bioanalysis 2014, 6, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, J.J.; Chung, M.K.; Fan, J.; Rabinowitz, J.D. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 24 June 2022).
- Wickham, H.; Hester, J. Readr: Read Rectangular Text Data 2021. Available online: https://readr.tidyverse.org/ (accessed on 24 June 2022).
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation 2021. Available online: https://dplyr.tidyverse.org/ (accessed on 24 June 2022).
- Wickham, H. Tidyr: Tidy Messy Data 2021. Available online: https://tidyr.tidyverse.org/ (accessed on 24 June 2022).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis. 2016. Available online: https://ggplot2.tidyverse.org/ (accessed on 24 June 2022).
- Winston, C. Extrafont: Tools for Using Fonts 2014. Available online: https://www.rdocumentation.org/packages/extrafont/versions/0.18 (accessed on 24 June 2022).
- Wickham, H. Forcats: Tools for Working with Categorical Variables (Factors) 2021. Available online: https://forcats.tidyverse.org/ (accessed on 24 June 2022).
- Hester, L.; Wickham, H. Vroom: Read and Write Rectangular Text Data Quickly 2021. Available online: https://vroom.r-lib.org/index.html (accessed on 24 June 2022).
- Chang, W.; Cheng, J.; Allaire, J.; Sievert, C.; Schloerke, B.; Xie, Y.; Allen, J.; McPherson, J.; Dipert, A.; Borges, B. Shiny: Web Application Framework for R 2021. Available online: https://shiny.rstudio.com/reference/shiny/1.4.0/shiny-package.html (accessed on 24 June 2022).
- Merlino, A.; Howard, P. ShinyFeedback: Display User Feedback in Shiny Apps 2021. Available online: https://rdrr.io/cran/shinyFeedback/ (accessed on 24 June 2022).
- Attali, D. shinyjs: Easily Improve the User Experience of Your Shiny Apps in Seconds 2021. Available online: https://cran.r-project.org/web/packages/shinyjs/index.html (accessed on 24 June 2022).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Craemer, S.; Driesen, K.; Ghesquière, B. TraVis Pies: A Guide for Stable Isotope Metabolomics Interpretation Using an Intuitive Visualization. Metabolites 2022, 12, 593. https://doi.org/10.3390/metabo12070593
De Craemer S, Driesen K, Ghesquière B. TraVis Pies: A Guide for Stable Isotope Metabolomics Interpretation Using an Intuitive Visualization. Metabolites. 2022; 12(7):593. https://doi.org/10.3390/metabo12070593
Chicago/Turabian StyleDe Craemer, Sam, Karen Driesen, and Bart Ghesquière. 2022. "TraVis Pies: A Guide for Stable Isotope Metabolomics Interpretation Using an Intuitive Visualization" Metabolites 12, no. 7: 593. https://doi.org/10.3390/metabo12070593
APA StyleDe Craemer, S., Driesen, K., & Ghesquière, B. (2022). TraVis Pies: A Guide for Stable Isotope Metabolomics Interpretation Using an Intuitive Visualization. Metabolites, 12(7), 593. https://doi.org/10.3390/metabo12070593