
Targeted Microchip Capillary Electrophoresis-Orbitrap Mass Spectrometry Metabolomics to Monitor Ovarian Cancer Progression

 , , ,
, , , 
Abstract
:1. Introduction
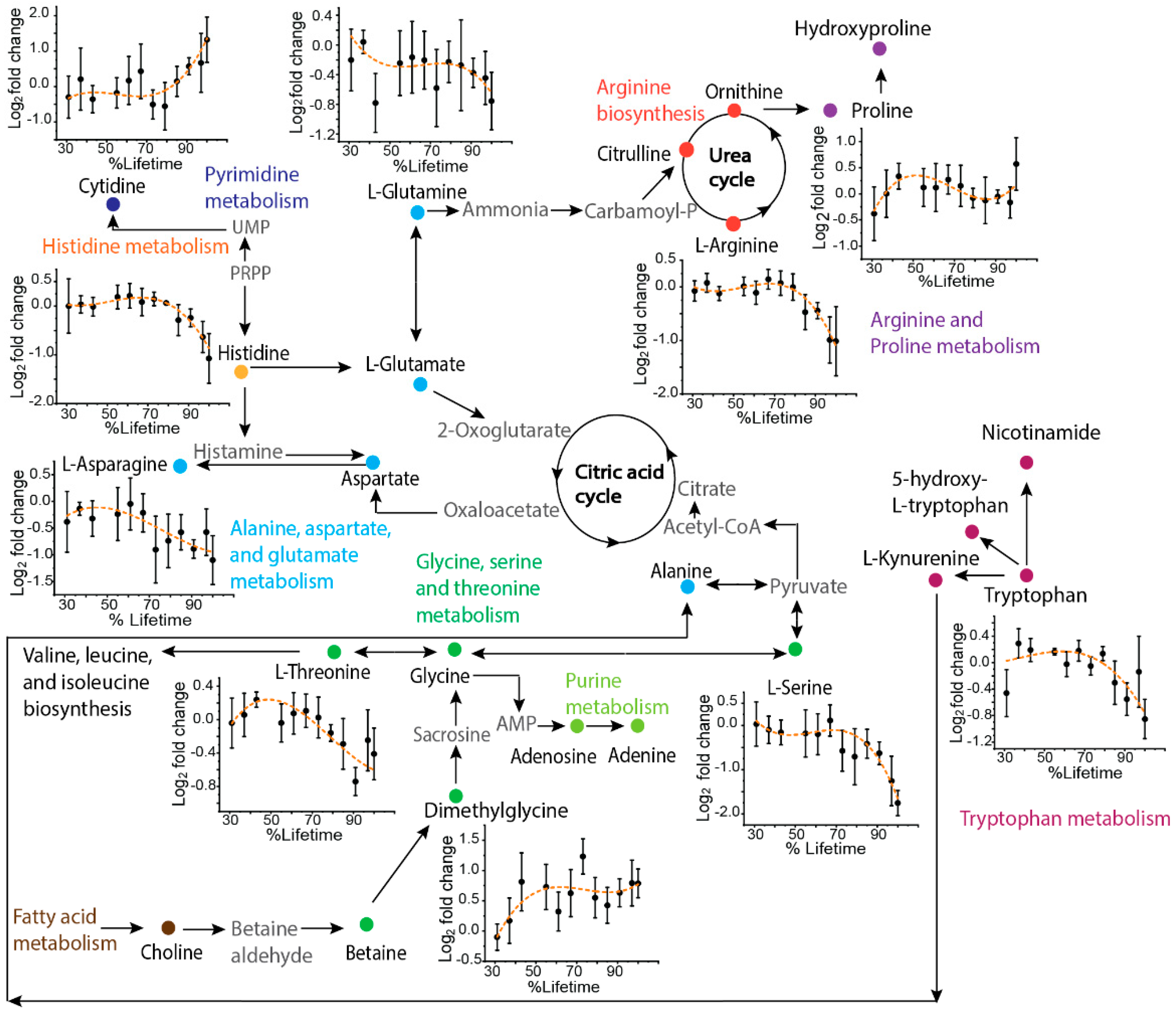
2. Results
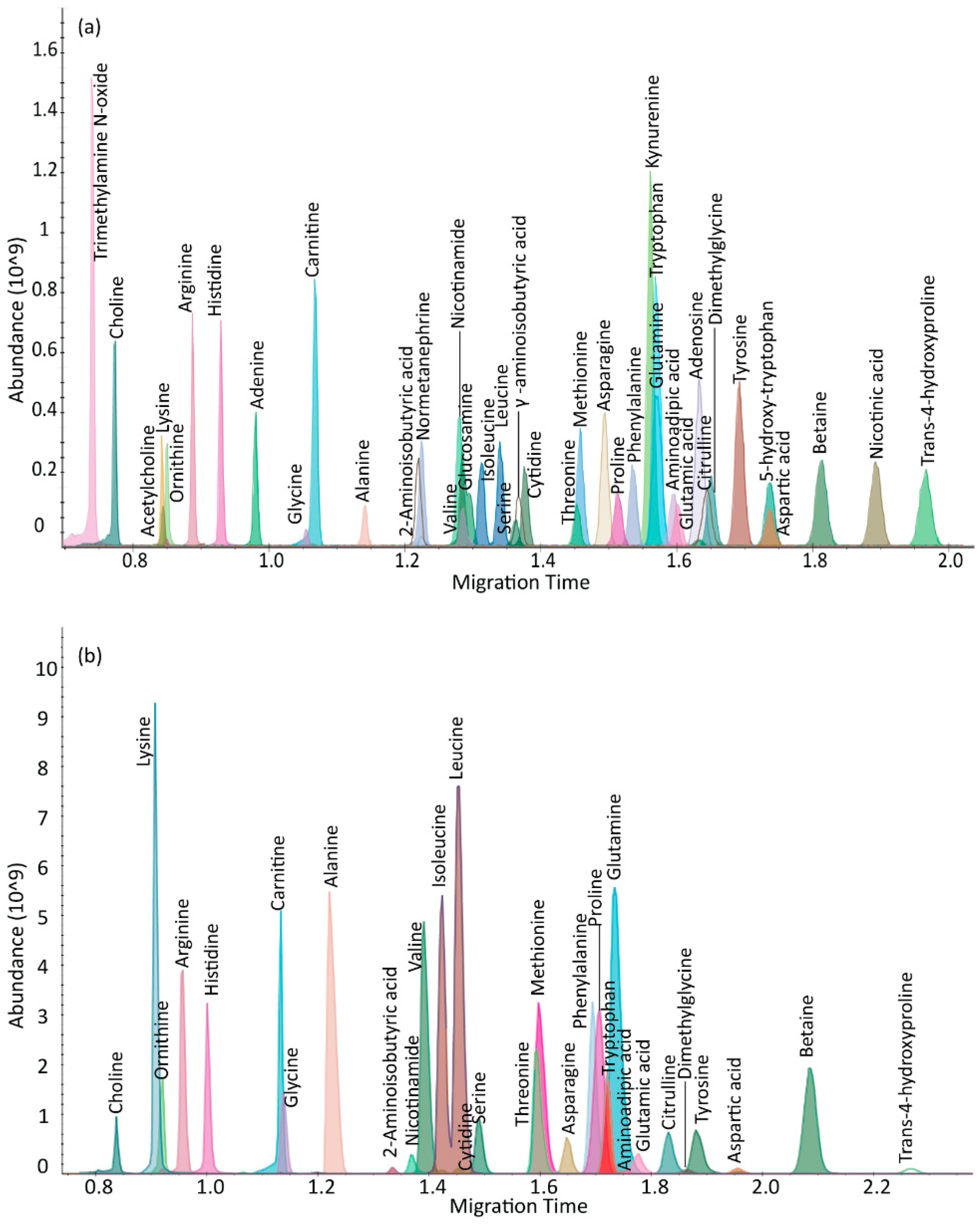
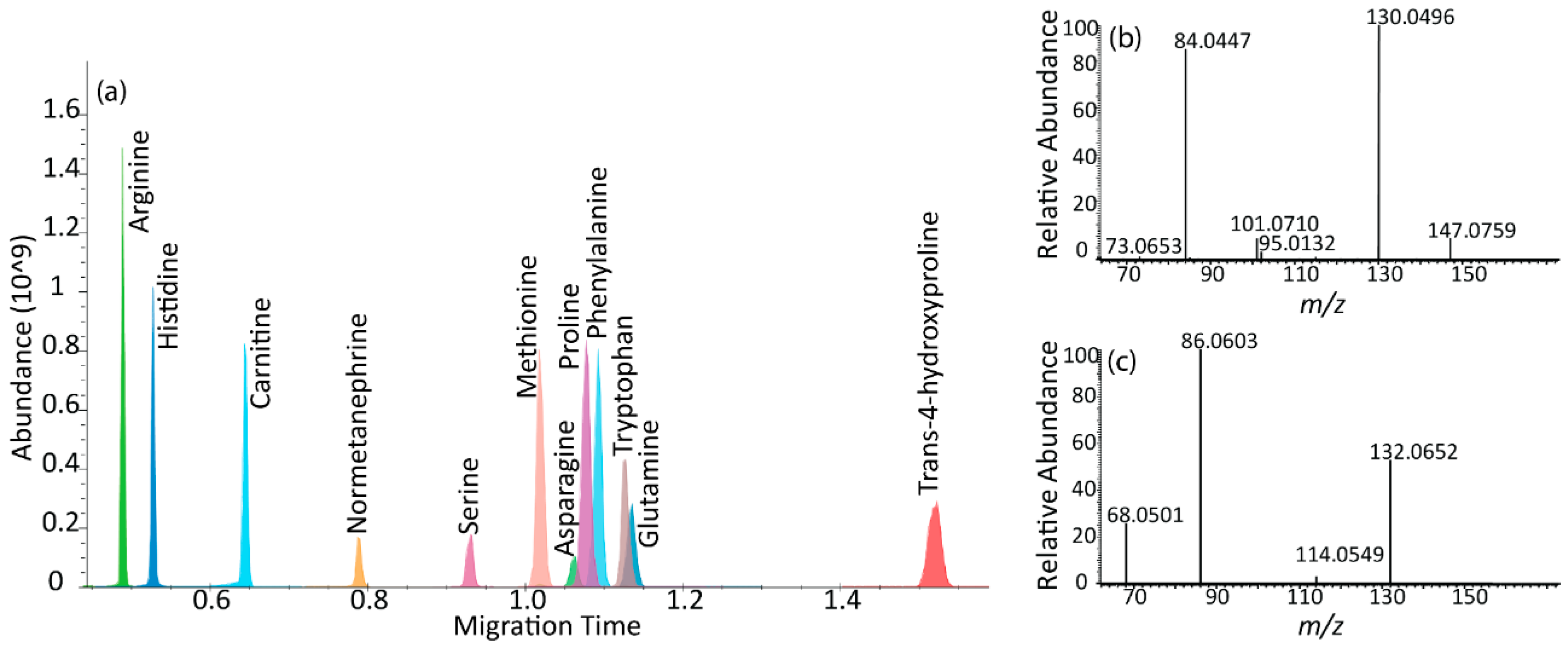
2.1. µCE-HRMS Method in Full Scan Mode
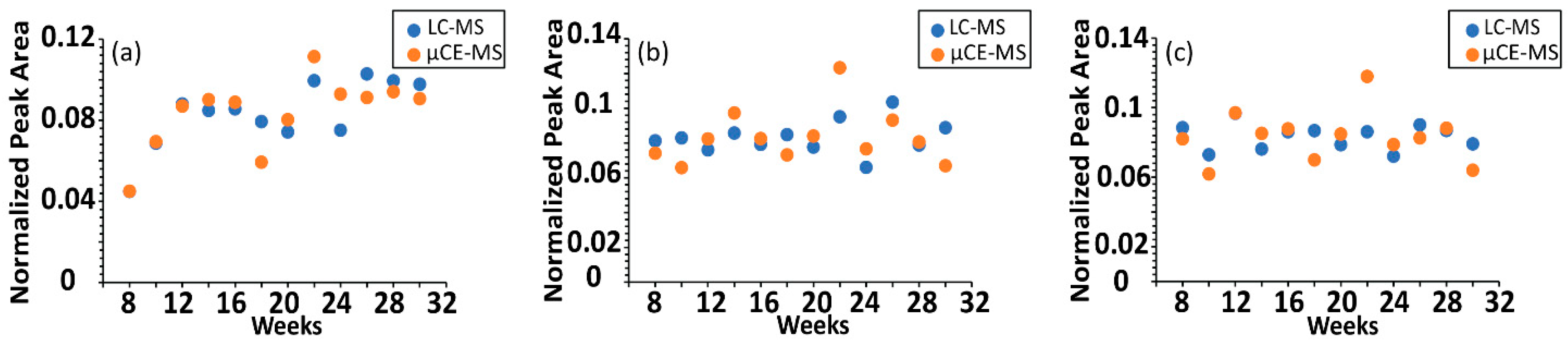
2.2. µCE-HRMS Method Validation in TKO Mouse Serum Samples
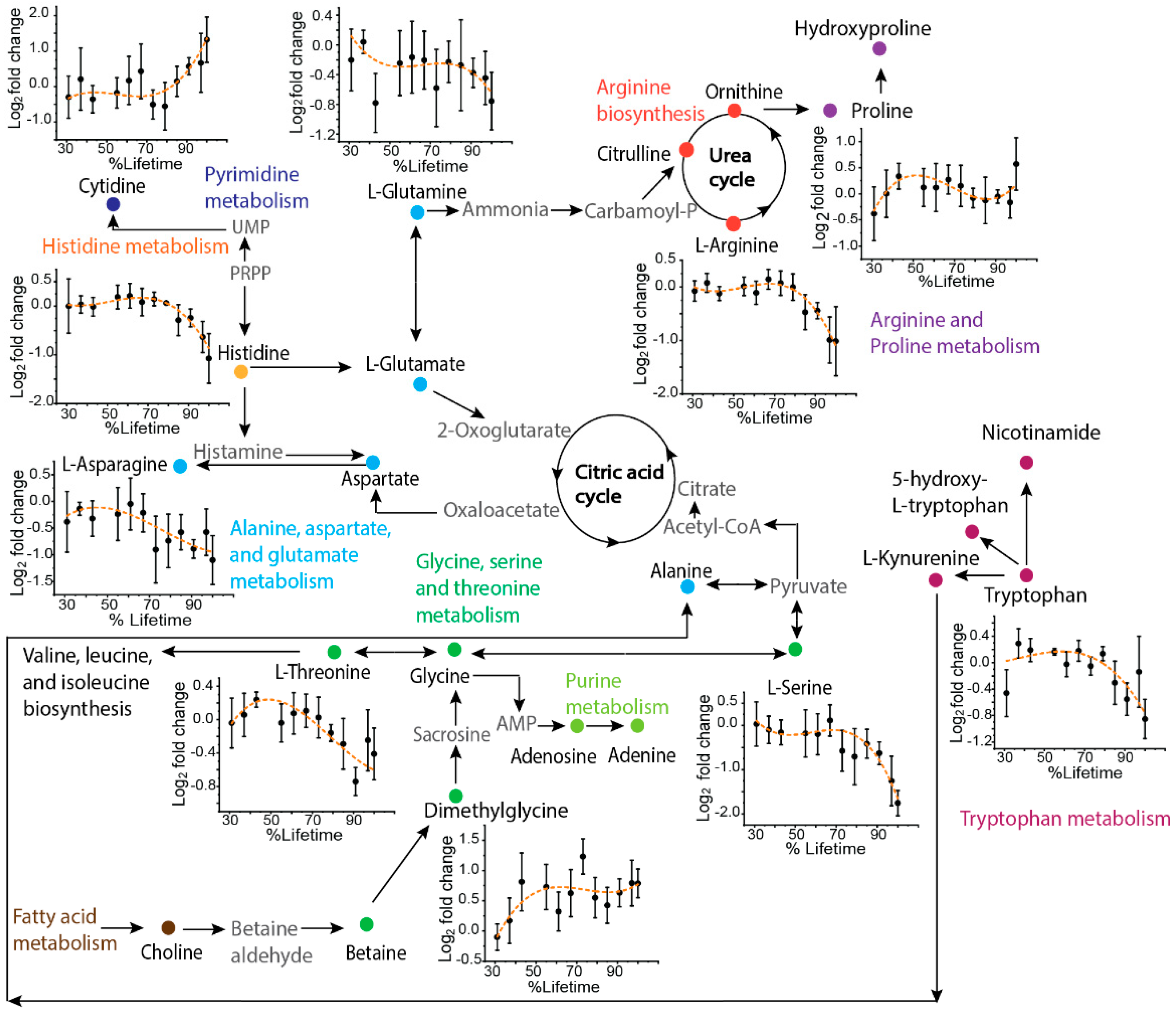
2.3. Application to Sequentially Collected TKO Mice Serum Samples
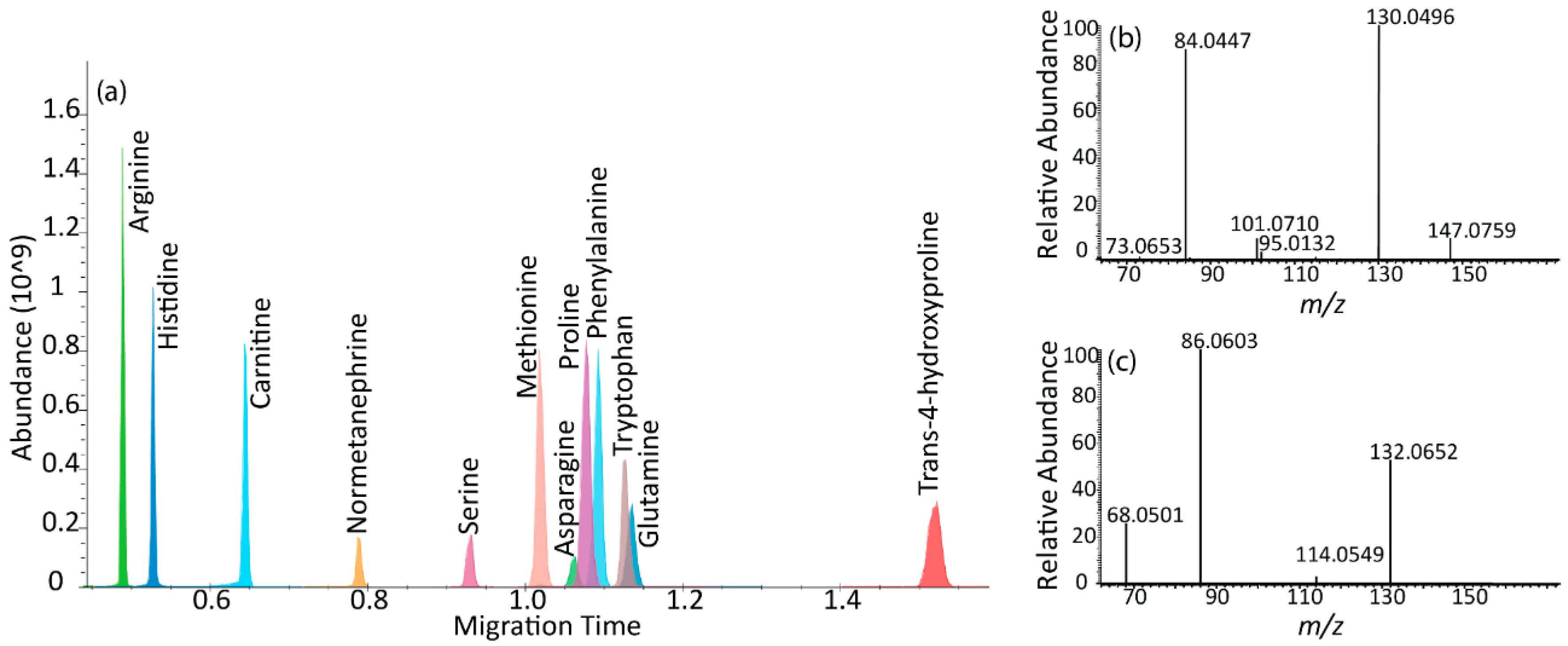
2.4. µCE-HRMS Coupled with Parallel Reaction Monitoring MS
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Triple Mutant (TKO) Mouse and Serum Sampling
4.3. Sample Preparation
4.3.1. Calibration Samples
4.3.2. Serum Sample Preparation
4.4. µCE-HRMS Analysis
4.5. Method Validation
4.6. Time-Resolved TKO Serum Metabolomics Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.H.; Bast, R.C., Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef] [Green Version]
- Nossov, V.; Amneus, M.; Su, F.; Lang, J.; Janco, J.M.T.; Reddy, S.T.; Farias-Eisner, R. The early detection of ovarian cancer: From traditional methods to proteomics. Can we really do better than serum CA-125? Am. J. Obstet. Gynecol. 2008, 199, 215–223. [Google Scholar] [CrossRef]
- Johnson, C.C.; Kessel, B.; Riley, T.L.; Ragard, L.R.; Williams, C.R.; Xu, J.L.; Buys, S.S. The epidemiology of CA-125 in women without evidence of ovarian cancer in the Prostate, Lung, Colorectal and Ovarian Cancer (PLCO) Screening Trial. Gynecol. Oncol. 2008, 110, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef]
- Mellors, J.S.; Gorbounov, V.; Ramsey, R.S.; Ramsey, J.M. Fully Integrated Glass Microfluidic Device for Performing High-Efficiency Capillary Electrophoresis and Electrospray Ionization Mass Spectrometry. Anal. Chem. 2008, 80, 6881–6887. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Ramautar, R. CE-MS for metabolomics: Developments and applications in the period 2018–2020. Electrophoresis 2021, 42, 381–401. [Google Scholar] [CrossRef]
- Soga, T.; Ohashi, Y.; Ueno, Y.; Naraoka, H.; Tomita, M.; Nishioka, T. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteome Res. 2003, 2, 488–494. [Google Scholar] [CrossRef]
- Mozafari, M.; Nachbar, M.; El Deeb, S. Precise small volume sample handling for capillary electrophoresis. Electrophoresis 2015, 36, 2665–2669. [Google Scholar] [CrossRef]
- Zhang, W.; Hankemeier, T.; Ramautar, R. Next-generation capillary electrophoresis–mass spectrometry approaches in metabolomics. Curr. Opin. Biotechnol. 2017, 43, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Harada, S.; Hirayama, A.; Chan, Q.; Kurihara, A.; Fukai, K.; Iida, M.; Kato, S.; Sugiyama, D.; Kuwabara, K.; Takeuchi, A.; et al. Reliability of plasma polar metabolite concentrations in a large-scale cohort study using capillary electrophoresis-mass spectrometry. PLoS ONE 2018, 13, e0191230. [Google Scholar] [CrossRef]
- Boizard, F.; Brunchault, V.; Moulos, P.; Breuil, B.; Klein, J.; Lounis, N.; Caubet, C.; Tellier, S.; Bascands, J.L.; Decramer, S.; et al. A capillary electrophoresis coupled to mass spectrometry pipeline for long term comparable assessment of the urinary metabolome. Sci. Rep. 2016, 6, 34453. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshitá, T.; SaiuM, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef] [Green Version]
- Austin Pickens, C.; Isenberg, S.L.; Cuthbert, C.; Petritis, K. Combining First and Second-Tier Newborn Screening in a Single Assay Using High-Throughput Chip-Based Capillary Electrophoresis Coupled to High-Resolution Mass Spectrometry. Clin. Chem. 2021, 67, 1709–1720. [Google Scholar] [CrossRef]
- Kim, J.; Coffey, D.M.; Ma, L.; Matzuk, M.M. The ovary is an alternative site of origin for high-grade serous ovarian cancer in mice. Endocrinology 2015, 156, 1975–1981. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Djukovic, D.; Deng, L.; Gu, H.; Himmati, F.; Chiorean, E.G.; Raftery, D. Colorectal cancer detection using targeted serum metabolic profiling. J. Proteome Res. 2014, 13, 4120–4130. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, S.; Wang, P.G. Matrix effects and application of matrix effect factor. Bioanalysis 2017, 9, 1839–1844. [Google Scholar] [CrossRef] [Green Version]
- Plewa, S.; Horała, A.; Dereziński, P.; Klupczynska, A.; Nowak-Markwitz, E.; Matysiak, J.; Kokot, Z.J. Usefulness of Amino Acid Profiling in Ovarian Cancer Screening with Special Emphasis on Their Role in Cancerogenesis. Int. J. Mol. Sci. 2017, 18, 2727. [Google Scholar] [CrossRef] [Green Version]
- Garg, G.; Yilmaz, A.; Kumar, P.; Turkoglu, O.; Mutch, D.G.; Powell, M.A.; Rosen, B.; Bahado-Singh, R.O.; Graham, S.F. Targeted metabolomic profiling of low and high grade serous epithelial ovarian cancer tissues: A pilot study. Metabolomics 2018, 14, 154. [Google Scholar] [CrossRef]
- Ahmed-Salim, Y.; Galazis, N.; Bracewell-Milnes, T.; Phelps, D.L.; Jones, B.P.; Chan, M.; Munoz-Gonzales, M.D.; Matsuzono, T.; Smith, J.R.; Yazbek, J. The application of metabolomics in ovarian cancer management: A systematic review. Int. J. Gynecol. Cancer 2021, 31, 754–774. [Google Scholar] [CrossRef] [PubMed]
- Frolkis, A.; Knox, C.; Lim, E.; Jewison, T.; Law, V.; Hau, D.D.; Liu, P.; Gautam, B.; Ly, S.; Guo, A.C.; et al. SMPDB: The Small Molecule Pathway Database. Nucleic Acids Res. 2009, 38, D480–D487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horala, A.; Plewa, S.; Derezinski, P.; Klupczynska, A.; Matysiak, J.; Nowak-Markwitz, E.; Kokot, Z.J. Serum Free Amino Acid Profiling in Differential Diagnosis of Ovarian Tumors—A Comparative Study with Review of the Literature. Int. J. Environ. Res. Public Health 2021, 18, 2167. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, Z.; Zhang, M.; Hu, H.; Li, J.; Zhang, D.; Guo, K.; Sha, H. Targeting the L-arginine-nitric oxide pathway for cancer treatment. Curr. Pharm. Des. 2010, 16, 392–410. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Sah, S.; Ma, X.; Botros, A.; Gaul, D.A.; Yun, S.R.; Park, E.Y.; Kim, O.; Moore, S.G.; Kim, J.; Fernández, F.M. Space- and Time-Resolved Metabolomics of a High-Grade Serous Ovarian Cancer Mouse Model. Cancers 2022, 14, 2262. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, H.; Liu, Y.; Liu, J.; Zhao, X.; Yin, Y. Development and Evaluation of a Parallel Reaction Monitoring Strategy for Large-Scale Targeted Metabolomics Quantification. Anal. Chem. 2016, 88, 4478–4486. [Google Scholar] [CrossRef]
- Reubsaet, L.; Sweredoski, M.J.; Moradian, A. Data-Independent Acquisition for the Orbitrap Q Exactive HF: A Tutorial. J. Proteome Res. 2019, 18, 803–813. [Google Scholar] [CrossRef]
- Bahga, S.S.; Bercovici, M.; Santiago, J.G. Ionic strength effects on electrophoretic focusing and separations. Electrophoresis 2010, 31, 910–919. [Google Scholar] [CrossRef]
- Adams, K.J.; Pratt, B.; Bose, N.; Dubois, L.G.; St John-Williams, L.; Perrott, K.M.; Ky, K.; Kapahi, P.; Sharma, V.; MacCoss, M.J.; et al. Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J. Proteome Res. 2020, 19, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
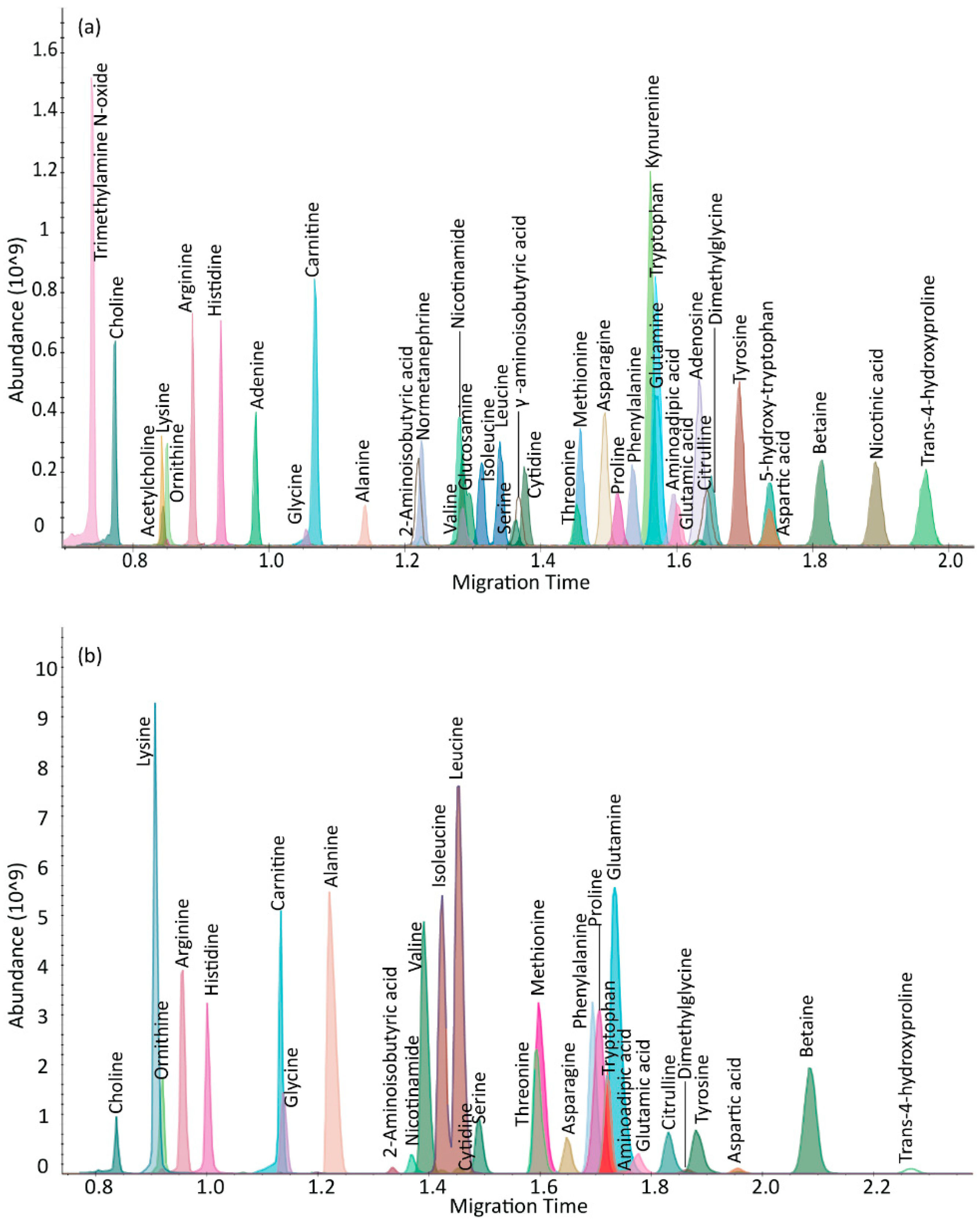


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite Name | Migration Time (min) | Migration Time % RSD (n = 18) | Detection Limit (nM) | Absolute limit of Detection (moles) | Limit of Quantification (nM) | R2 | Tested Concentration Range (µM) | Metabolite Classification |
|---|---|---|---|---|---|---|---|---|
| 5′-Hydroxy-L-tryptophan | 1.8 | 1.7 | 58.4 | 2.3 × 10−16 | 194.8 | 0.993 | 0.25–5 | Amino acid |
| Acetylcholine | 0.9 | 2.8 | 17.8 | 7.1 × 10−17 | 59.5 | 0.990 | 0.066–2 | Neurotransmitters |
| Alanine | 1.2 | 2.0 | 19.1 | 7.6 × 10−17 | 63.6 | 0.997 | 0.05–25 | Amino Acids |
| 2-Aminoisobutyric acid | 1.3 | 2.0 | 64.8 | 2.6 × 10−16 | 216.0 | 0.994 | 0.25–5 | Amino Acids |
| Arginine | 0.9 | 2.5 | 51.4 | 2.1 × 10−16 | 171.4 | 0.999 | 0.1–25 | Amino Acids |
| Asparagine | 1.5 | 1.8 | 34.8 | 1.4 × 10−16 | 116.1 | 0.982 | 0.1–25 | Amino Acids |
| Betaine | 1.9 | 1.6 | 11.5 | 4.6 × 10−17 | 38.2 | 0.999 | 0.033–10 | Amino Acids |
| Carnitine | 1.1 | 2.2 | 2.5 | 1.0 × 10−17 | 8.5 | 0.989 | 0.02–3 | Alkylamines |
| Choline | 0.8 | 3.0 | 6.8 | 2.7 × 10−17 | 22.5 | 0.999 | 0.05–25 | Vitamins |
| n,n-Dimethylglycine | 1.7 | 1.7 | 51.0 | 2.0 × 10−16 | 170.0 | 0.998 | 0.1–2 | Vitamins |
| Glucosamine | 1.3 | 2.1 | 29.6 | 1.2 × 10−16 | 98.7 | 0.994 | 0.16–5 | Amino Sugar |
| Glutamine | 1.6 | 1.8 | 69.3 | 2.8 × 10−16 | 231.1 | 0.997 | 0.25–25 | Amino Acids |
| Glycine | 1.1 | 2.0 | 27.0 | 1.1 × 10−16 | 89.9 | 0.998 | 0.25–50 | Amino Acids |
| Histidine | 1.0 | 2.4 | 19.5 | 7.8 × 10−17 | 65.1 | 0.996 | 0.1–25 | Amino Acids |
| Trans-4-hydroxy-L-proline | 2.0 | 1.3 | 55.5 | 2.2 × 10−16 | 185.0 | 0.991 | 0.1–2 | Amino Acids |
| Methionin × 10 | 1.5 | 1.9 | 23.3 | 9.3 × 10−17 | 77.8 | 0.999 | 0.1–25 | Amino Acids |
| Normetanephrine | 1.3 | 2.2 | 16.8 | 6.7 × 10−17 | 55.9 | 0.989 | 0.05–1 | Neurotransmitters |
| Phenylalanine | 1.6 | 1.9 | 17.4 | 7.0 × 10−17 | 58.1 | 0.997 | 0.25–25 | Amino Acids |
| Proline | 1.6 | 1.8 | 24.4 | 9.8 × 10−17 | 81.4 | 0.996 | 0.25–25 | Amino Acids |
| Serine | 1.4 | 1.9 | 31.0 | 1.2 × 10−16 | 103.3 | 0.991 | 0.1–25 | Amino Acids |
| Threonine | 1.5 | 1.8 | 45.0 | 1.8 × 10−16 | 150.1 | 0.999 | 0.1–25 | Amino Acids |
| Trimethylamine-n-oxide | 0.8 | 3.4 | 83.5 | 3.3 × 10−16 | 278.2 | 0.989 | 0.25–10 | Organic Oxoazanium Compounds |
| Tryptophan | 1.6 | 1.9 | 40.6 | 1.6 × 10−16 | 135.3 | 0.993 | 0.1–25 | Amino Acids |
| Tyrosine | 1.7 | 1.8 | 80.0 | 3.2 × 10−16 | 266.8 | 0.988 | 0.25–25 | Amino Acids |
| Valine | 1.3 | 2.0 | 7.5 | 3.0 × 10−17 | 24.9 | 0.997 | 0.05–25 | Amino Acids |
| Nicotinamide | 1.3 | 1.7 | 17.7 | 7.1 × 10−17 | 59.1 | 0.994 | 0.1–25 | Vitamins |
| Aspartic acid | 1.8 | 1.7 | 21.0 | 8.4 × 10−17 | 70.0 | 0.992 | 0.1–10 | Amino Acids |
| Nicotinic acid | 1.9 | 1.5 | 22.3 | 8.9 × 10−17 | 74.2 | 0.996 | 0.05–1 | Vitamins |
| γ-Aminobutyric acid | 1.0 | 2.3 | 65.2 | 2.6 × 10−16 | 217.3 | 0.994 | 0.25–5 | Amino Acids/Neurotransmitters |
| Aminoadipic acid | 1.6 | 1.8 | 30.3 | 1.2 × 10−16 | 101.0 | 0.998 | 0.1–2 | Amino acids |
| Cytidine | 1.4 | 2.0 | 13.2 | 5.3 × 10−17 | 44.0 | 0.998 | 0.05–25 | Pyrimidines |
| Citrulline | 1.7 | 1.8 | 29.3 | 1.2 × 10−16 | 97.8 | 0.994 | 0.1–25 | Amino Acids |
| Kynurenine | 1.6 | 1.8 | 154.8 | 6.2 × 10−16 | 516.0 | 0.998 | 0.5–5 | Amino acid |
| Isoleucine | 1.3 | 2.0 | 14.3 | 5.7 × 10−17 | 47.7 | 0.997 | 0.05–25 | Amino Acids |
| Leucine | 1.4 | 2.0 | 8.7 | 3.5 × 10−17 | 29.0 | 0.997 | 0.05–25 | Amino Acids |
| Ornithine | 0.9 | 2.6 | 17.9 | 7.2 × 10−17 | 59.5 | 0.998 | 0.05–25 | Amino Acid |
| Lysine | 0.9 | 2.0 | 10.6 | 4.2 × 10−17 | 35.5 | 0.994 | 0.25–25 | Amino Acids |
| Glutamic acid | 1.6 | 1.8 | 39.9 | 1.6 × 10−16 | 132.8 | 0.998 | 0.1–10 | Amino Acids |
| Adenosine | 1.7 | 1.6 | 96.3 | 3.9 × 10−16 | 321.0 | 0.984 | 0.25–5 | Purines |
| Adenine | 1.0 | 2.2 | 9.7 | 3.9 × 10−17 | 32.3 | 0.991 | 0.03–0.6 | Purines |
| Metabolite Name | Experimental m/z | Mass Error (ppm) | Migration Time (min) | %RSD Migration Time (n = 23) | Percent Recovery | %RSD for Peak Area without Internal Standard Correction (n = 23) | %RSD for Peak Area Corrected with Internal Standard (n = 23) |
|---|---|---|---|---|---|---|---|
| Alanine | 90.0553 | 2.22 | 1.2 | 0.94 | 98 | 17.24 | 6.07 |
| 2-Aminoisobutyric acid | 104.0709 | 2.88 | 1.3 | 0.84 | 108 | 21.92 | 5.02 |
| Arginine | 175.1190 | 0.57 | 1.0 | 0.13 | 109 | 20.86 | 2.85 |
| Asparagine | 133.0608 | 0.00 | 1.6 | 2.35 | 98 | 25.92 | 13.37 |
| Betaine | 118.0864 | 0.85 | 1.9 | 0.57 | 90 | 17.14 | 6.88 |
| Carnitine | 162.1123 | −0.62 | 1.2 | 1.32 | 96 | 15.12 | 11.59 |
| Choline | 104.1073 | 2.88 | 0.9 | 1.23 | 78 | 27.72 | 19.39 |
| Dimethylglycine | 104.0709 | 2.88 | 1.8 | 0.52 | 100 | 18.22 | 4.83 |
| Glutamine | 147.0762 | −1.36 | 1.7 | 2.53 | 93 | 20.47 | 10.95 |
| Glycine | 76.0397 | 5.26 | 1.2 | 1.66 | 120 | 20.38 | 8.15 |
| Histidine | 156.0767 | −0.64 | 1.1 | 0.38 | 119 | 22.35 | 9.59 |
| Trans-4-hydroxyproline | 132.0655 | 0.00 | 2.0 | 0.62 | 102 | 21.22 | 8.47 |
| Methionine | 150.0582 | −0.67 | 1.5 | 0.10 | 116 | 20.61 | 1.11 |
| Phenylalanine | 166.0862 | −0.60 | 1.6 | 0.10 | 107 | 20.65 | 1.45 |
| Proline | 116.0707 | 0.86 | 1.6 | 0.54 | 116 | 17.94 | 4.55 |
| Serine | 106.0501 | 1.89 | 1.5 | 2.06 | 120 | 27.30 | 13.09 |
| Threonine | 120.0657 | 1.67 | 1.5 | 2.32 | 117 | 23.20 | 8.34 |
| Tryptophan | 205.0971 | −0.49 | 1.6 | 0.31 | 94 | 24.01 | 7.27 |
| Tyrosine | 182.0812 | 0.00 | 1.8 | 0.49 | 110 | 25.05 | 10.09 |
| Valine | 118.0864 | 1.69 | 1.4 | 0.44 | 102 | 20.60 | 4.66 |
| Nicotinamide | 123.0556 | 2.44 | 1.5 | 0.70 | 105 | 21.28 | 4.48 |
| Aspartic acid | 134.0448 | 0.00 | 1.8 | 2.66 | 118 | 26.43 | 12.96 |
| Aminoadipic acid | 162.0759 | −1.23 | 1.7 | 0.28 | 97 | 21.31 | 8.25 |
| Cytidine | 244.0927 | −0.41 | 1.5 | 2.57 | 107 | 27.08 | 14.47 |
| Ornithine | 133.0971 | −0.75 | 1.0 | 0.35 | 78 | 24.58 | 12.45 |
| Citrulline | 176.1030 | 0.57 | 1.7 | 0.42 | 93 | 24.19 | 11.13 |
| Isoleucine | 132.1018 | −0.76 | 1.4 | 0.40 | 112 | 23.18 | 6.99 |
| Leucine | 132.1018 | −0.76 | 1.4 | 0.52 | 106 | 22.26 | 6.21 |
| Lysine | 147.1126 | −1.36 | 1.0 | 0.31 | 119 | 22.24 | 13.74 |
| Glutamic acid | 148.0603 | −0.68 | 1.7 | 0.26 | 115 | 23.87 | 11.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sah, S.; Yun, S.R.; Gaul, D.A.; Botros, A.; Park, E.Y.; Kim, O.; Kim, J.; Fernández, F.M. Targeted Microchip Capillary Electrophoresis-Orbitrap Mass Spectrometry Metabolomics to Monitor Ovarian Cancer Progression. Metabolites 2022, 12, 532. https://doi.org/10.3390/metabo12060532
Sah S, Yun SR, Gaul DA, Botros A, Park EY, Kim O, Kim J, Fernández FM. Targeted Microchip Capillary Electrophoresis-Orbitrap Mass Spectrometry Metabolomics to Monitor Ovarian Cancer Progression. Metabolites. 2022; 12(6):532. https://doi.org/10.3390/metabo12060532
Chicago/Turabian StyleSah, Samyukta, Sylvia R. Yun, David A. Gaul, Andro Botros, Eun Young Park, Olga Kim, Jaeyeon Kim, and Facundo M. Fernández. 2022. "Targeted Microchip Capillary Electrophoresis-Orbitrap Mass Spectrometry Metabolomics to Monitor Ovarian Cancer Progression" Metabolites 12, no. 6: 532. https://doi.org/10.3390/metabo12060532
APA StyleSah, S., Yun, S. R., Gaul, D. A., Botros, A., Park, E. Y., Kim, O., Kim, J., & Fernández, F. M. (2022). Targeted Microchip Capillary Electrophoresis-Orbitrap Mass Spectrometry Metabolomics to Monitor Ovarian Cancer Progression. Metabolites, 12(6), 532. https://doi.org/10.3390/metabo12060532