Guide to Metabolomics Analysis: A Bioinformatics Workflow


Abstract
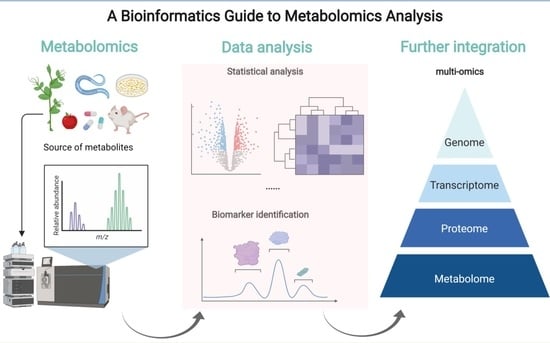
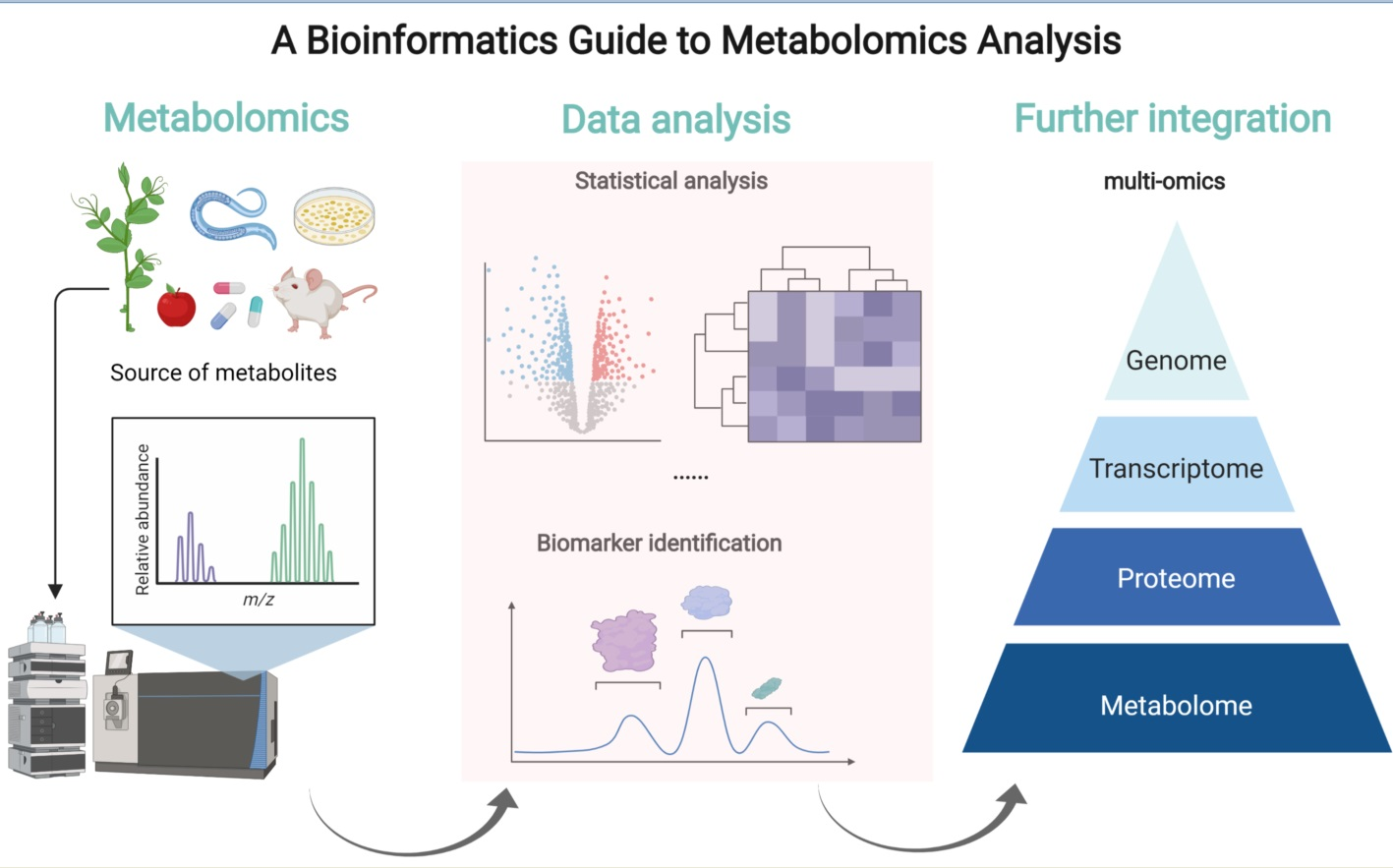
1. Introduction
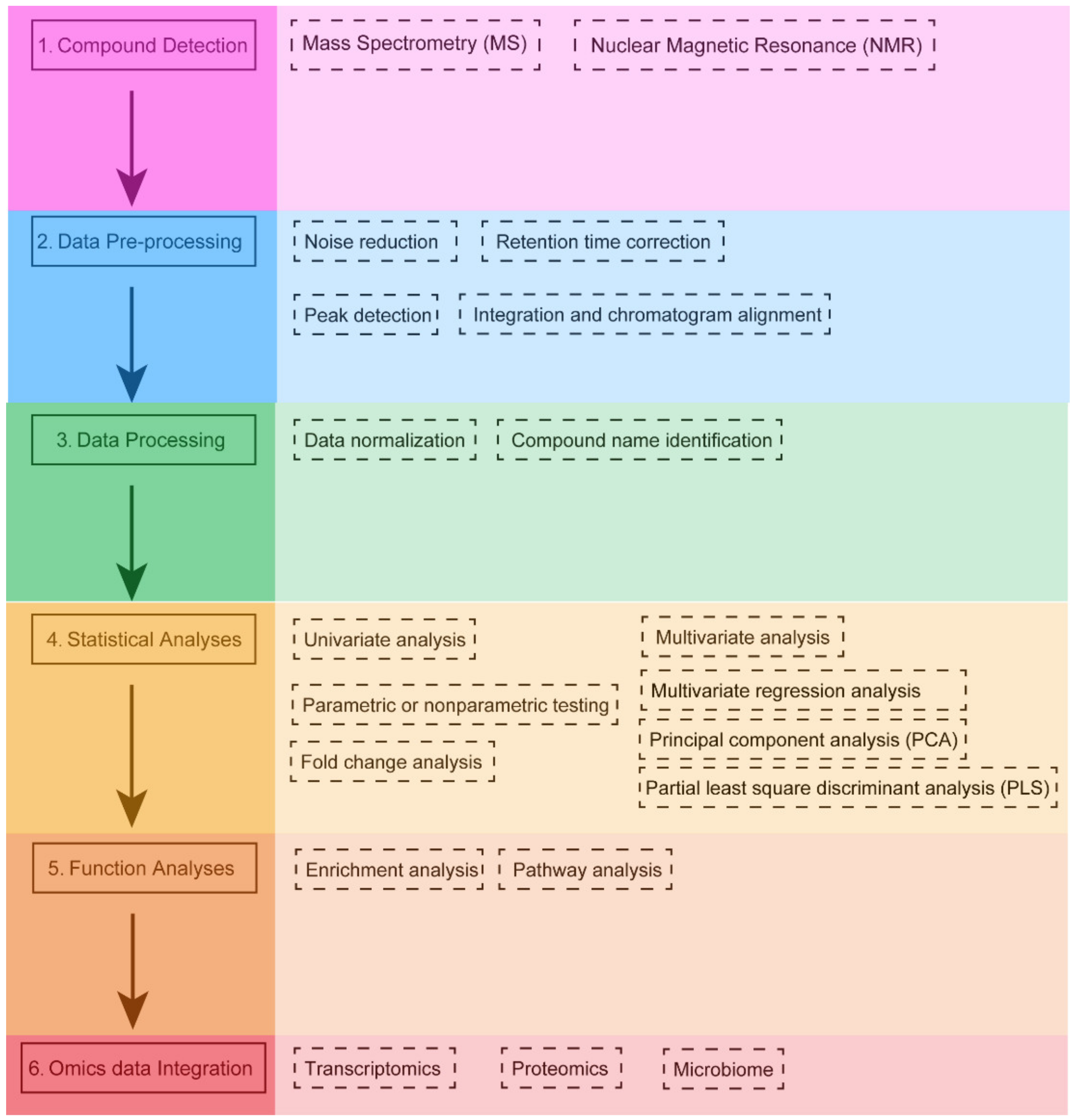
2. The Analysis Workflow of Metabolomics
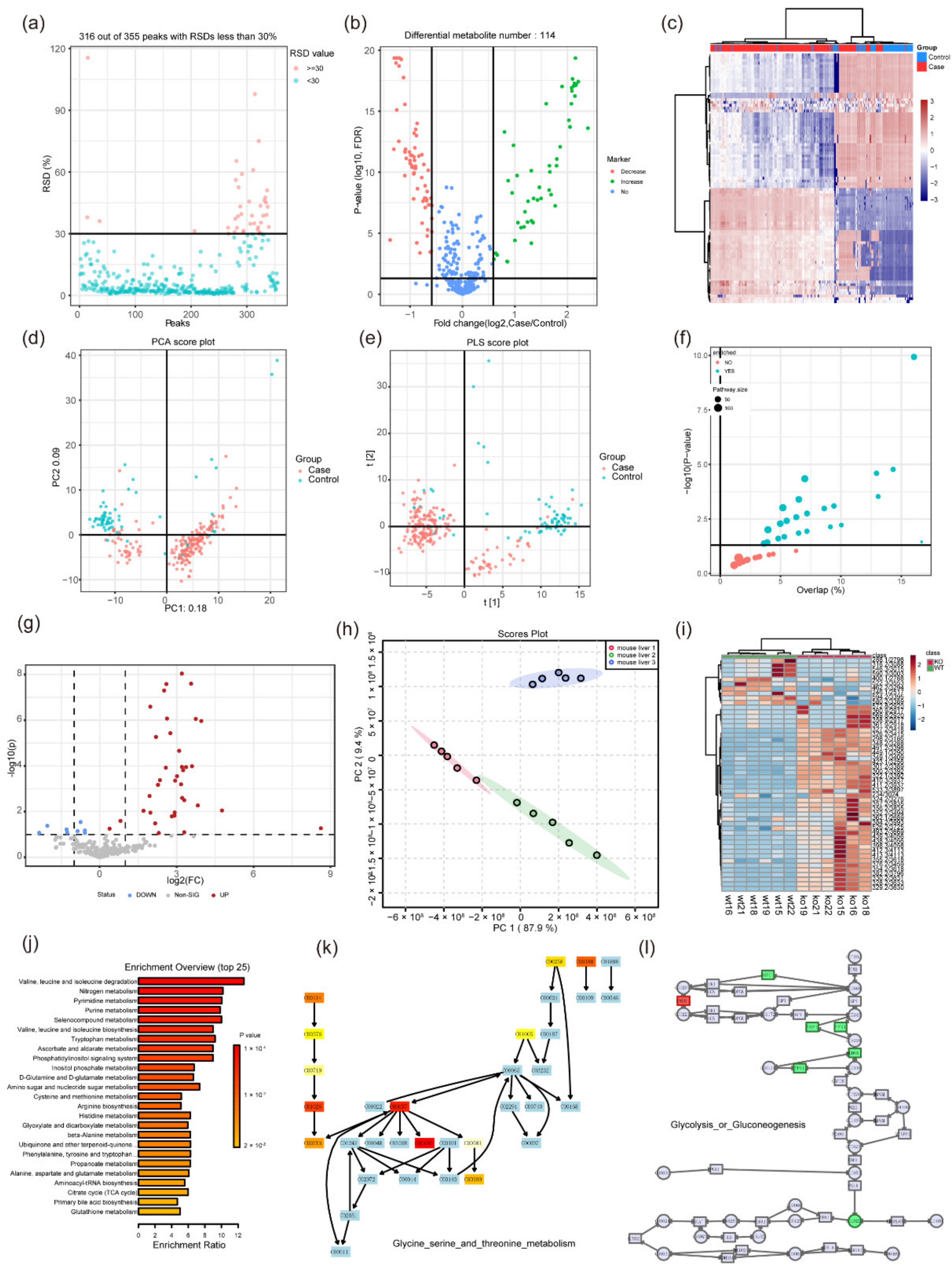
3. Statistical Analysis in Metabolomics
3.1. Univariate Analysis
3.2. Multivariate Analysis
4. Software Tools for Metabolomics Data Analysis and Integration
4.1. MS-DIAL
4.2. MZmine 3
4.3. El-MAVEN
4.4. LipidMatch
4.5. LipiDex
4.6. MetFlow
4.7. MetaboAnalyst 5.0
4.8. LipidSig
4.9. LION
4.10. METLIN
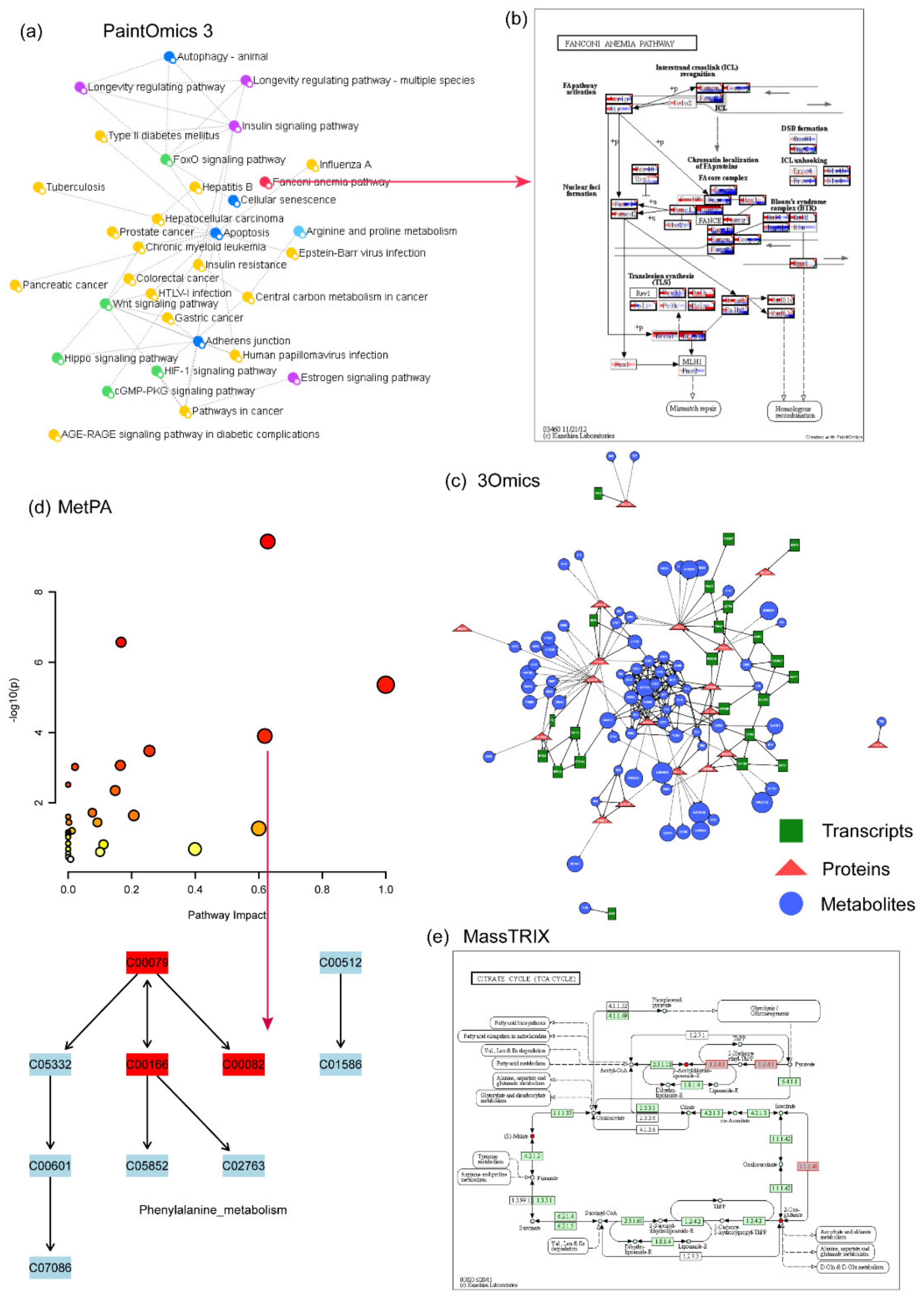
4.11. PaintOmics 3
4.12. 3Omics
4.13. IMPaLa
4.14. MetPA
4.15. MassTRIX
4.16. MetaCore™
4.17. OmicsNet
5. The Integration Algorithm of Multi-Omics Data
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Wang, X. Serum metabolomics as a novel diagnostic approach for disease: A systematic review. Anal. Bioanal. Chem. 2012, 404, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Gowda, G.A.; Zhang, S.; Gu, H.; Asiago, V.; Shanaiah, N.; Raftery, D. Metabolomics-based methods for early disease diagnostics. Expert Rev. Mol. Diagn. 2008, 8, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Turi, K.N.; Romick-Rosendale, L.; Ryckman, K.K.; Hartert, T.V. A review of metabolomics approaches and their application in identifying causal pathways of childhood asthma. J. Allergy Clin. Immunol. 2018, 141, 1191–1201. [Google Scholar] [CrossRef]
- Idle, J.R.; Gonzalez, F.J. Metabolomics. Cell Metab. 2007, 6, 348–351. [Google Scholar] [CrossRef]
- Pacchiarotta, T.; Deelder, A.M.; Mayboroda, O.A. Metabolomic investigations of human infections. Bioanalysis 2012, 4, 919–925. [Google Scholar] [CrossRef]
- Scrivo, R.; Casadei, L.; Valerio, M.; Priori, R.; Valesini, G.; Manetti, C. Metabolomics approach in allergic and rheumatic diseases. Curr. Allergy Asthma Rep. 2014, 14, 445. [Google Scholar] [CrossRef]
- Johnson, C.H.; Patterson, A.D.; Idle, J.R.; Gonzalez, F.J. Xenobiotic metabolomics: Major impact on the metabolome. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 37–56. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid. Res. 2005, 46, 839–861. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Kathiresan, S. Surprises From Genetic Analyses of Lipid Risk Factors for Atherosclerosis. Circ. Res. 2016, 118, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Kathiresan, S. Genetics of Common, Complex Coronary Artery Disease. Cell 2019, 177, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Gross, R.W. Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry: A bridge to lipidomics. J. Lipid. Res. 2003, 44, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Wenk, M.R. The emerging field of lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Zullig, T.; Kofeler, H.C. High Resolution Mass Spectrometry in Lipidomics. Mass Spectrom. Rev. 2021, 40, 162–176. [Google Scholar] [CrossRef]
- Pasikanti, K.K.; Esuvaranathan, K.; Hong, Y.; Ho, P.C.; Mahendran, R.; Raman Nee Mani, L.; Chiong, E.; Chan, E.C. Urinary metabotyping of bladder cancer using two-dimensional gas chromatography time-of-flight mass spectrometry. J. Proteome Res. 2013, 12, 3865–3873. [Google Scholar] [CrossRef]
- Pasikanti, K.K.; Esuvaranathan, K.; Ho, P.C.; Mahendran, R.; Kamaraj, R.; Wu, Q.H.; Chiong, E.; Chan, E.C. Noninvasive urinary metabonomic diagnosis of human bladder cancer. J. Proteome Res. 2010, 9, 2988–2995. [Google Scholar] [CrossRef]
- Huang, Z.; Lin, L.; Gao, Y.; Chen, Y.; Yan, X.; Xing, J.; Hang, W. Bladder cancer determination via two urinary metabolites: A biomarker pattern approach. Mol. Cell. Proteom. MCP 2011, 10, M111.007922. [Google Scholar] [CrossRef]
- Wittmann, B.M.; Stirdivant, S.M.; Mitchell, M.W.; Wulff, J.E.; McDunn, J.E.; Li, Z.; Dennis-Barrie, A.; Neri, B.P.; Milburn, M.V.; Lotan, Y.; et al. Bladder cancer biomarker discovery using global metabolomic profiling of urine. PLoS ONE 2014, 9, e115870. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Roy, R.; Singh, S.; Kumar, P.; Dalela, D.; Sankhwar, S.N.; Goel, A.; Sonkar, A.A. Taurine—A possible fingerprint biomarker in non-muscle invasive bladder cancer: A pilot study by 1H NMR spectroscopy. Cancer Biomark. Sect. A Dis. Markers 2010, 6, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, X.; Liu, X.; Guo, Z.; Sun, H.; Zhang, M.; Ji, Z.; Sun, W. Metabolomics of Non-muscle Invasive Bladder Cancer: Biomarkers for Early Detection of Bladder Cancer. Front. Oncol. 2018, 8, 494. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Xie, G.; Chen, T.; Qiu, Y.; Zou, X.; Zheng, M.; Tan, B.; Feng, B.; Dong, T.; He, P.; et al. Distinct urinary metabolic profile of human colorectal cancer. J. Proteome Res. 2012, 11, 1354–1363. [Google Scholar] [CrossRef]
- Chen, T.; Xie, G.; Wang, X.; Fan, J.; Qiu, Y.; Zheng, X.; Qi, X.; Cao, Y.; Su, M.; Wang, X.; et al. Serum and urine metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Mol. Cell. Proteom. MCP 2011, 10, M110.004945. [Google Scholar] [CrossRef]
- Shariff, M.I.; Gomaa, A.I.; Cox, I.J.; Patel, M.; Williams, H.R.; Crossey, M.M.; Thillainayagam, A.V.; Thomas, H.C.; Waked, I.; Khan, S.A.; et al. Urinary metabolic biomarkers of hepatocellular carcinoma in an Egyptian population: A validation study. J. Proteome Res. 2011, 10, 1828–1836. [Google Scholar] [CrossRef]
- Ladep, N.G.; Dona, A.C.; Lewis, M.R.; Crossey, M.M.; Lemoine, M.; Okeke, E.; Shimakawa, Y.; Duguru, M.; Njai, H.F.; Fye, H.K.; et al. Discovery and validation of urinary metabotypes for the diagnosis of hepatocellular carcinoma in West Africans. Hepatology 2014, 60, 1291–1301. [Google Scholar] [CrossRef]
- Cox, I.J.; Aliev, A.E.; Crossey, M.M.; Dawood, M.; Al-Mahtab, M.; Akbar, S.M.; Rahman, S.; Riva, A.; Williams, R.; Taylor-Robinson, S.D. Urinary nuclear magnetic resonance spectroscopy of a Bangladeshi cohort with hepatitis-B hepatocellular carcinoma: A biomarker corroboration study. World J. Gastroenterol. 2016, 22, 4191–4200. [Google Scholar] [CrossRef]
- Chen, J.; Wang, W.; Lv, S.; Yin, P.; Zhao, X.; Lu, X.; Zhang, F.; Xu, G. Metabonomics study of liver cancer based on ultra performance liquid chromatography coupled to mass spectrometry with HILIC and RPLC separations. Anal. Chim. Acta 2009, 650, 3–9. [Google Scholar] [CrossRef]
- Shariff, M.I.; Ladep, N.G.; Cox, I.J.; Williams, H.R.; Okeke, E.; Malu, A.; Thillainayagam, A.V.; Crossey, M.M.; Khan, S.A.; Thomas, H.C.; et al. Characterization of urinary biomarkers of hepatocellular carcinoma using magnetic resonance spectroscopy in a Nigerian population. J. Proteome Res. 2010, 9, 1096–1103. [Google Scholar] [CrossRef]
- Liang, Q.; Liu, H.; Wang, C.; Li, B. Phenotypic Characterization Analysis of Human Hepatocarcinoma by Urine Metabolomics Approach. Sci. Rep. 2016, 6, 19763. [Google Scholar] [CrossRef] [PubMed]
- Osman, D.; Ali, O.; Obada, M.; El-Mezayen, H.; El-Said, H. Chromatographic determination of some biomarkers of liver cirrhosis and hepatocellular carcinoma in Egyptian patients. Biomed. Chromatogr. BMC 2017, 31, e3893. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xue, R.; Dong, L.; Liu, T.; Deng, C.; Zeng, H.; Shen, X. Metabolomic profiling of human urine in hepatocellular carcinoma patients using gas chromatography/mass spectrometry. Anal. Chim. Acta 2009, 648, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Salek, R.M.; Maguire, M.L.; Bentley, E.; Rubtsov, D.V.; Hough, T.; Cheeseman, M.; Nunez, D.; Sweatman, B.C.; Haselden, J.N.; Cox, R.D.; et al. A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol. Genom. 2007, 29, 99–108. [Google Scholar] [CrossRef]
- Adams, S.H.; Hoppel, C.L.; Lok, K.H.; Zhao, L.; Wong, S.W.; Minkler, P.E.; Hwang, D.H.; Newman, J.W.; Garvey, W.T. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr. 2009, 139, 1073–1081. [Google Scholar] [CrossRef]
- Mihalik, S.J.; Goodpaster, B.H.; Kelley, D.E.; Chace, D.H.; Vockley, J.; Toledo, F.G.; DeLany, J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 2010, 18, 1695–1700. [Google Scholar] [CrossRef]
- Ha, C.Y.; Kim, J.Y.; Paik, J.K.; Kim, O.Y.; Paik, Y.H.; Lee, E.J.; Lee, J.H. The association of specific metabolites of lipid metabolism with markers of oxidative stress, inflammation and arterial stiffness in men with newly diagnosed type 2 diabetes. Clin. Endocrinol. 2012, 76, 674–682. [Google Scholar] [CrossRef]
- Li, X.; Xu, Z.; Lu, X.; Yang, X.; Yin, P.; Kong, H.; Yu, Y.; Xu, G. Comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry for metabonomics: Biomarker discovery for diabetes mellitus. Anal. Chim. Acta 2009, 633, 257–262. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Guan, C.; Li, K.; Wang, C.; Feng, R.; Sun, C. Free fatty acid metabolic profile and biomarkers of isolated post-challenge diabetes and type 2 diabetes mellitus based on GC-MS and multivariate statistical analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 2817–2825. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Cao, H.; King, I.B.; Lemaitre, R.N.; Song, X.; Siscovick, D.S.; Hotamisligil, G.S. Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. Am. J. Clin. Nutr. 2010, 92, 1350–1358. [Google Scholar] [CrossRef]
- Lee, Y.; Pamungkas, A.D.; Medriano, C.A.D.; Park, J.; Hong, S.; Jee, S.H.; Park, Y.H. High-resolution metabolomics determines the mode of onset of type 2 diabetes in a 3-year prospective cohort study. Int. J. Mol. Med. 2018, 41, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Messana, I.; Forni, F.; Ferrari, F.; Rossi, C.; Giardina, B.; Zuppi, C. Proton nuclear magnetic resonance spectral profiles of urine in type II diabetic patients. Clin. Chem. 1998, 44, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Meisinger, C.; Doring, A.; Altmaier, E.; Belcredi, P.; Gieger, C.; Chang, D.; Milburn, M.V.; Gall, W.E.; Weinberger, K.M.; et al. Metabolic footprint of diabetes: A multiplatform metabolomics study in an epidemiological setting. PLoS ONE 2010, 5, e13953. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhou, J.; Bao, Y.; Chen, T.; Zhang, Y.; Zhao, A.; Qiu, Y.; Xie, G.; Wang, C.; Jia, W.; et al. Serum metabolic signatures of fulminant type 1 diabetes. J. Proteome Res. 2012, 11, 4705–4711. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Natali, A.; Camastra, S.; Nannipieri, M.; Mari, A.; Adam, K.P.; Milburn, M.V.; Kastenmuller, G.; Adamski, J.; Tuomi, T.; et al. Early metabolic markers of the development of dysglycemia and type 2 diabetes and their physiological significance. Diabetes 2013, 62, 1730–1737. [Google Scholar] [CrossRef]
- Floegel, A.; Stefan, N.; Yu, Z.; Muhlenbruch, K.; Drogan, D.; Joost, H.G.; Fritsche, A.; Haring, H.U.; Hrabe de Angelis, M.; Peters, A.; et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 2013, 62, 639–648. [Google Scholar] [CrossRef]
- Fiehn, O.; Garvey, W.T.; Newman, J.W.; Lok, K.H.; Hoppel, C.L.; Adams, S.H. Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PLoS ONE 2010, 5, e15234. [Google Scholar] [CrossRef]
- Hodge, A.M.; English, D.R.; O’Dea, K.; Sinclair, A.J.; Makrides, M.; Gibson, R.A.; Giles, G.G. Plasma phospholipid and dietary fatty acids as predictors of type 2 diabetes: Interpreting the role of linoleic acid. Am. J. Clin. Nutr. 2007, 86, 189–197. [Google Scholar] [CrossRef]
- Chow, L.S.; Li, S.; Eberly, L.E.; Seaquist, E.R.; Eckfeldt, J.H.; Hoogeveen, R.C.; Couper, D.J.; Steffen, L.M.; Pankow, J.S. Estimated plasma stearoyl co-A desaturase-1 activity and risk of incident diabetes: The Atherosclerosis Risk in Communities (ARIC) study. Metab. Clin. Exp. 2013, 62, 100–108. [Google Scholar] [CrossRef]
- Nakashima, K. Glycolytic and gluconeogenic metabolites and enzymes in the liver of obese-hyperglycemic mice (KK) and alloxan diabetic mice. Nagoya J. Med. Sci. 1969, 32, 143–158. [Google Scholar]
- Wood, I.S.; Stezhka, T.; Trayhurn, P. Modulation of adipokine production, glucose uptake and lactate release in human adipocytes by small changes in oxygen tension. Pflug. Arch. Eur. J. Physiol. 2011, 462, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Bohm, A.; Halama, A.; Meile, T.; Zdichavsky, M.; Lehmann, R.; Weigert, C.; Fritsche, A.; Stefan, N.; Konigsrainer, A.; Haring, H.U.; et al. Metabolic signatures of cultured human adipocytes from metabolically healthy versus unhealthy obese individuals. PLoS ONE 2014, 9, e93148. [Google Scholar] [CrossRef] [PubMed]
- Lillefosse, H.H.; Clausen, M.R.; Yde, C.C.; Ditlev, D.B.; Zhang, X.; Du, Z.Y.; Bertram, H.C.; Madsen, L.; Kristiansen, K.; Liaset, B. Urinary loss of tricarboxylic acid cycle intermediates as revealed by metabolomics studies: An underlying mechanism to reduce lipid accretion by whey protein ingestion? J. Proteome Res. 2014, 13, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.E.; Larson, M.G.; Ghorbani, A.; Cheng, S.; Chen, M.H.; Keyes, M.; Rhee, E.P.; Clish, C.B.; Vasan, R.S.; Gerszten, R.E.; et al. Metabolomic Profiles of Body Mass Index in the Framingham Heart Study Reveal Distinct Cardiometabolic Phenotypes. PLoS ONE 2016, 11, e0148361. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Moon, J.S.; Kang, J.H.; Jang, H.B.; Lee, H.J.; Park, S.I.; Yu, K.S.; Cho, J.Y. Combined untargeted and targeted metabolomic profiling reveals urinary biomarkers for discriminating obese from normal-weight adolescents. Pediatric Obes. 2017, 12, 93–101. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef]
- Graham, S.F.; Chevallier, O.P.; Elliott, C.T.; Holscher, C.; Johnston, J.; McGuinness, B.; Kehoe, P.G.; Passmore, A.P.; Green, B.D. Untargeted metabolomic analysis of human plasma indicates differentially affected polyamine and L-arginine metabolism in mild cognitive impairment subjects converting to Alzheimer’s disease. PLoS ONE 2015, 10, e0119452. [Google Scholar] [CrossRef]
- Toledo, J.B.; Arnold, M.; Kastenmuller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2017, 13, 965–984. [Google Scholar] [CrossRef]
- Proitsi, P.; Kim, M.; Whiley, L.; Pritchard, M.; Leung, R.; Soininen, H.; Kloszewska, I.; Mecocci, P.; Tsolaki, M.; Vellas, B.; et al. Plasma lipidomics analysis finds long chain cholesteryl esters to be associated with Alzheimer’s disease. Transl. Psychiatry 2015, 5, e494. [Google Scholar] [CrossRef]
- Kim, M.; Nevado-Holgado, A.; Whiley, L.; Snowden, S.G.; Soininen, H.; Kloszewska, I.; Mecocci, P.; Tsolaki, M.; Vellas, B.; Thambisetty, M.; et al. Association between Plasma Ceramides and Phosphatidylcholines and Hippocampal Brain Volume in Late Onset Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 60, 809–817. [Google Scholar] [CrossRef]
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of blood lipids with Alzheimer’s disease: A comprehensive lipidomics analysis. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2017, 13, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.P.; Montoliu, I.; Da Silva, L.; Dayon, L.; Galindo, A.N.; Corthesy, J.; Kussmann, M.; Martin, F.P. High-throughput and simultaneous quantitative analysis of homocysteine-methionine cycle metabolites and co-factors in blood plasma and cerebrospinal fluid by isotope dilution LC-MS/MS. Anal. Bioanal. Chem. 2017, 409, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Stocchero, M.; Cacciatore, S.; Lai, S.; Angel, P.; Alam, M.T.; Keller, M.; Ralser, M.; Astarita, G. Unbiased Metabolomic Investigation of Alzheimer’s Disease Brain Points to Dysregulation of Mitochondrial Aspartate Metabolism. J. Proteome Res. 2016, 15, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Koal, T.; Klavins, K.; Seppi, D.; Kemmler, G.; Humpel, C. Sphingomyelin SM(d18:1/18:0) is significantly enhanced in cerebrospinal fluid samples dichotomized by pathological amyloid-beta42, tau, and phospho-tau-181 levels. J. Alzheimer’s Dis. JAD 2015, 44, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Mamas, M.; Dunn, W.B.; Neyses, L.; Goodacre, R. The role of metabolites and metabolomics in clinically applicable biomarkers of disease. Arch. Toxicol. 2011, 85, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Puchades-Carrasco, L.; Lecumberri, R.; Martinez-Lopez, J.; Lahuerta, J.J.; Mateos, M.V.; Prosper, F.; San-Miguel, J.F.; Pineda-Lucena, A. Multiple myeloma patients have a specific serum metabolomic profile that changes after achieving complete remission. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4770–4779. [Google Scholar] [CrossRef]
- O’Shea, K.; Misra, B.B. Software tools, databases and resources in metabolomics: Updates from 2018 to 2019. Metabolomics 2020, 16, 36. [Google Scholar] [CrossRef]
- Mattoli, L.; Gianni, M.; Burico, M. Mass Spectrometry Based Metabolomic Analysis as a Tool for Quality Control of Natural Complex Products. Mass Spectrom. Rev. 2022. [Google Scholar] [CrossRef]
- Beisken, S.; Eiden, M.; Salek, R.M. Getting the right answers: Understanding metabolomics challenges. Expert Rev. Mol. Diagn. 2015, 15, 97–109. [Google Scholar] [CrossRef]
- Burnap, R.L. Systems and photosystems: Cellular limits of autotrophic productivity in cyanobacteria. Front. Bioeng. Biotechnol. 2015, 3, 1. [Google Scholar] [CrossRef]
- Ma, S.; Huang, J. Regularized gene selection in cancer microarray meta-analysis. BMC Bioinform. 2009, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, G.; Gika, H.G.; Wilson, I.D. Mass spectrometry-based holistic analytical approaches for metabolite profiling in systems biology studies. Mass Spectrom. Rev. 2011, 30, 884–906. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.S.; Lam, C.W.; Chan, M.H.; Cheung, R.C.; Law, L.K.; Lit, L.C.; Ng, K.F.; Suen, M.W.; Tai, H.L. Electrospray ionisation mass spectrometry: Principles and clinical applications. Clin. Biochem. Rev. 2003, 24, 3–12. [Google Scholar] [PubMed]
- Pan, Z.; Raftery, D. Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal. Bioanal. Chem. 2007, 387, 525–527. [Google Scholar] [CrossRef]
- Veenstra, T.D. Metabolomics: The final frontier? Genome Med. 2012, 4, 40. [Google Scholar] [CrossRef]
- Ebbels, T.M.; Lindon, J.C.; Coen, M. Processing and modeling of nuclear magnetic resonance (NMR) metabolic profiles. Methods Mol. Biol. 2011, 708, 365–388. [Google Scholar] [CrossRef]
- DeFeo, E.M.; Wu, C.L.; McDougal, W.S.; Cheng, L.L. A decade in prostate cancer: From NMR to metabolomics. Nat. Rev. Urol. 2011, 8, 301–311. [Google Scholar] [CrossRef]
- Vignoli, A.; Ghini, V.; Meoni, G.; Licari, C.; Takis, P.G.; Tenori, L.; Turano, P.; Luchinat, C. High-Throughput Metabolomics by 1D NMR. Angew. Chem. Int. Ed. Engl. 2019, 58, 968–994. [Google Scholar] [CrossRef]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef]
- Lai, Z.; Fiehn, O. Mass spectral fragmentation of trimethylsilylated small molecules. Mass Spectrom. Rev. 2018, 37, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Xiang, L.; Cai, Z. Emerging environmental pollutants hydroxylated polybrominated diphenyl ethers: From analytical methods to toxicology research. Mass Spectrom. Rev. 2021, 40, 255–279. [Google Scholar] [CrossRef] [PubMed]
- Castle, A.L.; Fiehn, O.; Kaddurah-Daouk, R.; Lindon, J.C. Metabolomics Standards Workshop and the development of international standards for reporting metabolomics experimental results. Brief. Bioinform. 2006, 7, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X. Modern analytical techniques in metabolomics analysis. Analyst 2012, 137, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Clasquin, M.F.; Melamud, E.; Rabinowitz, J.D. LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr. Protoc. Bioinform. 2012, 37, 14.11.1–14.11.23. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Salek, R.M.; Steinbeck, C.; Viant, M.R.; Goodacre, R.; Dunn, W.B. The role of reporting standards for metabolite annotation and identification in metabolomic studies. Gigascience 2013, 2, 13. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef]
- Sugimoto, M.; Kawakami, M.; Robert, M.; Soga, T.; Tomita, M. Bioinformatics Tools for Mass Spectroscopy-Based Metabolomic Data Processing and Analysis. Curr. Bioinform. 2012, 7, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Reshetova, P.; Smilde, A.K.; van Kampen, A.H.; Westerhuis, J.A. Use of prior knowledge for the analysis of high-throughput transcriptomics and metabolomics data. BMC Syst. Biol. 2014, 8 (Suppl. S2), S2. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Noble, W.S. How does multiple testing correction work? Nat. Biotechnol. 2009, 27, 1135–1137. [Google Scholar] [CrossRef]
- Antonelli, J.; Claggett, B.L.; Henglin, M.; Kim, A.; Ovsak, G.; Kim, N.; Deng, K.; Rao, K.; Tyagi, O.; Watrous, J.D.; et al. Statistical Workflow for Feature Selection in Human Metabolomics Data. Metabolites 2019, 9, 143. [Google Scholar] [CrossRef]
- Chen, T.; Cao, Y.; Zhang, Y.; Liu, J.; Bao, Y.; Wang, C.; Jia, W.; Zhao, A. Random forest in clinical metabolomics for phenotypic discrimination and biomarker selection. Evid.-Based Complementary Altern. Med. 2013, 2013, 298183. [Google Scholar] [CrossRef]
- Gromski, P.S.; Xu, Y.; Kotze, H.L.; Correa, E.; Ellis, D.I.; Armitage, E.G.; Turner, M.L.; Goodacre, R. Influence of missing values substitutes on multivariate analysis of metabolomics data. Metabolites 2014, 4, 433–452. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Kumar, S.; Sehgal, R.; George, S.; Gupta, R.; Poddar, S.; Jha, A.; Pathak, S. El-MAVEN: A Fast, Robust, and User-Friendly Mass Spectrometry Data Processing Engine for Metabolomics. Methods Mol. Biol. 2019, 1978, 301–321. [Google Scholar] [CrossRef] [PubMed]
- Koelmel, J.P.; Kroeger, N.M.; Ulmer, C.Z.; Bowden, J.A.; Patterson, R.E.; Cochran, J.A.; Beecher, C.W.W.; Garrett, T.J.; Yost, R.A. LipidMatch: An automated workflow for rule-based lipid identification using untargeted high-resolution tandem mass spectrometry data. BMC Bioinform. 2017, 18, 331. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, P.D.; Russell, J.D.; Coon, J.J. LipiDex: An Integrated Software Package for High-Confidence Lipid Identification. Cell Syst. 2018, 6, 621–625.e625. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhu, Z.J. MetFlow: An interactive and integrated workflow for metabolomics data cleaning and differential metabolite discovery. Bioinformatics 2019, 35, 2870–2872. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef]
- Xia, J.; Mandal, R.; Sinelnikov, I.V.; Broadhurst, D.; Wishart, D.S. MetaboAnalyst 2.0—A comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012, 40, W127–W133. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.E.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Lin, W.J.; Shen, P.C.; Liu, H.C.; Cho, Y.C.; Hsu, M.K.; Lin, I.C.; Chen, F.H.; Yang, J.C.; Ma, W.L.; Cheng, W.C. LipidSig: A web-based tool for lipidomic data analysis. Nucleic Acids Res. 2021, 49, W336–W345. [Google Scholar] [CrossRef]
- Molenaar, M.R.; Jeucken, A.; Wassenaar, T.A.; van de Lest, C.H.A.; Brouwers, J.F.; Helms, J.B. LION/web: A web-based ontology enrichment tool for lipidomic data analysis. Gigascience 2019, 8, giz061. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Cho, K.; Uritboonthai, W.; Zhu, Z.; Patti, G.J.; Siuzdak, G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat. Biotechnol. 2012, 30, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-de-Diego, R.; Tarazona, S.; Martinez-Mira, C.; Balzano-Nogueira, L.; Furio-Tari, P.; Pappas, G.J., Jr.; Conesa, A. PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018, 46, W503–W509. [Google Scholar] [CrossRef]
- Kuo, T.C.; Tian, T.F.; Tseng, Y.J. 3Omics: A web-based systems biology tool for analysis, integration and visualization of human transcriptomic, proteomic and metabolomic data. BMC Syst. Biol. 2013, 7, 64. [Google Scholar] [CrossRef]
- Fernandez, J.M.; Hoffmann, R.; Valencia, A. iHOP web services. Nucleic Acids Res. 2007, 35, W21–W26. [Google Scholar] [CrossRef][Green Version]
- Kamburov, A.; Cavill, R.; Ebbels, T.M.; Herwig, R.; Keun, H.C. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 2011, 27, 2917–2918. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef]
- Suhre, K.; Schmitt-Kopplin, P. MassTRIX: Mass translator into pathways. Nucleic Acids Res. 2008, 36, W481–W484. [Google Scholar] [CrossRef]
- Zhou, G.; Xia, J. OmicsNet: A web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 2018, 46, W514–W522. [Google Scholar] [CrossRef]
- Altmae, S.; Esteban, F.J.; Stavreus-Evers, A.; Simon, C.; Giudice, L.; Lessey, B.A.; Horcajadas, J.A.; Macklon, N.S.; D’Hooghe, T.; Campoy, C.; et al. Guidelines for the design, analysis and interpretation of ‘omics’ data: Focus on human endometrium. Hum. Reprod. Update 2014, 20, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.K.; Forslund, S.K.; Gudmundsdottir, V.; Petersen, A.O.; Hildebrand, F.; Hyotylainen, T.; Nielsen, T.; Hansen, T.; Bork, P.; Ehrlich, S.D.; et al. A computational framework to integrate high-throughput ‘-omics’ datasets for the identification of potential mechanistic links. Nat. Protoc. 2018, 13, 2781–2800. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, D.A.; Piccolo, B.D.; Marco, M.L.; Kim, E.B.; Goodson, M.L.; Keenan, M.J.; Dunn, T.N.; Knudsen, K.E.; Martin, R.J.; Adams, S.H. Mice Fed a High-Fat Diet Supplemented with Resistant Starch Display Marked Shifts in the Liver Metabolome Concurrent with Altered Gut Bacteria. J. Nutr. 2016, 146, 2476–2490. [Google Scholar] [CrossRef] [PubMed]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J.; et al. Integrated Analyses of Microbiome and Longitudinal Metabolome Data Reveal Microbial-Host Interactions on Sulfur Metabolism in Parkinson’s Disease. Cell Rep. 2019, 29, 1767–1777.e1768. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105, 975–991.e977. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Year | Description | Functions | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Data Pre-Processing | Data Processing | Statistical Analyses | Pathway Enrichment Analysis | Omics Data Integration | ||||||
| Normalization | Compound Name Identification | Transcriptomics | Proteomics | Microbiome | ||||||
| Mzmine3 | 2022 | MZmine3 builds on the success of MZmine 2 with many features focused on improving the user-friendly graphical | Y | Y | Y | Y | - | - | - | - |
| MetaboAnalyst 5.0 | 2021 | Comprehensive web-based tool for comprehensive metabolomics data analysis, interpretation, and integration with other omics data. | Y | Y | Y | Y | Y | Y | - | - |
| LipidSig | 2021 | Web-based tool for lipidomic data analysis | Y | Y | Y | Y | - | - | - | - |
| MS-DIAL 4.0 | 2020 | Lipidome atlas in MS-DIAL 4.0 | Y | Y | Y | Y | - | - | - | - |
| El-MAVEN | 2019 | Fast, Robust, and User-Friendly Mass Spectrometry Data Processing Engine for Metabolomics | Y | Y | Y | - | - | - | - | - |
| MetFlow | 2019 | Interactive and integrated web server for metabolomics data cleaning and differential metabolite discovery. | Y | Y | Y | Y | Y | - | - | - |
| LION | 2019 | Web-based ontology enrichment tool for lipidomic data analysis. | - | Y | Y | Y | Y | - | - | - |
| Omicsnet | 2018 | Web-based tool for creation and visual analysis of biological networks in 3D space | - | - | - | Y | Y | Y | Y | Y |
| METLIN | 2018 | Technology platform for the identification of known and unknown metabolites and other chemical entities. | - | - | Y | - | - | - | - | - |
| PaintOmics 3 | 2018 | Web-based resource for the integrated visualization of multiple omics data types onto KEGG pathway diagrams. | - | - | - | - | Y | Y | Y | - |
| LipiDex | 2018 | Integrated Software Package for High-Confidence Lipid Identification | Y | - | Y | - | - | - | - | - |
| LipidMatch | 2017 | Automated workflow for rule-based lipid identification using untargeted high-resolution tandem mass spectrometry data | Y | - | Y | - | - | - | - | - |
| 3Omics | 2013 | One-click web tool for fast analysis and visualization of multi-omics data. | Y | Y | - | Y | Y | Y | Y | - |
| IMPaLa | 2011 | Pathway analysis of transcriptomics or proteomics and metabolomics data. | - | - | - | - | Y | Y | Y | - |
| MetPA | 2010 | Pathway analysis for metabolomics data. | Y | - | - | - | Y | - | - | - |
| MassTRIX | 2008 | Tool for high precision MS data annotation. | Y | - | Y | - | Y | - | - | - |
| MetaCoreTM | 2004 | Commercial tool for functional analysis and integrated analysis of multi-omics data. | Y | - | - | - | Y | Y | Y | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Li, E.-M.; Xu, L.-Y. Guide to Metabolomics Analysis: A Bioinformatics Workflow. Metabolites 2022, 12, 357. https://doi.org/10.3390/metabo12040357
Chen Y, Li E-M, Xu L-Y. Guide to Metabolomics Analysis: A Bioinformatics Workflow. Metabolites. 2022; 12(4):357. https://doi.org/10.3390/metabo12040357
Chicago/Turabian StyleChen, Yang, En-Min Li, and Li-Yan Xu. 2022. "Guide to Metabolomics Analysis: A Bioinformatics Workflow" Metabolites 12, no. 4: 357. https://doi.org/10.3390/metabo12040357
APA StyleChen, Y., Li, E.-M., & Xu, L.-Y. (2022). Guide to Metabolomics Analysis: A Bioinformatics Workflow. Metabolites, 12(4), 357. https://doi.org/10.3390/metabo12040357