
Modeling the Amino Acid Effect on Glucagon Secretion from Pancreatic Alpha Cells





Abstract
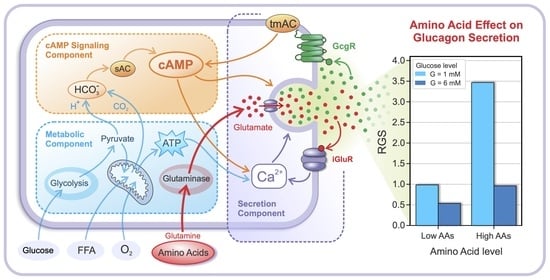
:
1. Introduction
2. Results
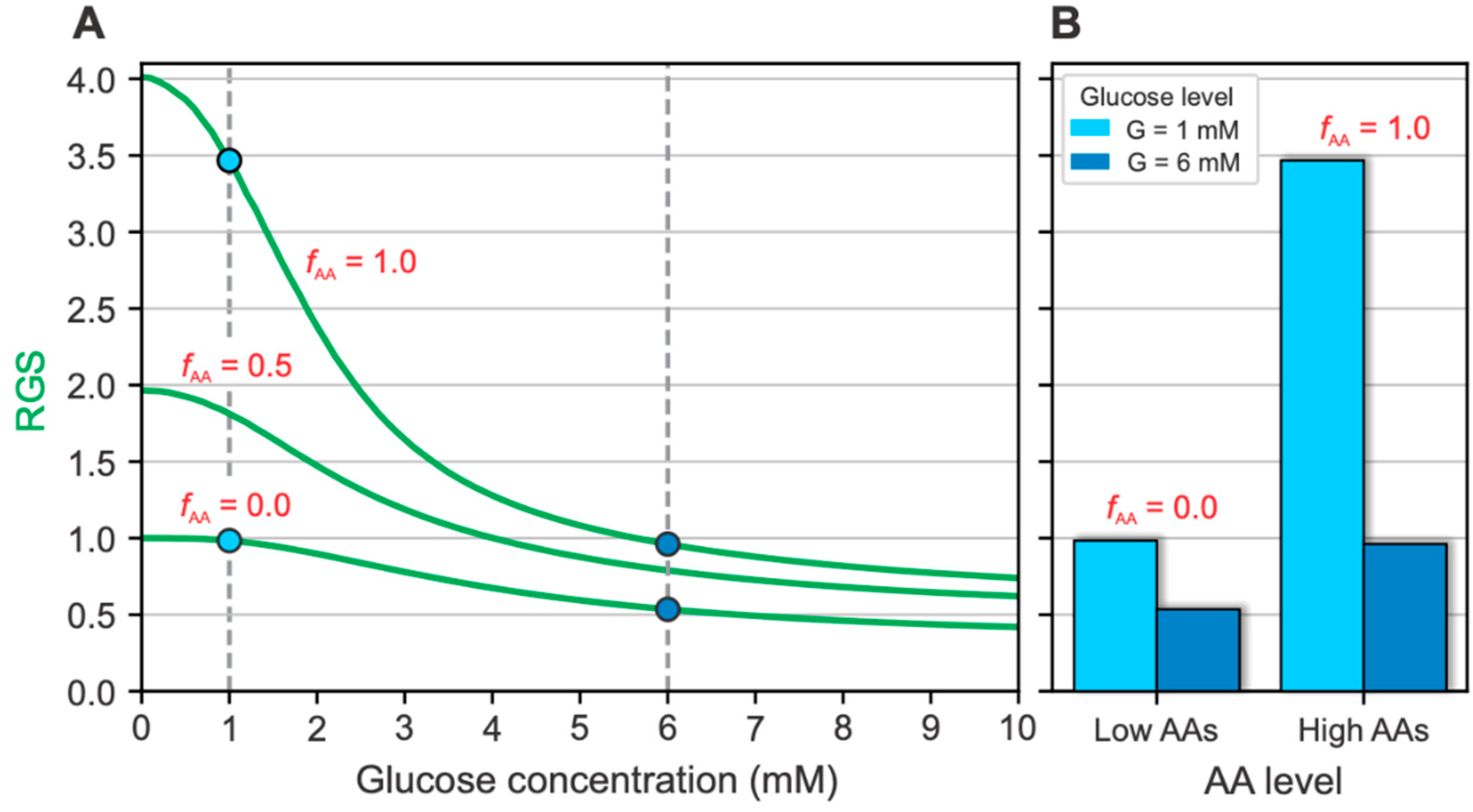
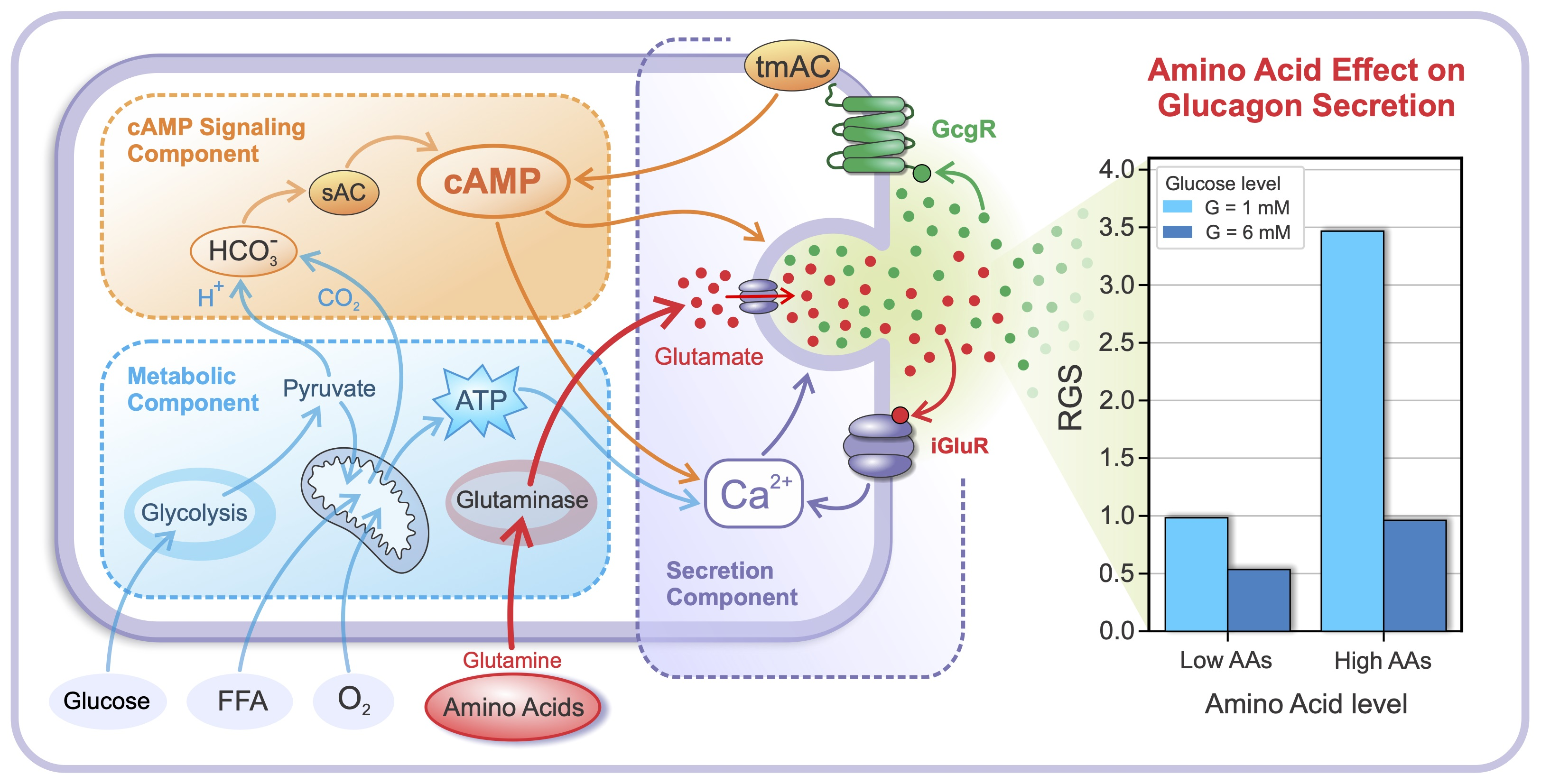
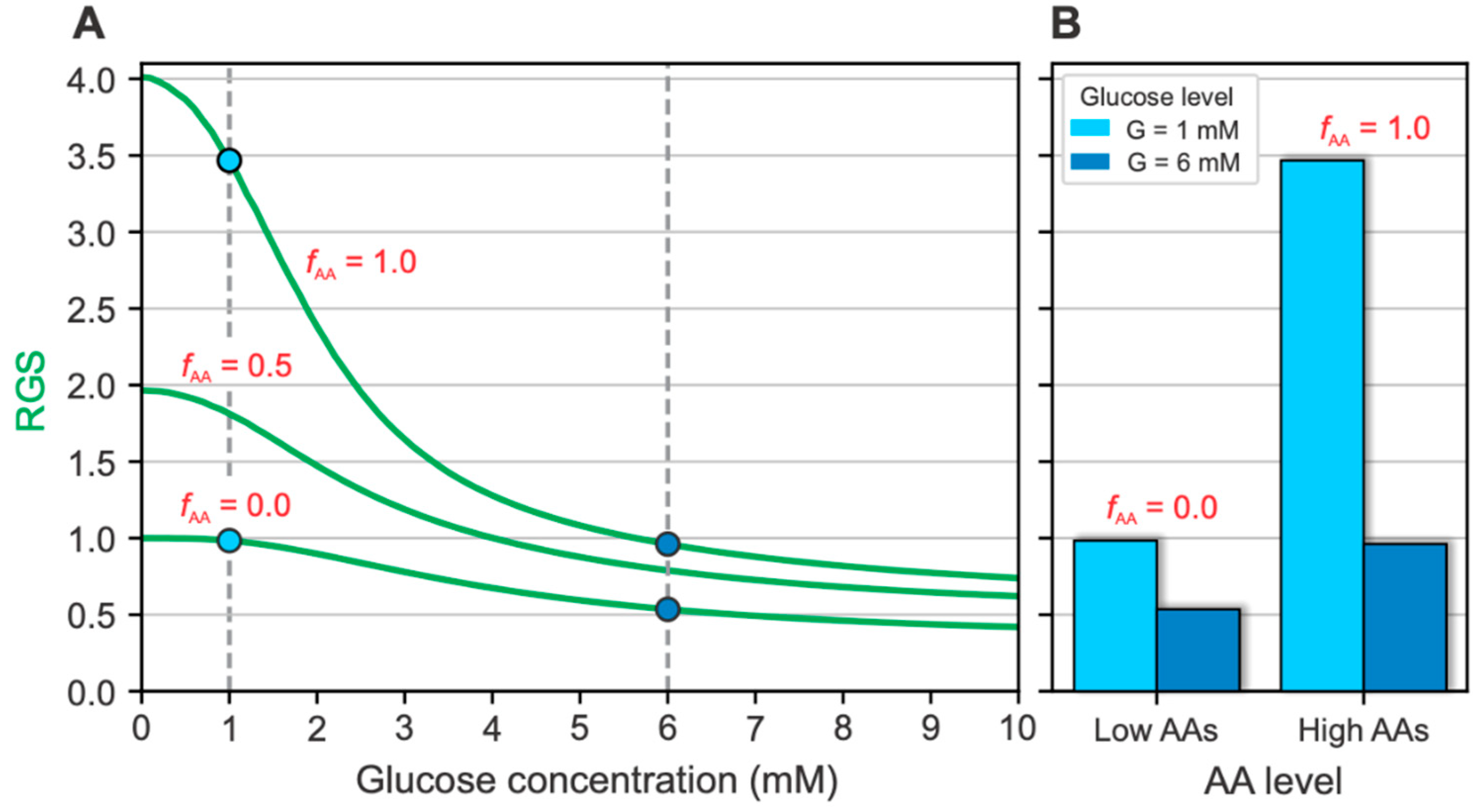
2.1. Glucose- and AA-Dependent RGS
2.2. The Intracellular Mechanisms behind Glucose- and AA-Dependent RGS
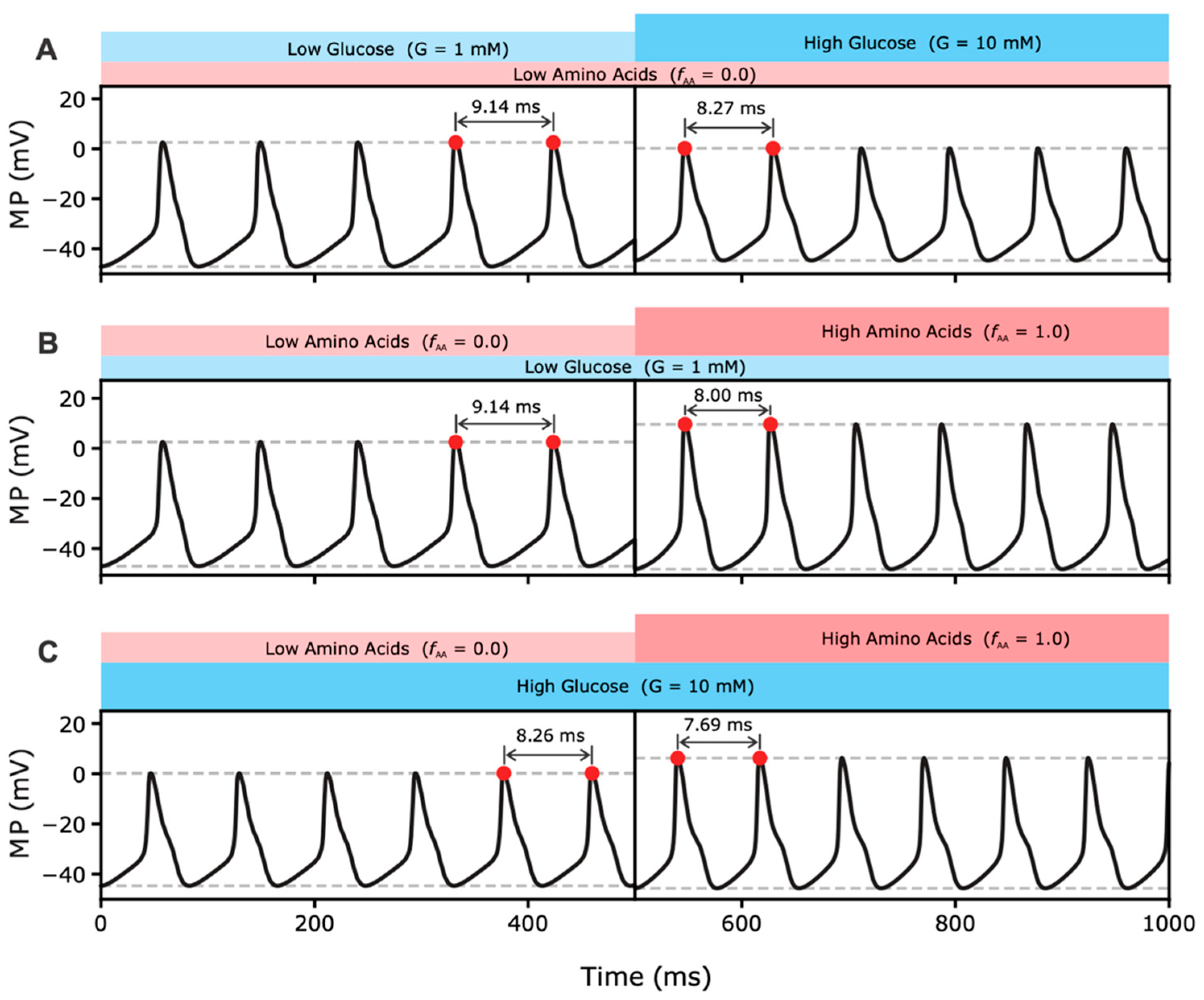
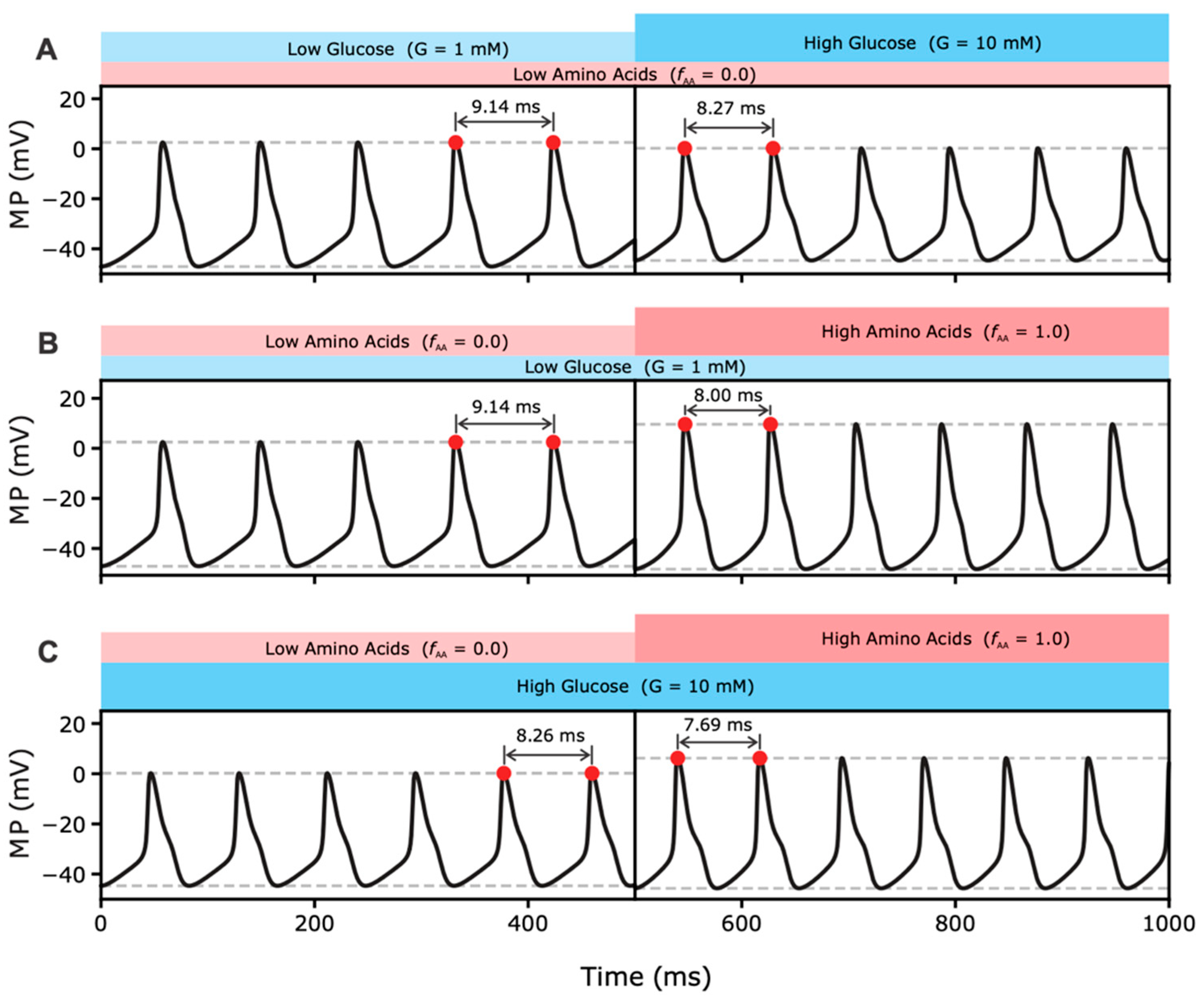
2.2.1. Effects of MP Oscillations on RGS
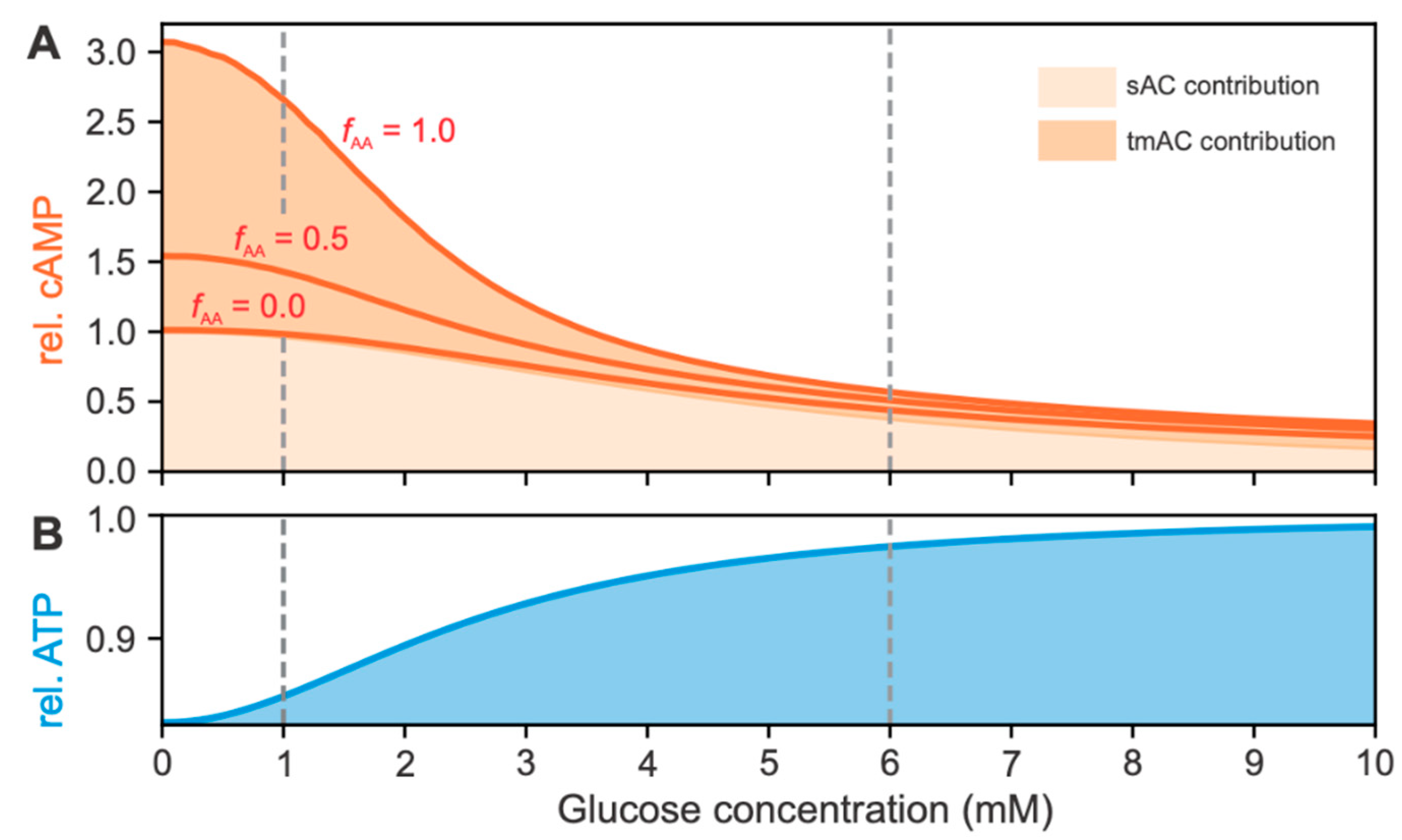
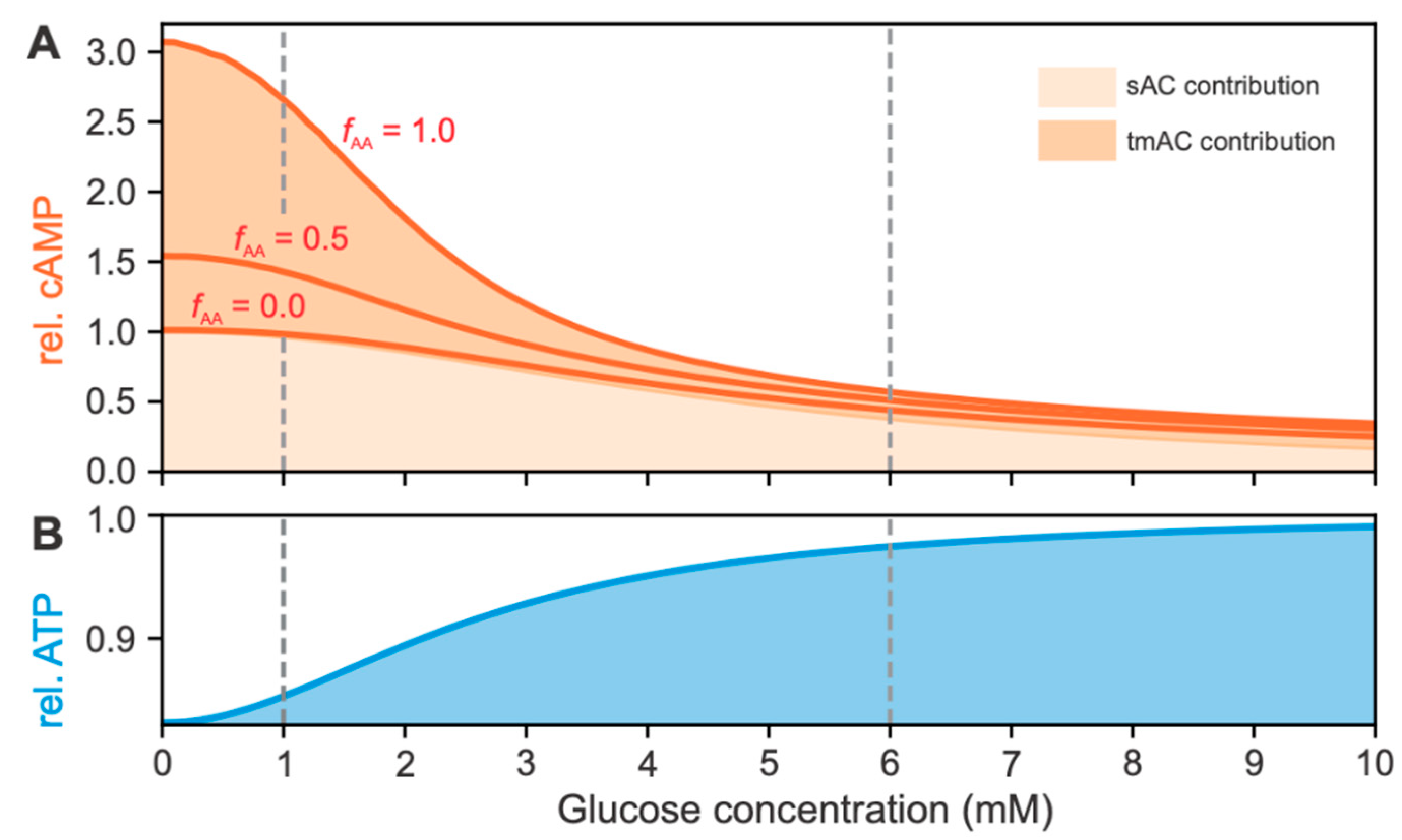
2.2.2. Effects of cAMP and ATP Concentrations on RGS
3. Discussion
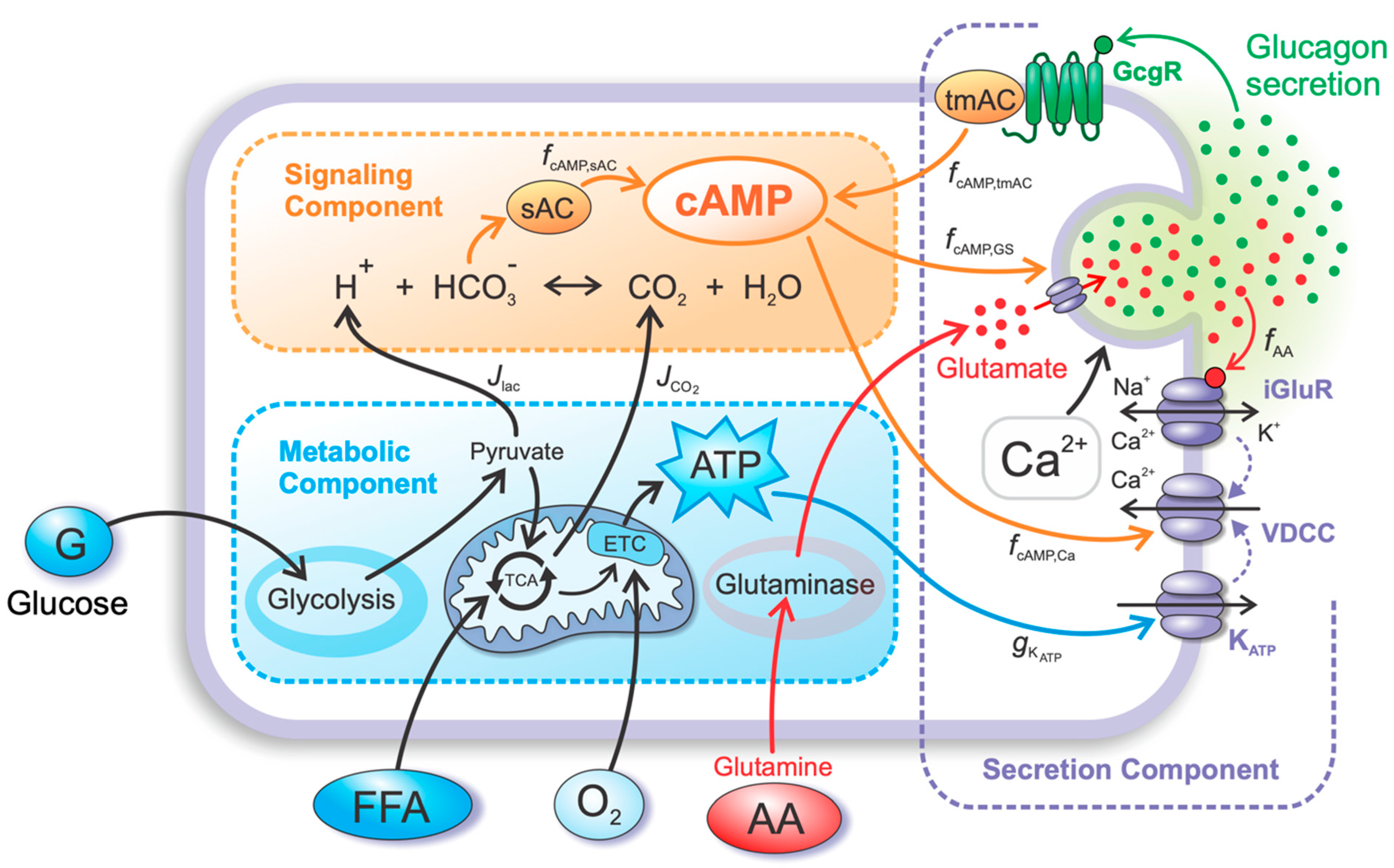
4. Materials and Methods
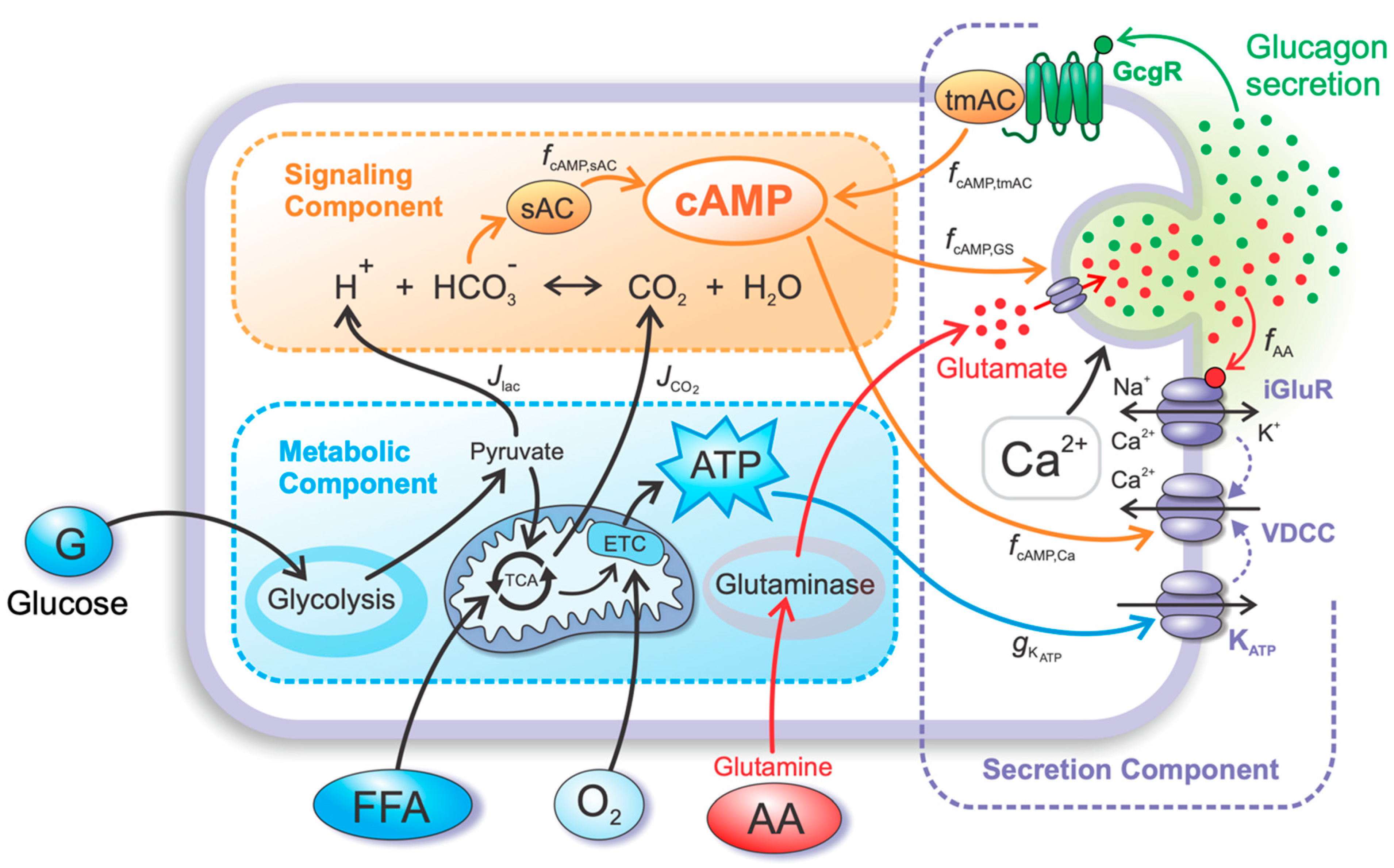
4.1. Metabolic Component
4.2. Signaling Component
4.3. Secretion Component
4.3.1. The Effects of Glutamate and cAMP on the MP and Glucagon Granule Exocytosis
4.3.2. Autocrine Effects of Glucagon on cAMP Concentration
4.3.3. Net Glucose- and AAs-Dependent RGS
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Girard, J. Glucagon, a key factor in the pathophysiology of type 2 diabetes. Biochimie 2017, 143, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Gilon, P. The Role of α-Cells in Islet Function and Glucose Homeostasis in Health and Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1367–1394. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Braun, M.; Zhang, Q. Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium 2012, 51, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Bosco, D.; Armanet, M.; Morel, P.; Niclauss, N.; Sgroi, A.; Muller, Y.D.; Giovannoni, L.; Parnaud, G.; Berney, T. Unique Arrangement of α- and β-Cells in Human Islets of Langerhans. Diabetes 2010, 59, 1202–1210. [Google Scholar] [CrossRef] [Green Version]
- Vieira, E.; Salehi, A.; Gylfe, E. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Diabetologia 2007, 50, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Heimberg, H.; De Vos, A.; Moens, K.; Quartier, E.; Bouwens, L.; Pipeleers, D.; Van Schaftingen, E.; Madsen, O.; Schuit, F. The glucose sensor protein glucokinase is expressed in glucagon-producing alpha-cells. Proc. Natl. Acad. Sci. USA 1996, 93, 7036–7041. [Google Scholar] [CrossRef] [Green Version]
- Basco, D.; Zhang, Q.; Salehi, A.; Tarasov, A.; Dolci, W.; Herrera, P.; Spiliotis, I.; Berney, X.; Tarussio, D.; Rorsman, P.; et al. α-cell glucokinase suppresses glucose-regulated glucagon secretion. Nat. Commun. 2018, 9, 546. [Google Scholar] [CrossRef] [Green Version]
- Bokvist, K.; Olsen, H.; Høy, M.; Gotfredsen, C.; Holmes, W.; Buschard, K.; Rorsman, P.; Gromada, J. Characterisation of sulphonylurea and ATP-regulated K+ channels in rat pancreatic A-cells. Pflügers Arch. Eur. J. Physiol. 1999, 438, 428–436. [Google Scholar] [CrossRef]
- Ramracheya, R.; Ward, C.; Shigeto, M.; Walker, J.N.; Amisten, S.; Zhang, Q.; Johnson, P.R.; Rorsman, P.; Braun, M. Membrane Potential-Dependent Inactivation of Voltage-Gated Ion Channels in α-Cells Inhibits Glucagon Secretion From Human Islets. Diabetes 2010, 59, 2198–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Modelling of energy-driven switch for glucagon and insulin secretion. J. Theor. Biol. 2020, 493, 110213. [Google Scholar] [CrossRef] [PubMed]
- Grubelnik, V.; Markovič, R.; Lipovšek, S.; Leitinger, G.; Gosak, M.; Dolenšek, J.; Valladolid-Acebes, I.; Berggren, P.-O.; Stožer, A.; Perc, M.; et al. Modelling of dysregulated glucagon secretion in type 2 diabetes by considering mitochondrial alterations in pancreatic α-cells. R. Soc. Open Sci. 2020, 7, 191171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Mitochondrial Dysfunction in Pancreatic α and β Cells Associated with Type 2 Diabetes Mellitus. Life 2020, 10, 348. [Google Scholar] [CrossRef]
- Zmazek, J.; Grubelnik, V.; Markovič, R.; Marhl, M. Role of cAMP in Double Switch of Glucagon Secretion. Cells 2021, 10, 896. [Google Scholar] [CrossRef]
- Schuit, F.; De Vos, A.; Farfari, S.; Moens, K.; Pipeleers, D.; Brun, T.; Prentki, M. Metabolic Fate of Glucose in Purified Islet Cells. J. Biol. Chem. 1997, 272, 18572–18579. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.W.; Ustione, A.; Lavagnino, Z.; Piston, D.W. Regulation of islet glucagon secretion: Beyond calcium. Diabetes Obes. Metab. 2018, 20, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Shuai, H.; Ahooghalandari, P.; Gylfe, E.; Tengholm, A. Glucose controls glucagon secretion by directly modulating cAMP in alpha cells. Diabetologia 2019, 62, 1212–1224. [Google Scholar] [CrossRef] [Green Version]
- Tengholm, A.; Gylfe, E. cAMP signalling in insulin and glucagon secretion. Diabetes Obes. Metab. 2017, 19, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Elliott, A.D.; Ustione, A.; Piston, D.W. Somatostatin and insulin mediate glucose-inhibited glucagon secretion in the pancreatic α-cell by lowering cAMP. Am. J. Physiol. Metab. 2015, 308, E130–E143. [Google Scholar] [CrossRef] [Green Version]
- Zippin, J.H.; Levin, L.; Buck, J. CO2/HCO3−—responsive soluble adenylyl cyclase as a putative metabolic sensor. Trends Endocrinol. Metab. 2001, 12, 366–370. [Google Scholar] [CrossRef]
- Litvin, T.N.; Kamenetsky, M.; Zarifyan, A.; Buck, J.; Levin, L. Kinetic Properties of “Soluble” Adenylyl Cyclase. J. Biol. Chem. 2003, 278, 15922–15926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, J.J.; Albrechtsen, N.J.W.; Pedersen, J.; Knop, F.K. Glucagon and Amino Acids Are Linked in a Mutual Feedback Cycle: The Liver—α-Cell Axis. Diabetes 2017, 66, 235–240. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-K.; Shin, H.M.; Jung, H.; Lee, E.; Kim, T.K.; Kwon, M.J.; Lee, S.H.; Rhee, B.D.; Park, J.H. Comparison of pancreatic beta cells and alpha cells under hyperglycemia: Inverse coupling in pAkt-FoxO1. Diabetes Res. Clin. Pract. 2017, 131, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gutierrez, G.D.; Xin, Y.; Cavino, K.; Sung, B.; Sipos, B.; Kloeppel, G.; Gromada, J.; Okamoto, H. Increased SLC38A4 Amino Acid Transporter Expression in Human Pancreatic α-Cells After Glucagon Receptor Inhibition. Endocrinology 2019, 160, 979–988. [Google Scholar] [CrossRef]
- Solloway, M.J.; Madjidi, A.; Gu, C.; Eastham-Anderson, J.; Clarke, H.J.; Kljavin, N.; Zavala-Solorio, J.; Kates, L.; Friedman, B.; Brauer, M.; et al. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of α-Cell Mass. Cell Rep. 2015, 12, 495–510. [Google Scholar] [CrossRef] [Green Version]
- Galsgaard, K.D.; Winther-Sørensen, M.; Ørskov, C.; Kissow, H.; Poulsen, S.S.; Vilstrup, H.; Prehn, C.; Adamski, J.; Jepsen, S.L.; Hartmann, B.; et al. Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am. J. Physiol. Metab. 2018, 314, E93–E103. [Google Scholar] [CrossRef] [Green Version]
- Wewer Albrechtsen, N.J.; Pedersen, J.; Galsgaard, K.D.; Winther-Sørensen, M.; Suppli, M.P.; Janah, L.; Gromada, J.; Vilstrup, H.; Knop, F.K.; Holst, J.J. The Liver–α-Cell Axis and Type 2 Diabetes. Endocr. Rev. 2019, 40, 1353–1366. [Google Scholar] [CrossRef] [Green Version]
- Galsgaard, K.D.; Jepsen, S.L.; Kjeldsen, S.; Pedersen, J.; Albrechtsen, N.J.W.; Holst, J.J. Alanine, arginine, cysteine, and proline, but not glutamine, are substrates for, and acute mediators of, the liver-α-cell axis in female mice. Am. J. Physiol. Metab. 2020, 318, E920–E929. [Google Scholar] [CrossRef]
- Dean, E.D. A Primary Role for α-Cells as Amino Acid Sensors. Diabetes 2019, 69, 542–549. [Google Scholar] [CrossRef]
- Finan, B.; Capozzi, M.E.; Campbell, J.E. Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes 2019, 69, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, D.M.; Faloona, G.R.; Unger, R.H. Glucagon-stimulating activity of 20 amino acids in dogs. J. Clin. Investig. 1972, 51, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Pipeleers, D.G.; Schuit, F.C.; in’t Veld, P.A.; Maes, E.; Hooghe-Peters, E.L.; Van De Winkel, M.; Gepts, W. Interplay of Nutrients and Hormones in the Regulation of Insulin Release. Endocrinology 1985, 117, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.J. Medical Biochemistry (4th ed.): Bhagavan, N.V. Biochem. Mol. Biol. Educ. 2002, 30, 270–271. [Google Scholar] [CrossRef]
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; Braun, M.; Brereton, M.; Collins, S.; Galvanovskis, J.; et al. Role of KATP Channels in Glucose-Regulated Glucagon Secretion and Impaired Counterregulation in Type 2 Diabetes. Cell Metab. 2013, 18, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Capozzi, M.E.; Svendsen, B.; Encisco, S.E.; Lewandowski, S.L.; Martin, M.D.; Lin, H.; Jaffe, J.L.; Coch, R.W.; Haldeman, J.M.; MacDonald, P.E.; et al. β Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 2019, 4, e126742. [Google Scholar] [CrossRef]
- Tong, B.C.; Barbul, A. Cellular and physiological effects of arginine. Mini-Rev. Med. Chem. 2004, 4, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, A.B.; Strack, I. Amino Acid Stimulation of Glucagon Secretion by Perifused Islets of High-protein-fed Rats. Diabetes 1978, 27, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Östenson, C.-G.; Grebing, C. Evidence for metabolic regulation of pancreatic glucagon secretion by l-glutamine. Eur. J. Endocrinol. 1985, 108, 386–391. [Google Scholar] [CrossRef]
- Li, C.; Liu, C.; Nissim, I.; Chen, J.; Chen, P.; Doliba, N.; Zhang, T.; Nissim, I.; Daikhin, Y.; Stokes, D.; et al. Regulation of Glucagon Secretion in Normal and Diabetic Human Islets by γ-Hydroxybutyrate and Glycine. J. Biol. Chem. 2013, 288, 3938–3951. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Buettger, C.; Kwagh, J.; Matter, A.; Daikhin, Y.; Nissim, I.B.; Collins, H.W.; Yudkoff, M.; Stanley, C.; Matschinsky, F.M. A Signaling Role of Glutamine in Insulin Secretion. J. Biol. Chem. 2004, 279, 13393–13401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, F.; Baglietto-Vargas, D.; Moreno-González, I.; López-Tellez, J.; Cuesta-Munoz, A.; Gutiérrez, A.; Aledo, J. Glutaminase activity is confined to the mantle of the islets of Langerhans. Biochimie 2007, 89, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Yokoi, N.; Seino, S. Glutamate as intracellular and extracellular signals in pancreatic islet functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 246–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Yamada, H.; Uehara, S.; Morimoto, R.; Muroyama, A.; Yatsushiro, S.; Takeda, J.; Yamamoto, A.; Moriyama, Y. Secretory Granule-mediated Co-secretion ofl-Glutamate and Glucagon Triggers Glutamatergic Signal Transmission in Islets of Langerhans. J. Biol. Chem. 2003, 278, 1966–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, O.; Jacques-Silva, M.C.; Speier, S.; Yang, S.-N.; Köhler, M.; Fachado, A.; Vieira, E.; Zierath, J.R.; Kibbey, R.; Berman, D.M.; et al. Glutamate Is a Positive Autocrine Signal for Glucagon Release. Cell Metab. 2008, 7, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, F.A.; Reimer, R.J.; Krizaj, D.; Barber, D.; Storm-Mathisen, J.; Copenhagen, D.R.; Edwards, R.H. Molecular Analysis of System N Suggests Novel Physiological Roles in Nitrogen Metabolism and Synaptic Transmission. Cell 1999, 99, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Gammelsaeter, R.; Jenstad, M.; Bredahl, M.; Gundersen, V.; Chaudhry, F. Complementary expression of SN1 and SAT2 in the islets of Langerhans suggests concerted action of glutamine transport in the regulation of insulin secretion. Biochem. Biophys. Res. Commun. 2009, 381, 378–382. [Google Scholar] [CrossRef]
- Schmid, R.; Schusdziarra, V.; Schulte-Frohlinde, E.; Maier, V.; Classen, M. Role of Amino Acids in Stimulation of Postprandial Insulin, Glucagon, and Pancreatic Polypeptide in Humans. Pancreas 1989, 4, 305–314. [Google Scholar] [CrossRef]
- Rudman, D.; Mattson, D.E.; Feller, A.G.; Cotter, R.; Johnson, R.C. Fasting plasma amino acids in elderly men. Am. J. Clin. Nutr. 1989, 49, 559–566. [Google Scholar] [CrossRef]
- Montefusco, F.; Pedersen, M.G. Mathematical modelling of local calcium and regulated exocytosis during inhibition and stimulation of glucagon secretion from pancreatic alpha-cells. J. Physiol. 2015, 593, 4519–4530. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, P.E.; De Marinis, Y.Z.; Ramracheya, R.; Salehi, A.; Ma, X.; Johnson, P.R.V.; Cox, R.; Eliasson, L.; Rorsman, P. A KATP Channel-Dependent Pathway within α Cells Regulates Glucagon Release from Both Rodent and Human Islets of Langerhans. PLoS Biol. 2007, 5, e143. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhang, Y.; Gromada, J.; Sewing, S.; Berggren, P.-O.; Buschard, K.; Salehi, A.; Vikman, J.; Rorsman, P.; Eliasson, L. Glucagon Stimulates Exocytosis in Mouse and Rat Pancreatic α-Cells by Binding to Glucagon Receptors. Mol. Endocrinol. 2005, 19, 198–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, G.; Sandler, S.; Gylfe, E.; Tengholm, A. Glucose- and Hormone-Induced cAMP Oscillations in α- and β-Cells within Intact Pancreatic Islets. Diabetes 2011, 60, 1535–1543. [Google Scholar] [CrossRef] [Green Version]
- Briant, L.; Salehi, A.; Vergari, E.; Zhang, Q.; Rorsman, P. Glucagon secretion from pancreatic α-cells. Upsala J. Med. Sci. 2016, 121, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendt, A.; Eliasson, L. Pancreatic α-cells—The unsung heroes in islet function. Semin. Cell Dev. Biol. 2020, 103, 41–50. [Google Scholar] [CrossRef]
- Watts, M.; Sherman, A. Modeling the Pancreatic α-Cell: Dual Mechanisms of Glucose Suppression of Glucagon Secretion. Biophys. J. 2014, 106, 741–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alford, F.; Bloom, S.; Nabarro, J.; Hall, R.; Besser, G.; Coy, D.; Kastin, A.; Schally, A. Glucagon control of fasting glucose in man. Lancet 1974, 304, 974–977. [Google Scholar] [CrossRef]
- Lee, Y.; Berglund, E.D.; Yu, X.; Wang, M.-Y.; Evans, M.R.; Scherer, P.E.; Holland, W.L.; Charron, M.J.; Roth, M.G.; Unger, R.H. Hyperglycemia in rodent models of type 2 diabetes requires insulin-resistant alpha cells. Proc. Natl. Acad. Sci. USA 2014, 111, 13217–13222. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, D.H.; Cherrington, A.D. Hepatic fuel metabolism during muscular work: Role and regulation. Am. J. Physiol. Metab. 1991, 260, E811–E824. [Google Scholar] [CrossRef]
- Marroquí, L.; Alonso-Magdalena, P.; Merino, B.; Fuentes, E.; Nadal, A.; Quesada, I. Nutrient regulation of glucagon secretion: Involvement in metabolism and diabetes. Nutr. Res. Rev. 2014, 27, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Lang, V.; Bellisle, F.; Alamowitch, C.; Craplet, C.; Bornet, F.; Slama, G.; Guy-Grand, B. Varying the protein source in mixed meal modifies glucose, insulin and glucagon kinetics in healthy men, has weak effects on subjective satiety and fails to affect food intake. Eur. J. Clin. Nutr. 1999, 53, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Markova, M.; Hornemann, S.; Sucher, S.; Wegner, K.; Pivovarova, O.R.; Rudovich, N.; Thomann, R.; Schneeweiss, R.; Rohn, S.; Pfeiffer, A.F.H. Rate of appearance of amino acids after a meal regulates insulin and glucagon secretion in patients with type 2 diabetes: A randomized clinical trial. Am. J. Clin. Nutr. 2018, 108, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El, K.; Capozzi, M.E.; Campbell, J.E. Repositioning the Alpha Cell in Postprandial Metabolism. Endocrinology 2020, 161, bqaa169. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, L.M.; Parris, E.E.; Cahill, G.F. Starvation in Man. N. Engl. J. Med. 1970, 282, 668–675. [Google Scholar] [CrossRef]
- Korenfeld, N.; Finkel, M.; Buchshtab, N.; Bar-Shimon, M.; Charni-Natan, M.; Goldstein, I. Fasting Hormones Synergistically Induce Amino Acid Catabolism Genes to Promote Gluconeogenesis. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1021–1036. [Google Scholar] [CrossRef]
- Bertrand, G.; Gross, R.; Puech, R.; Loubatières-Mariani, M.-M.; Bockaert, J. Glutamate stimulates glucagon secretion via an excitatory amino acid receptor of the AMPA subtype in rat pancreas. Eur. J. Pharmacol. 1993, 237, 45–50. [Google Scholar] [CrossRef]
- Seifert, G.; Steinhäuser, C. Ionotropic glutamate receptors in astrocytes. Prog. Brain Res. 2001, 132, 287–299. [Google Scholar] [CrossRef]
- Gaisano, H.Y.; Leung, Y.M. Pancreatic Islet α Cell Commands Itself: Secrete More Glucagon! Cell Metab. 2008, 7, 474–475. [Google Scholar] [CrossRef] [Green Version]
- Bettler, B.; Mulle, C. AMPA and kainate receptors. Neuropharmacology 1995, 34, 123–139. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| 0.2 | |
| 0.5 | |
| 0.4 | |
| 0.2 | |
| 0.16 | |
| 0.25 | |
| 175 | |
| 2.8 | |
| 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zmazek, J.; Grubelnik, V.; Markovič, R.; Marhl, M. Modeling the Amino Acid Effect on Glucagon Secretion from Pancreatic Alpha Cells. Metabolites 2022, 12, 348. https://doi.org/10.3390/metabo12040348
Zmazek J, Grubelnik V, Markovič R, Marhl M. Modeling the Amino Acid Effect on Glucagon Secretion from Pancreatic Alpha Cells. Metabolites. 2022; 12(4):348. https://doi.org/10.3390/metabo12040348
Chicago/Turabian StyleZmazek, Jan, Vladimir Grubelnik, Rene Markovič, and Marko Marhl. 2022. "Modeling the Amino Acid Effect on Glucagon Secretion from Pancreatic Alpha Cells" Metabolites 12, no. 4: 348. https://doi.org/10.3390/metabo12040348
APA StyleZmazek, J., Grubelnik, V., Markovič, R., & Marhl, M. (2022). Modeling the Amino Acid Effect on Glucagon Secretion from Pancreatic Alpha Cells. Metabolites, 12(4), 348. https://doi.org/10.3390/metabo12040348