
PNPLA6/NTE, an Evolutionary Conserved Phospholipase Linked to a Group of Complex Human Diseases

Abstract
1. Introduction
2. PNPLA6/NTE and Organophosphate-Induced Delayed Neuropathy
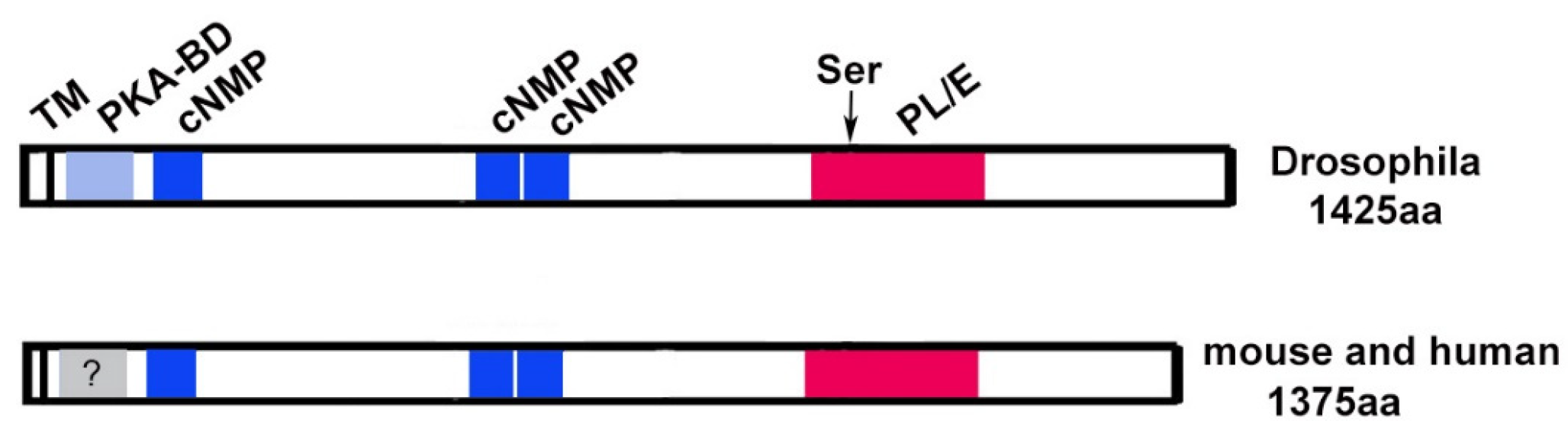
3. PNPLA6/NTE Is an Evolutionarily Conserved Protein with Several Functional Domains
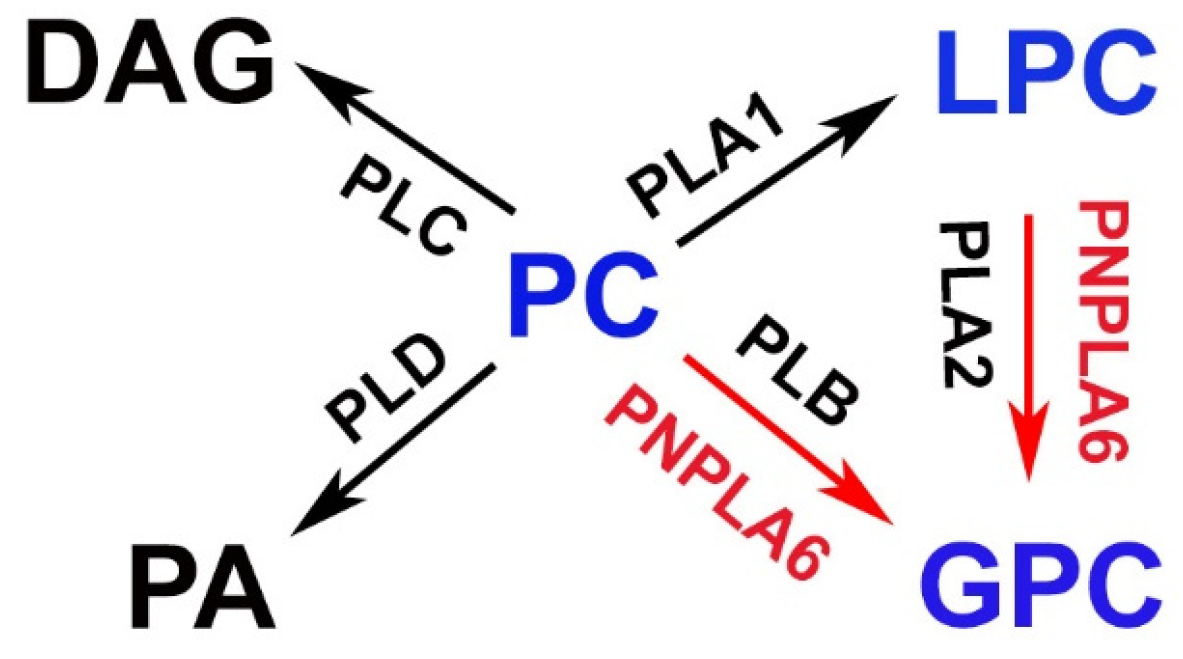
4. Phospholipase Function of PNPLA6/NTE
5. Expression Pattern of PNPLA6/NTE
6. Phenotypic Consequences Due to the Loss of PNPLA6/NTE in Model Systems
6.1. Drosophila Melanogaster
6.2. Danio Rerio
6.3. Mus Musculus
7. PNPLA6 in Human Disease
8. Future Directions
Funding
Conflicts of Interest
References
- Kretzschmar, D.; Hasan, G.; Sharma, S.; Heisenberg, M.; Benzer, S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J. Neurosci. 1997, 17, 7425–7432. [Google Scholar] [CrossRef] [PubMed]
- Lush, M.J.; Li, Y.; Read, D.J.; Willis, A.C.; Glynn, P. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem. J. 1998, 332, 1–4. [Google Scholar] [CrossRef]
- Melentev, P.A.; Agranovich, O.E.; Sarantseva, S.V. Human diseases associated with NTE gene. Ecol. Genet. 2020, 18, 229–242. [Google Scholar] [CrossRef]
- Synofzik, M.; Kernstock, C.; Haack, T.B.; Schols, L. Ataxia meets chorioretinal dystrophy and hypogonadism: Boucher-Neuhauser syndrome due to PNPLA6 mutations. J. Neurol. Neurosurg. Psychiatry 2015, 86, 580–581. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Stempfl, T.; Li, Y.; Glynn, P.; Buttner, R.; Kretzschmar, D. Cloning and expression of the murine sws/NTE gene. Mech. Dev. 2000, 90, 279–282. [Google Scholar] [CrossRef]
- Zaccheo, O.; Dinsdale, D.; Meacock, P.A.; Glynn, P. Neuropathy target esterase and its yeast homologue degrade phosphatidylcholine to glycerophosphocholine in living cells. J. Biol. Chem. 2004, 279, 24024–24033. [Google Scholar] [CrossRef]
- Read, D.J.; Li, Y.; Chao, M.V.; Cavanagh, J.B.; Glynn, P. Neuropathy target esterase is required for adult vertebrate axon maintenance. J. Neurosci. 2009, 29, 11594–11600. [Google Scholar] [CrossRef] [PubMed]
- Kmoch, S.; Majewski, J.; Ramamurthy, V.; Cao, S.; Fahiminiya, S.; Ren, H.; MacDonald, I.M.; Lopez, I.; Sun, V.; Keser, V.; et al. Mutations in PNPLA6 are linked to photoreceptor degeneration and various forms of childhood blindness. Nat. Commun. 2015, 6, 5614. [Google Scholar] [CrossRef]
- Bettencourt da Cruz, A.; Wentzell, J.; Kretzschmar, D. Swiss Cheese, a protein involved in progressive neurodegeneration, acts as a noncanonical regulatory subunit for PKA-C3. J. Neurosci. 2008, 28, 10885–10892. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.I. The pharmacological action of certain phenol esters, with special reference to the etiology of so-called ginger paralysis (second report). Public Health Rep. 1930, 45, 2509–2524. [Google Scholar] [CrossRef]
- Aldridge, W.N.; Barnes, J.M.; Johnson, M.K. Studies on delayed neurotoxicity produced by some organophosphorus compounds. Ann. N. Y. Acad. Sci. 1969, 160, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.K. An enzyme in hen brain hydrolysing phenyl phenylacetate: A possible connection with the delayed neurotoxic effect of some organophosphorus compounds. Biochem. J. 1968, 110, 13P. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.K. A phosphorylation site in brain and the delayed neurotoxic effect of some organophosphorus compounds. Biochem. J. 1969, 111, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.K. Organophosphorus and other inhibitors of brain ‘neurotoxic esterase’ and the development of delayed neurotoxicity in hens. Biochem. J. 1970, 120, 523–531. [Google Scholar] [CrossRef]
- Johnson, M.K.; Lauwerys, R. Protection by some Carbamates against the Delayed Neurotoxic Effects of Di-isopropyl Phosphorofluoridate. Nature 1969, 222, 1066–1067. [Google Scholar] [CrossRef]
- Clothier, B.; Johnson, M.K. Rapid aging of neurotoxic esterase after inhibition by di-isopropyl phosphorofluoridate. Biochem. J. 1979, 177, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Nayak, P.; Mallick, A.K.; Mishra, S.; Panigrahy, D. Organophosphorus-Induced Toxic Myeloneuropathy: Series of Three Adolescent Patients with Short Review. J. Pediatr. Neurosci. 2019, 14, 42–45. [Google Scholar] [CrossRef]
- Davis, C.S.; Richardson, R.J. Organophosphorus compounds. Exp. Clin. Neurotoxicol. 1980, 1, 527–544. [Google Scholar]
- Richardson, R.J.; Fink, J.K.; Glynn, P.; Hufnagel, R.B.; Makhaeva, G.F.; Wijeyesakere, S.J. Neuropathy target esterase (NTE/PNPLA6) and organophosphorus compound-induced delayed neurotoxicity (OPIDN). In Advances in Neurotoxicology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 4, pp. 1–78. [Google Scholar]
- Lotti, M.; Moretto, A. Organophosphate-induced delayed polyneuropathy. Toxicol. Rev. 2005, 24, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Emerick, G.L.; Peccinini, R.G.; de Oliveira, G.H. Organophosphorus-induced delayed neuropathy: A simple and efficient therapeutic strategy. Toxicol. Lett. 2010, 192, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Emerick, G.L.; DeOliveira, G.H.; dos Santos, A.C.; Ehrich, M. Mechanisms for consideration for intervention in the development of organophosphorus-induced delayed neuropathy. Chem. Biol. Interact. 2012, 199, 177–184. [Google Scholar] [CrossRef]
- Gupta, R.C. Handbook of toxicology of chemical warfare agents; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Cavanagh, J. The toxic effects of tri-ortho-cresyl phosphate on the nervous system: An experimental study in hens. J. Neurol. Neurosurg. Psychiatry 1954, 17, 163. [Google Scholar] [CrossRef] [PubMed]
- Bouldin, T.; Cavanagh, J. Organophosphorous neuropathy. II. A fine-structural study of the early stages of axonal degeneration. Am. J. Pathol. 1979, 94, 253. [Google Scholar]
- Bouldin, T.; Cavanagh, J. Organophosphorous neuropathy. I. A teased-fiber study of the spatio-temporal spread of axonal degeneraion. Am. J. Pathol. 1979, 94, 241. [Google Scholar]
- Song, M.; Kang, K.; Song, F. SARM1-mediated wallerian degeneration: A possible mechanism underlying organophosphorus-induced delayed neuropathy. Med. Hypotheses 2021, 155, 110666. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.; Glynn, P. Membrane association of and critical residues in the catalytic domain of human neuropathy target esterase. J. Biol. Chem. 2000, 275, 24477–24483. [Google Scholar] [CrossRef]
- Li, Y.; Dinsdale, D.; Glynn, P. Protein domains, catalytic activity, and subcellular distribution of neuropathy target esterase in mammalian cells. J. Biol. Chem. 2003, 278, 8820–8825. [Google Scholar] [CrossRef] [PubMed]
- Wijeyesakere, S.J.; Richardson, R.J.; Stuckey, J.A. Modeling the tertiary structure of the patatin domain of neuropathy target esterase. Protein J. 2007, 26, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Muhlig-Versen, M.; da Cruz, A.B.; Tschape, J.A.; Moser, M.; Buttner, R.; Athenstaedt, K.; Glynn, P.; Kretzschmar, D. Loss of Swiss cheese/neuropathy target esterase activity causes disruption of phosphatidylcholine homeostasis and neuronal and glial death in adult Drosophila. J. Neurosci. 2005, 25, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kim, C.; Vigil, D.; Haste, N.M.; Yang, J.; Wu, J.; Anand, G.S. Dynamics of signaling by PKA. Biochim. Biophys. Acta Proteins Proteom. 2005, 1754, 25–37. [Google Scholar] [CrossRef]
- Francis, S.H.; Poteet-Smith, C.; Busch, J.L.; Richie-Jannetta, R.; Corbin, J.D. Mechanisms of autoinhibition in cyclic nucleotide-dependent protein kinases. Front Biosci 2002, 7, d580–d592. [Google Scholar] [CrossRef] [PubMed]
- Wentzell, J.S.; Cassar, M.; Kretzschmar, D. Organophosphate-induced changes in the PKA regulatory function of Swiss Cheese/NTE lead to behavioral deficits and neurodegeneration. PLoS ONE 2014, 9, e87526. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-X.; Long, D.-X.; Hou, W.-Y.; Li, W.; Wu, Y.-J. Regulation of neuropathy target esterase by the cAMP/protein kinase A signal. Pharmacol. Res. 2010, 62, 259–264. [Google Scholar] [CrossRef]
- Kienesberger, P.C.; Oberer, M.; Lass, A.; Zechner, R. Mammalian patatin domain containing proteins: A family with diverse lipolytic activities involved in multiple biological functions. J. Lipid Res. 2009, 50 Suppl, S63–S68. [Google Scholar] [CrossRef]
- Quistad, G.B.; Barlow, C.; Winrow, C.J.; Sparks, S.E.; Casida, J.E. Evidence that mouse brain neuropathy target esterase is a lysophospholipase. Proc. Natl. Acad. Sci. USA 2003, 100, 7983–7987. [Google Scholar] [CrossRef] [PubMed]
- van Tienhoven, M.; Atkins, J.; Li, Y.; Glynn, P. Human Neuropathy Target Esterase Catalyzes Hydrolysis of Membrane Lipids. J. Biol. Chem. 2002, 277, 20942–20948. [Google Scholar] [CrossRef]
- Glynn, P. Neuronal phospholipid deacylation is essential for axonal and synaptic integrity. Biochim. Biophys. Acta 2013, 1831, 633–641. [Google Scholar] [CrossRef]
- Aloulou, A.; Ali, Y.B.; Bezzine, S.; Gargouri, Y.; Gelb, M.H. Phospholipases: An Overview. In Lipases and Phospholipases: Methods and Protocols; Sandoval, G., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 63–85. [Google Scholar]
- Davis, C.S.; Richardson, R.J. Neurotoxic esterase: Characterization of the solubilized enzyme and the conditions for its solubilization from chicken brain microsomal membranes with ionic, zwitterionic, or nonionic detergents. Biochem. Pharmacol. 1987, 36, 1393–1399. [Google Scholar] [CrossRef]
- Pope, C.N.; Padilla, S. Modulation of neurotoxic esterase activity in vitro by phospholipids. Toxicol. Appl. Pharmacol. 1989, 97, 272–278. [Google Scholar] [CrossRef]
- Vose, S.C.; Fujioka, K.; Gulevich, A.G.; Lin, A.Y.; Holland, N.T.; Casida, J.E. Cellular function of neuropathy target esterase in lysophosphatidylcholine action. Toxicol. Appl. Pharmacol. 2008, 232, 376–383. [Google Scholar] [CrossRef]
- Greiner, A.J.; Richardson, R.J.; Worden, R.M.; Ofoli, R.Y. Influence of lysophospholipid hydrolysis by the catalytic domain of neuropathy target esterase on the fluidity of bilayer lipid membranes. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1533–1539. [Google Scholar] [CrossRef]
- Chang, P.; He, L.; Wang, Y.; Heier, C.; Wu, Y.; Huang, F. Characterization of the interaction of neuropathy target esterase with the endoplasmic reticulum and lipid droplets. Biomolecules 2019, 9, 848. [Google Scholar] [CrossRef] [PubMed]
- Read, D.J.; Langford, L.; Barbour, H.R.; Forshaw, P.J.; Glynn, P. Phospholipase B activity and organophosphorus compound toxicity in cultured neural cells. Toxicol. Appl. Pharmacol. 2007, 219, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, P.; Sun, Y.-J.; Xu, M.-Y.; Wu, Y.-J. Disturbed phospholipid homeostasis in endoplasmic reticulum initiates tri-o-cresyl phosphate-induced delayed neurotoxicity. Sci. Rep. 2016, 6, 37574. [Google Scholar] [CrossRef]
- Hou, W.-Y.; Long, D.-X.; Wang, H.-P.; Wang, Q.; Wu, Y.-J. The homeostasis of phosphatidylcholine and lysophosphatidylcholine was not disrupted during tri-o-cresyl phosphate-induced delayed neurotoxicity in hens. Toxicology 2008, 252, 56–63. [Google Scholar] [CrossRef]
- Hou, W.-Y.; Long, D.-X.; Wu, Y.-J. The homeostasis of phosphatidylcholine and lysophosphatidylcholine in nervous tissues of mice was not disrupted after administration of tri-o-cresyl phosphate. Toxicol. Sci. 2009, 109, 276–285. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tkachuk, M.; Matiytsiv, N. Exposure to an organophosphate tocp drosophila melanogaster flies with altered activity of sws and its micrornas genes. Biopolym. Cell 2019, 35, 390. [Google Scholar]
- Glynn, P.; Holton, J.L.; Nolan, C.C.; Read, D.J.; Brown, L.; Hubbard, A.; Cavanagh, J.B. Neuropathy target esterase: Immunolocalization to neuronal cell bodies and axons. Neuroscience 1998, 83, 295–302. [Google Scholar] [CrossRef]
- Chang, P.-A.; Sun, Q.; Ni, X.-M.; Qv, F.-Q.; Wu, Y.-J.; Song, F.-Z. Molecular cloning and expression analysis of cDNA ends of chicken neuropathy target esterase. Chem. Biol. Interact. 2008, 172, 54–62. [Google Scholar] [CrossRef]
- Chang, P.-A.; Long, D.-X.; Wu, Y.-J.; Sun, Q.; Song, F.-Z. Identification and characterization of chicken neuropathy target esterase. Gene 2009, 435, 45–52. [Google Scholar] [CrossRef]
- Ryabova, E.V.; Melentev, P.A.; Komissarov, A.E.; Surina, N.V.; Ivanova, E.A.; Matiytsiv, N.; Shcherbata, H.R.; Sarantseva, S.V. Morpho-Functional Consequences of Swiss Cheese Knockdown in Glia of Drosophila melanogaster. Cells 2021, 10, 529. [Google Scholar] [CrossRef]
- Li, S.; Yu, X.; Feng, Q. Fat Body Biology in the Last Decade. Annu. Rev. Entomol. 2019, 64, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.; Kühnlein, R.P. Triacylglycerol Metabolism in Drosophila melanogaster. Genetics 2018, 210, 1163–1184. [Google Scholar] [CrossRef]
- Nocelli, R.; Cintra-Socolowski, P.; Roat, T.; Silva-Zacarin, E.; Malaspina, O. Comparative physiology of Malpighian tubules: Form and function. Insect Physiol 2016, 2016, 13–23. [Google Scholar]
- Melentev, P.A.; Sharapenkov, E.G.; Surina, N.V.; Ivanova, E.A.; Ryabova, E.V.; Sarantseva, S.V. Drosophila Lysophospholipase Gene swiss cheese Is Required for Survival and Reproduction. Insects 2022, 13, 14. [Google Scholar] [CrossRef]
- Hufnagel, R.B.; Arno, G.; Hein, N.D.; Hersheson, J.; Prasad, M.; Anderson, Y.; Krueger, L.A.; Gregory, L.C.; Stoetzel, C.; Jaworek, T.J.; et al. Neuropathy target esterase impairments cause Oliver–McFarlane and Laurence–Moon syndromes. J. Med. Genet. 2015, 52, 85. [Google Scholar] [CrossRef]
- McFerrin, J.; Patton, B.L.; Sunderhaus, E.R.; Kretzschmar, D. NTE/PNPLA6 is expressed in mature Schwann cells and is required for glial ensheathment of Remak fibers. Glia 2017, 65, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.; Hunt, J.; Ehrich, M. Inhibition of calcium-stimulated ATPase in the hen brain P2 synaptosomal fraction by organophosphorus esters: Relevance to delayed neuropathy. J. Toxicol. Environ. Health Part A 2001, 63, 101–113. [Google Scholar] [CrossRef]
- Huggins, D.J. Central Nervous System Endogenous Phosphorylation in Organophosphorus Delayed Neurotoxicity. Ph.D. Thesis, University of Michigan, Ann Arbor, Michigan, 1982. [Google Scholar]
- Richardson, R.J.; Hein, N.D.; Wijeyesakere, S.J.; Fink, J.K.; Makhaeva, G.F. Neuropathy target esterase (NTE): Overview and future. Chem. Biol. Interact. 2013, 203, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Masoud, A.; Sandhir, R. Increased oxidative stress is associated with the development of organophosphate-induced delayed neuropathy. Hum. Exp. Toxicol. 2012, 31, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, B.; Padilla, S. Phenylmethylsulfonyl fluoride protects rats from Mipafox-induced delayed neuropathy. Toxicol. Appl. Pharmacol. 1985, 81, 258–264. [Google Scholar] [CrossRef]
- Moretto, A.; Gardiman, G.; Panfilo, S.; Colle, M.-A.; Lock, E.A.; Lotti, M. Effects of S-ethyl hexahydro-1H-azepine-1-carbothioate (molinate) on di-n-butyl dichlorovinyl phosphate (DBDCVP) neuropathy. Toxicol. Sci. 2001, 62, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Heisenberg, M.; Böhl, K. Isolation of anatomical brain mutants of Drosophila by histological means. Z. Für Nat. C 1979, 34, 143–147. [Google Scholar] [CrossRef]
- Sunderhaus, E.R.; Law, A.D.; Kretzschmar, D. ER responses play a key role in Swiss-Cheese/Neuropathy Target Esterase-associated neurodegeneration. Neurobiol. Dis. 2019, 130, 104520. [Google Scholar] [CrossRef] [PubMed]
- Melentev, P.A.; Ryabova, E.V.; Surina, N.V.; Zhmujdina, D.R.; Komissarov, A.E.; Ivanova, E.A.; Boltneva, N.P.; Makhaeva, G.F.; Sliusarenko, M.I.; Yatsenko, A.S. Loss of swiss cheese in neurons contributes to neurodegeneration with mitochondria abnormalities, reactive oxygen species acceleration and accumulation of lipid droplets in Drosophila brain. Int. J. Mol. Sci. 2021, 22, 8275. [Google Scholar] [CrossRef]
- Dutta, S.; Rieche, F.; Eckl, N.; Duch, C.; Kretzschmar, D. Glial expression of Swiss cheese (SWS), the Drosophila orthologue of neuropathy target esterase (NTE), is required for neuronal ensheathment and function. Dis. Models Mech. 2016, 9, 283–294. [Google Scholar] [CrossRef]
- Sujkowski, A.; Rainier, S.; Fink, J.K.; Wessells, R.J. Delayed induction of human NTE (PNPLA6) rescues neurodegeneration and mobility defects of Drosophila swiss cheese (sws) mutants. PLoS ONE 2015, 10, e0145356. [Google Scholar] [CrossRef] [PubMed]
- Sunderhaus, E.R.; Law, A.D.; Kretzschmar, D. Disease-associated PNPLA6 mutations maintain partial functions when analyzed in Drosophila. Front. Neurosci. 2019, 13, 1207. [Google Scholar] [CrossRef]
- Song, Y.; Wang, M.; Mao, F.; Shao, M.; Zhao, B.; Song, Z.; Shao, C.; Gong, Y. Knockdown of Pnpla6 protein results in motor neuron defects in zebrafish. Dis. Models Mech. 2013, 6, 404–413. [Google Scholar] [CrossRef]
- Moser, M.; Li, Y.; Vaupel, K.; Kretzschmar, D.; Kluge, R.; Glynn, P.; Buettner, R. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice. Mol Cell Biol. 2004, 24, 1667–1679. [Google Scholar] [CrossRef]
- Pamies, D.; Vilanova, E.; Sogorb, M.A. Functional pathways altered after silencing Pnpla6 (the codifying gene of neuropathy target esterase) in mouse embryonic stem cells under differentiation. Vitr. Cell. Dev. Biol. Anim. 2014, 50, 261–273. [Google Scholar] [CrossRef]
- Pamies, D.; Reig, J.A.; Vilanova, E.; Sogorb, M.A. Expression of neuropathy target esterase in mouse embryonic stem cells during differentiation. Arch. Toxicol. 2010, 84, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Sogorb, M.A.; Pamies, D.; Estevan, C.; Estévez, J.; Vilanova, E. Roles of NTE protein and encoding gene in development and neurodevelopmental toxicity. Chem. Biol. Interact. 2016, 259, 352–357. [Google Scholar] [CrossRef]
- Pamies, D.; Bal-Price, A.; Fabbri, M.; Gribaldo, L.; Scelfo, B.; Harris, G.; Collotta, A.; Vilanova, E.; Sogorb, M. Silencing of PNPLA6, the neuropathy target esterase (NTE) codifying gene, alters neurodifferentiation of human embryonal carcinoma stem cells (NT2). Neuroscience 2014, 281, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Sogorb, M.A.; Fuster, E.; Del Río, E.; Estévez, J.; Vilanova, E. Effects of mipafox, paraoxon, chlorpyrifos and its metabolite chlorpyrifos-oxon on the expression of biomarker genes of differentiation in D3 mouse embryonic stem cells. Chem. Biol. Interact. 2016, 259, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Winrow, C.J.; Hemming, M.L.; Allen, D.M.; Quistad, G.B.; Casida, J.E.; Barlow, C. Loss of neuropathy target esterase in mice links organophosphate exposure to hyperactivity. Nat Genet 2003, 33, 477–485. [Google Scholar] [CrossRef]
- Akassoglou, K.; Malester, B.; Xu, J.; Tessarollo, L.; Rosenbluth, J.; Chao, M.V. Brain-specific deletion of neuropathy target esterase/swisscheese results in neurodegeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 5075–5080. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-X.; Xu, L.-L.; Mei, J.-H.; Yu, X.-B.; Kuang, H.-B.; Liu, H.-Y.; Wu, Y.-J.; Wang, J.-L. Involvement of neuropathy target esterase in tri-ortho-cresyl phosphate-induced testicular spermatogenesis failure and growth inhibition of spermatogonial stem cells in mice. Toxicol. Lett. 2012, 211, 54–61. [Google Scholar] [CrossRef]
- Rainier, S.; Bui, M.; Mark, E.; Thomas, D.; Tokarz, D.; Ming, L.; Delaney, C.; Richardson, R.J.; Albers, J.W.; Matsunami, N.; et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am. J. Hum. Genet. 2008, 82, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Gonzalez, M.A.; Lourenco, C.M.; Coutelier, M.; Haack, T.B.; Rebelo, A.; Hannequin, D.; Strom, T.M.; Prokisch, H.; Kernstock, C.; et al. PNPLA6 mutations cause Boucher-Neuhäuser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014, 137, 69–77. [Google Scholar] [CrossRef]
- Suchowersky, O.; Ashtiani, S.; Au, P.-Y.B.; McLeod, S.; Estiar, M.A.; Gan-Or, Z.; Rouleau, G.A. Hereditary spastic paraplegia initially diagnosed as cerebral palsy. Clin. Parkinsonism Relat. Disord. 2021, 5, 100114. [Google Scholar] [CrossRef] [PubMed]
- Sen, K.; Finau, M.; Ghosh, P. Bi-allelic variants in PNPLA6 possibly associated with Parkinsonian features in addition to spastic paraplegia phenotype. J. Neurol. 2020, 267, 2749–2753. [Google Scholar] [CrossRef] [PubMed]
- Nanetti, L.; Di, D.B.; Magri, S.; Fichera, M.; Sarto, E.; Castaldo, A.; Mongelli, A.; Baratta, S.; Fenu, S.; Moscatelli, M. Multifaceted and Age-Dependent Phenotypes Associated With Biallelic PNPLA6 Gene Variants: Eight Novel Cases and Review of the Literature. Front. Neurol. 2021, 12, 793547. [Google Scholar] [CrossRef] [PubMed]
- Tarnutzer, A.A.; Gerth-Kahlert, C.; Timmann, D.; Chang, D.I.; Harmuth, F.; Bauer, P.; Straumann, D.; Synofzik, M. Boucher-Neuhauser syndrome: Cerebellar degeneration, chorioretinal dystrophy and hypogonadotropic hypogonadism: Two novel cases and a review of 40 cases from the literature. J. Neurol. 2015, 262, 194–202. [Google Scholar] [CrossRef] [PubMed]
- DeNaro, B.B.; Dhrami-Gavazi, E.; Rubaltelli, D.M.; Freund, K.B.; Lee, W.; Yannuzzi, L.A.; Tsang, S.H.; Kang, J.J. Chorioretinal changes in a genetically confirmed case of Boucher-Neuhäuser syndrome. Retin. Cases Brief Rep. 2018, 40, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Doğan, M.; Eröz, R.; Öztürk, E. Chorioretinal dystrophy, hypogonadotropic hypogonadism, and cerebellar ataxia: Boucher-Neuhauser syndrome due to a homozygous (c. 3524C> G (p. Ser1175Cys)) variant in PNPLA6 gene. Ophthalmic Genet. 2021, 42, 276–282. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, E.; Serrano, L.; Scoles, D.; Cunningham, K.E.; Han, G.; Chiang, J.; Bennett, J.; Aleman, T.S. Detailed retinal phenotype of Boucher-Neuhäuser syndrome associated with mutations in PNPLA6 mimicking choroideremia. Ophthalmic Genet. 2019, 40, 267–275. [Google Scholar] [CrossRef]
- Teive, H.A.G.; Camargo, C.H.F.; Sato, M.T.; Shiokawa, N.; Boguszewski, C.L.; Raskin, S.; Buck, C.; Seminara, S.B.; Munhoz, R.P. Different cerebellar ataxia phenotypes associated with mutations of the PNPLA6 gene in Brazilian patients with recessive ataxias. Cerebellum 2018, 17, 380–385. [Google Scholar] [CrossRef]
- Koh, K.; Kobayashi, F.; Miwa, M.; Shindo, K.; Isozaki, E.; Ishiura, H.; Tsuji, S.; Takiyama, Y. Novel mutations in the PNPLA6 gene in Boucher-Neuhäuser syndrome. J. Hum. Genet. 2015, 60, 217–220. [Google Scholar] [CrossRef]
- Langdahl, J.H.; Frederiksen, A.L.; Nguyen, N.; Brusgaard, K.; Juhl, C.B. Boucher Neuhäuser Syndrome–a rare cause of inherited hypogonadotropic hypogonadism. A case of two adult siblings with two novel mutations in PNPLA6. Eur. J. Med. Genet. 2017, 60, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Zhao, Y.; Wu, J.; Wang, Y.; Liu, J.L.; Zhou, Z.L.; Zhou, X.T.; Chen, D.N.; Liao, W.H.; Li, J.D. A novel PNPLA6 compound heterozygous mutation identified in a Chinese patient with Boucher-Neuhäuser syndrome. Mol. Med. Rep. 2018, 18, 261–267. [Google Scholar] [PubMed]
- Deik, A.; Johannes, B.; Rucker, J.C.; Sánchez, E.; Brodie, S.E.; Deegan, E.; Landy, K.; Kajiwara, Y.; Scelsa, S.; Saunders-Pullman, R.; et al. Compound heterozygous PNPLA6 mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia. J. Neurol. 2014, 261, 2411–2423. [Google Scholar] [CrossRef]
- Chung, E.J.; You, E.; Oh, S.H.; Seo, G.H.; Chung, W.Y.; Kim, Y.J.; Kim, S.J. The First Korean Family with Boucher-Neuhäuser Syndrome Carrying a Novel Mutation in PNPLA6. J. Clin. Neurol. 2022, 18, 233–234. [Google Scholar] [CrossRef]
- He, J.; Liu, X.; Liu, L.; Zeng, S.; Shan, S.; Liao, Z. Identification of Novel Compound Heterozygous Variants of the PNPLA6 Gene in Boucher-Neuhäuser Syndrome. Front. Genet. 2022, 13, 810537. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, A.K.; Lomniczi, A.; Kretzschmar, D.; Dissen, G.A.; Kotan, L.D.; McArdle, C.A.; Koc, A.F.; Hamel, B.C.; Guclu, M.; Papatya, E.D.; et al. Loss-of-function mutations in PNPLA6 encoding neuropathy target esterase underlie pubertal failure and neurological deficits in Gordon Holmes syndrome. J. Clin. Endocrinol. Metab. 2014, 99, E2067–E2075. [Google Scholar] [CrossRef] [PubMed]
- Salgado, P.; Carvalho, R.; Brandão, A.F.; Jorge, P.; Ramos, C.; Dias, D.; Alonso, I.; Magalhães, M. Gordon Holmes syndrome due to compound heterozygosity of two new PNPLA6 variants–A diagnostic challenge. eNeurologicalSci 2019, 14, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Locci, S.; Bianchi, S.; Tessa, A.; Santorelli, F.M.; Mignarri, A. Gordon Holmes syndrome caused by two novel mutations in the PNPLA6 gene. Clin. Neurol. Neurosurg. 2021, 106763. [Google Scholar] [CrossRef] [PubMed]
- Emekli, A.S.; Samanci, B.; Şimşir, G.; Hanagasi, H.A.; Gürvit, H.; Bilgiç, B.; Başak, A.N. A novel PNPLA6 mutation in a Turkish family with intractable Holmes tremor and spastic ataxia. Neurol. Sci. 2021, 42, 1535–1539. [Google Scholar] [CrossRef]
- Liu, F.; Ji, Y.; Li, G.; Xu, C.; Sun, Y. Identification of Oliver-McFarlane syndrome caused by novel compound heterozygous variants of PNPLA6. Gene 2020, 761, 145027. [Google Scholar] [CrossRef] [PubMed]
- Patsi, O.; De Beaufort, C.; Kerschen, P.; Cardillo, S.; Soehn, A.; Rautenberg, M.; Diederich, N. A new PNPLA6 mutation presenting as Oliver McFarlane syndrome. J. Neurol. Sci. 2018, 392, 1–2. [Google Scholar] [CrossRef]
- Lisbjerg, K.; Andersen, M.K.G.; Bertelsen, M.; Brost, A.G.; Buchvald, F.F.; Jensen, R.B.; Bisgaard, A.-M.; Rosenberg, T.; Tümer, Z.; Kessel, L. Oliver McFarlane syndrome: Two new cases and a review of the literature. Ophthalmic Genet. 2021, 42, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Cassar, M.; Sunderhaus, E.; Wentzell, J.S.; Kuntz, S.; Strauss, R.; Kretzschmar, D. The PKA-C3 catalytic subunit is required in two pairs of interneurons for successful mating of Drosophila. Sci. Rep. 2018, 8, 2458. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, Q.; Alberts, I.; Li, X. PRKX, a Novel cAMP-Dependent Protein Kinase Member, Plays an Important Role in Development. J. Cell. Biochem. 2016, 117, 566–573. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ataxia | Hypogonadism/ Delay in Sexual Development | Chorioretinal Dystrophy | Trichomegaly/ Alopecia | Spastic Paraplegia | Intellectual Disabilities | Dwarfism/ Short Statue | |
|---|---|---|---|---|---|---|---|
| SPG 39 | +/− | + | |||||
| Boucher–Neuhäuser | + | + | +/- | +/− | +/− mild | ||
| Gordon Holmes | + | + | +/− | +/− mild | |||
| Oliver–McFarlane | +/− | + | + severe | + | +/− | + | + |
| Laurence–Moon | +/− | + | + | +/− | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretzschmar, D. PNPLA6/NTE, an Evolutionary Conserved Phospholipase Linked to a Group of Complex Human Diseases. Metabolites 2022, 12, 284. https://doi.org/10.3390/metabo12040284
Kretzschmar D. PNPLA6/NTE, an Evolutionary Conserved Phospholipase Linked to a Group of Complex Human Diseases. Metabolites. 2022; 12(4):284. https://doi.org/10.3390/metabo12040284
Chicago/Turabian StyleKretzschmar, Doris. 2022. "PNPLA6/NTE, an Evolutionary Conserved Phospholipase Linked to a Group of Complex Human Diseases" Metabolites 12, no. 4: 284. https://doi.org/10.3390/metabo12040284
APA StyleKretzschmar, D. (2022). PNPLA6/NTE, an Evolutionary Conserved Phospholipase Linked to a Group of Complex Human Diseases. Metabolites, 12(4), 284. https://doi.org/10.3390/metabo12040284