
Correlating Mass Spectrometry Imaging and Liquid Chromatography-Tandem Mass Spectrometry for Tissue-Based Pharmacokinetic Studies

 ,
,  ,
,
Abstract
:1. Introduction
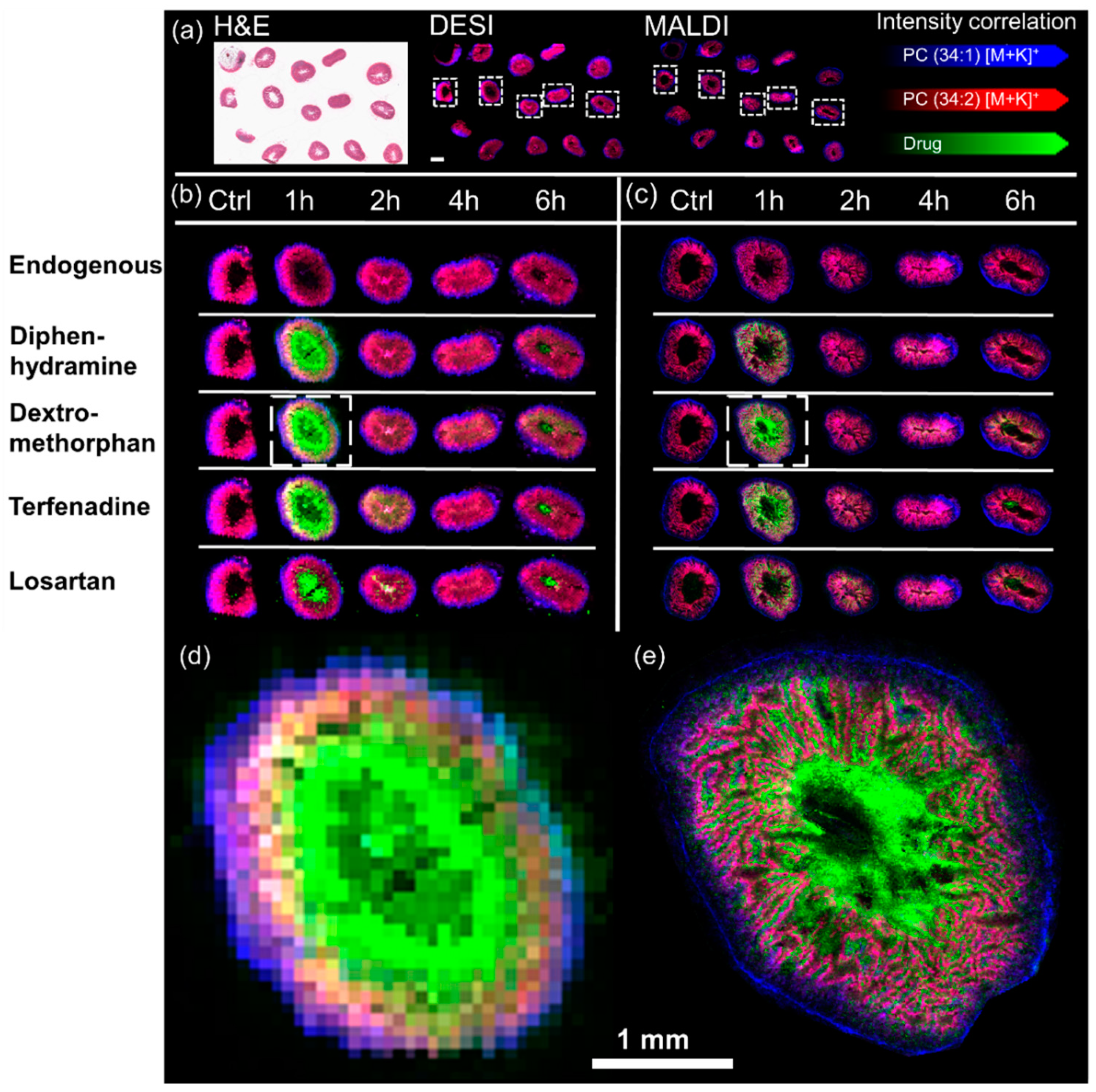
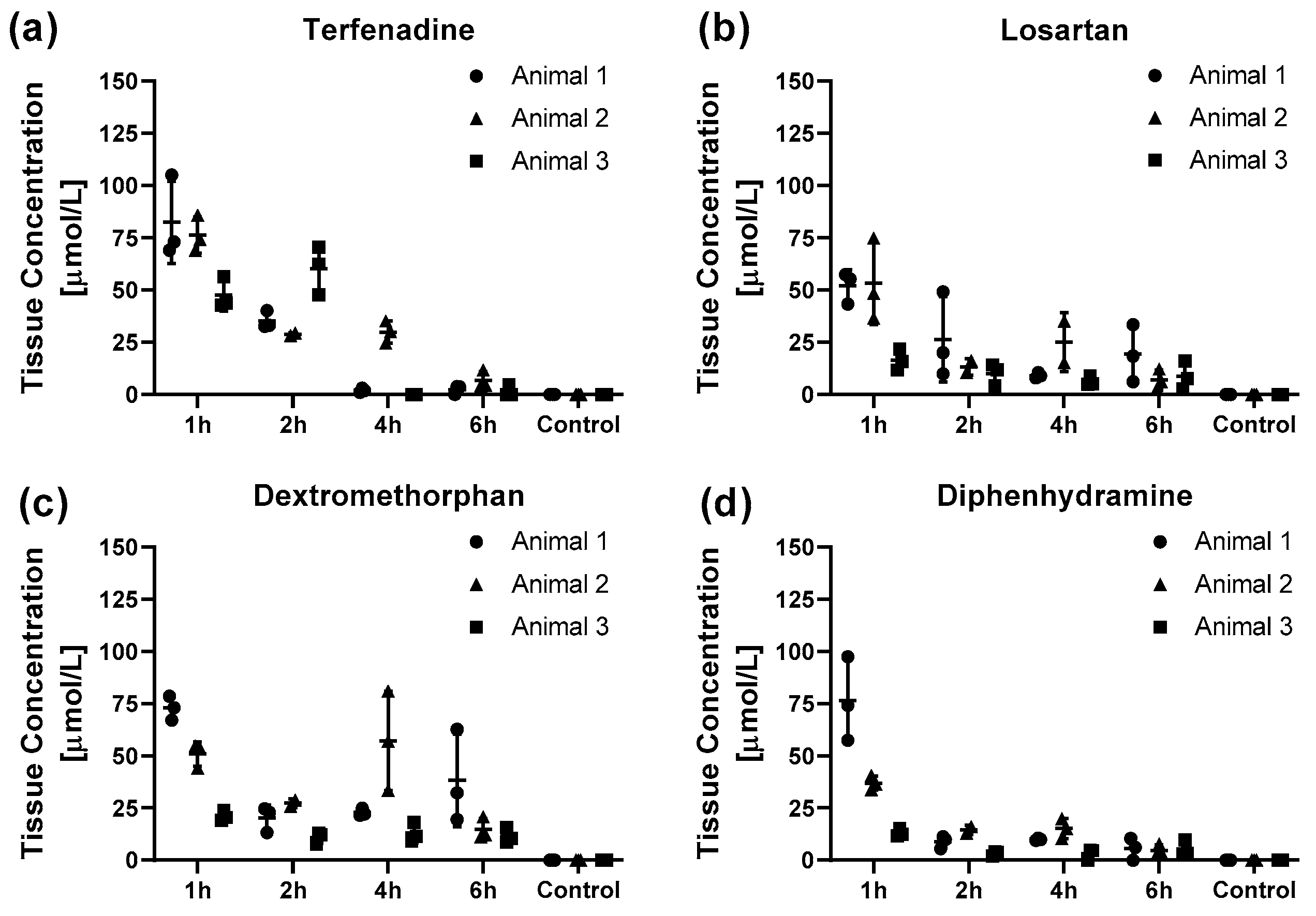
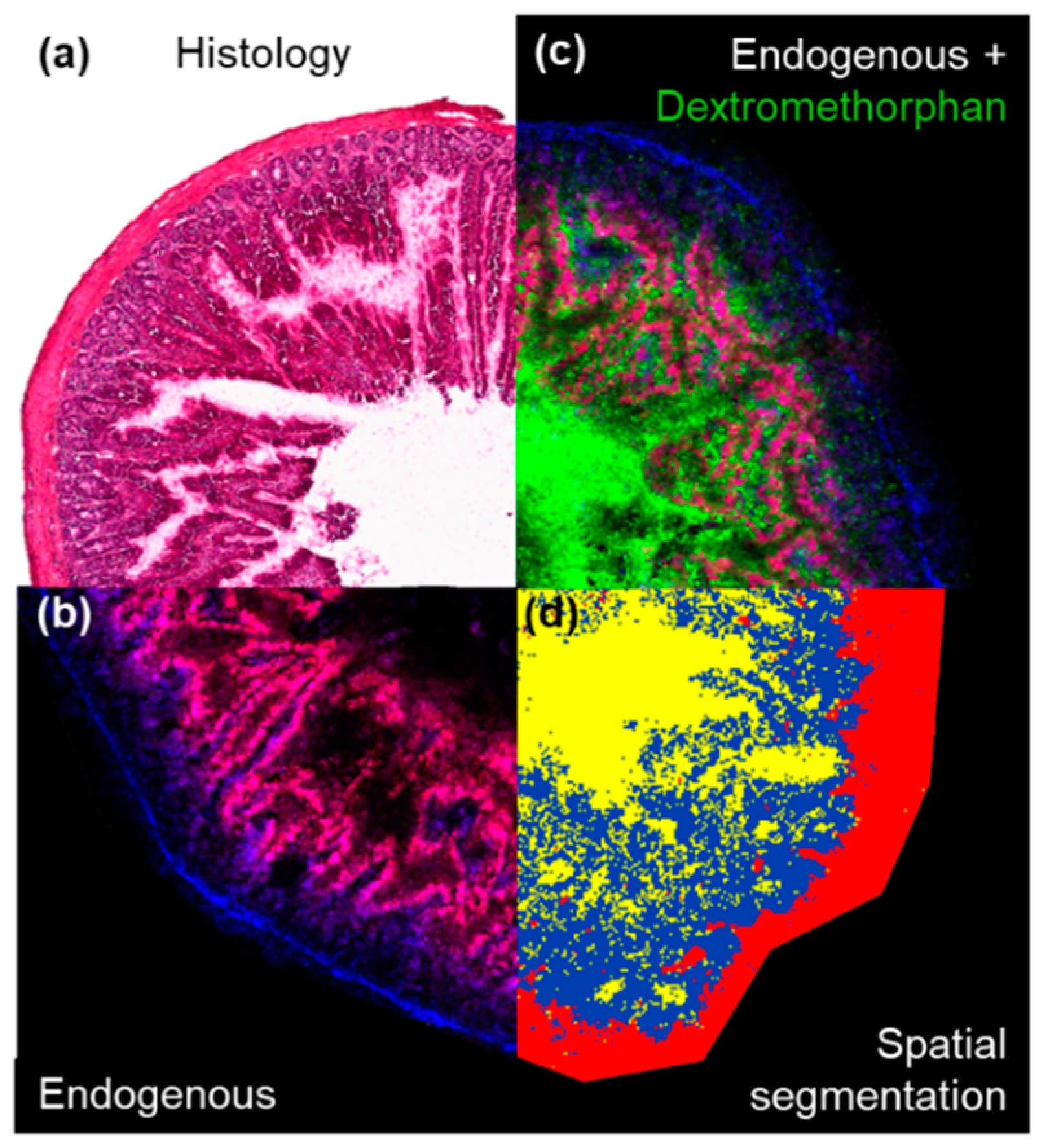
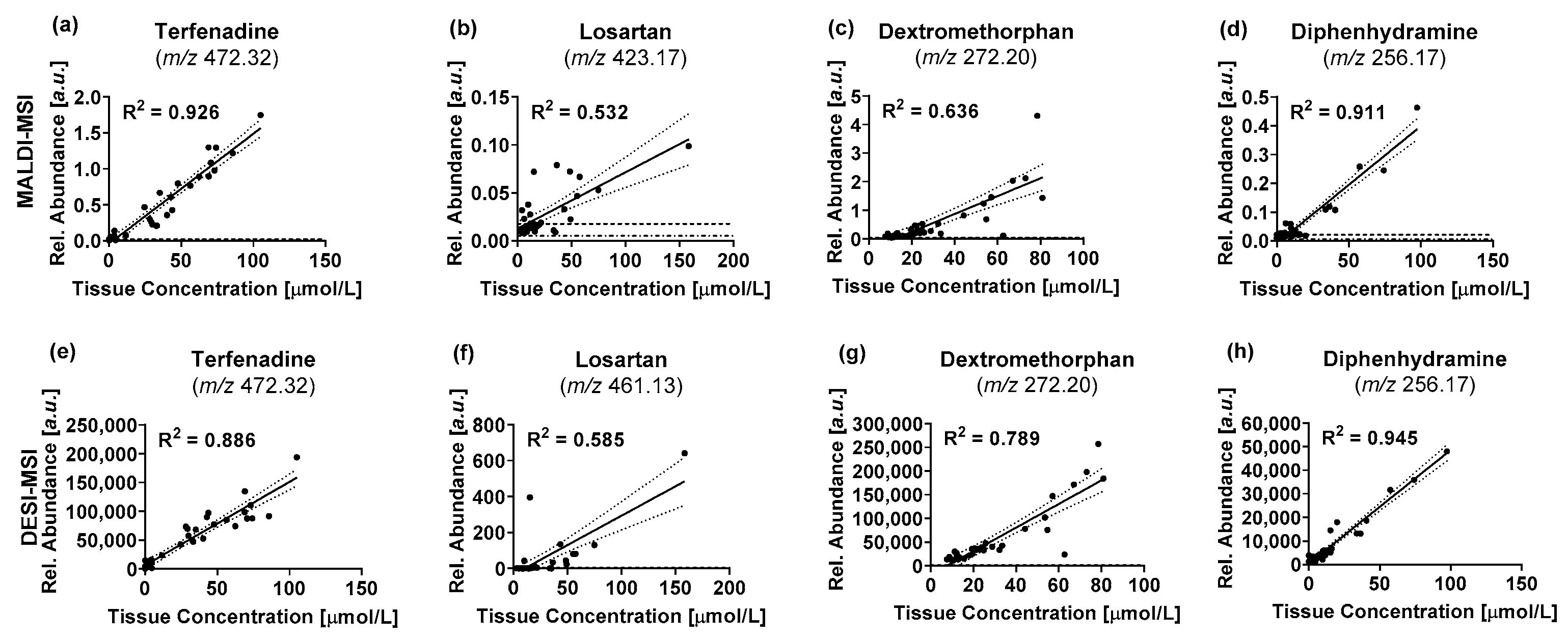
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animals and Dosing
4.3. Preparation of Intestine Specimens for MSI Analysis
4.4. DESI-MSI
4.5. MALDI-MSI
4.6. LC-MS/MS Sample Preparation and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patterson, K.B.; Prince, H.A.; Kraft, E.; Jenkins, A.J.; Shaheen, N.J.; Rooney, J.F.; Cohen, M.S.; Kashuba, A.D. Penetration of tenofovir and emtricitabine in mucosal tissues: Implications for prevention of HIV-1 transmission. Sci. Transl. Med. 2011, 3, 112re114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prathipati, P.K.; Mandal, S.; Destache, C.J. Simultaneous quantification of tenofovir, emtricitabine, rilpivirine, elvitegravir and dolutegravir in mouse biological matrices by LC-MS/MS and its application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2016, 129, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, N.; Roy, U.; Balkundi, S.; Puligujja, P.; Guo, D.; Smith, N.; Liu, X.M.; Lamberty, B.; Morsey, B.; Fox, H.S.; et al. Preclinical pharmacokinetics and tissue distribution of long-acting nanoformulated antiretroviral therapy. Antimicrob. Agents Chemother. 2013, 57, 3110–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groseclose, M.R.; Castellino, S. A mimetic tissue model for the quantification of drug distributions by MALDI imaging mass spectrometry. Anal. Chem. 2013, 85, 10099–10106. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.; Fehniger, T.E.; Gustavsson, L.; Andersson, M.; Kenne, K.; Marko-Varga, G.; Andren, P.E. Fine mapping the spatial distribution and concentration of unlabeled drugs within tissue micro-compartments using imaging mass spectrometry. PLoS ONE 2010, 5, e11411. [Google Scholar] [CrossRef]
- Prentice, B.M.; Chumbley, C.W.; Caprioli, R.M. Absolute Quantification of Rifampicin by MALDI Imaging Mass Spectrometry Using Multiple TOF/TOF Events in a Single Laser Shot. J. Am. Soc. Mass Spectrom. 2017, 28, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Pirman, D.A.; Reich, R.F.; Kiss, A.; Heeren, R.M.; Yost, R.A. Quantitative MALDI tandem mass spectrometric imaging of cocaine from brain tissue with a deuterated internal standard. Anal. Chem. 2013, 85, 1081–1089. [Google Scholar] [CrossRef]
- Huizing, L.R.S.; McDuffie, J.; Cuyckens, F.; van Heerden, M.; Koudriakova, T.; Heeren, R.M.A.; Vreeken, R.J. Quantitative Mass Spectrometry Imaging to Study Drug Distribution in the Intestine Following Oral Dosing. Anal. Chem. 2021, 93, 2144–2151. [Google Scholar] [CrossRef]
- Groseclose, M.R.; Laffan, S.B.; Frazier, K.S.; Hughes-Earle, A.; Castellino, S. Imaging MS in Toxicology: An Investigation of Juvenile Rat Nephrotoxicity Associated with Dabrafenib Administration. J. Am. Soc. Mass Spectrom. 2015, 26, 887–898. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, L.A.; Corthals, G.L.; Willems, S.M.; van Remoortere, A.; van Zeijl, R.J.; Deelder, A.M. Peptide and protein imaging mass spectrometry in cancer research. J. Proteom. 2010, 73, 1921–1944. [Google Scholar] [CrossRef]
- Kompauer, M.; Heiles, S.; Spengler, B. Atmospheric pressure MALDI mass spectrometry imaging of tissues and cells at 1.4-mum lateral resolution. Nat. Methods 2017, 14, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, M.; Iwamoto, N.; Kawaguchi-Sakita, N.; Sugimoto, M.; Ueno, T.; Mikami, Y.; Terasawa, K.; Sato, T.A.; Tanaka, K.; Shimizu, K.; et al. High-resolution imaging mass spectrometry reveals detailed spatial distribution of phosphatidylinositols in human breast cancer. Cancer Sci. 2013, 104, 1372–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavalin, A.; Todd, E.M.; Rawhouser, P.D.; Yang, J.; Norris, J.L.; Caprioli, R.M. Direct imaging of single cells and tissue at sub-cellular spatial resolution using transmission geometry MALDI MS. J. Mass Spectrom. 2012, 47, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ide, J.L.; Norton, I.; Marchionni, M.A.; Ebling, M.C.; Wang, L.Y.; Davis, E.; Sauvageot, C.M.; Kesari, S.; Kellersberger, K.A.; et al. Molecular imaging of drug transit through the blood-brain barrier with MALDI mass spectrometry imaging. Sci. Rep. 2013, 3, 2859. [Google Scholar] [CrossRef]
- Nilsson, A.; Peric, A.; Strimfors, M.; Goodwin, R.J.A.; Hayes, M.A.; Andren, P.E.; Hilgendorf, C. Mass Spectrometry Imaging proves differential absorption profiles of well-characterised permeability markers along the crypt-villus axis. Sci. Rep. 2017, 7, 6352. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, M.; Lestner, J.; Prideaux, B.; O’Brien, P.; Dias-Freedman, I.; Chen, C.; Dietzold, J.; Daudelin, I.; Kaya, F.; Blanc, L.; et al. Ethambutol Partitioning in Tuberculous Pulmonary Lesions Explains Its Clinical Efficacy. Antimicrob. Agents Chemother. 2017, 61, e00924-17. [Google Scholar] [CrossRef] [Green Version]
- Dilillo, M.; Pellegrini, D.; Ait-Belkacem, R.; de Graaf, E.L.; Caleo, M.; McDonnell, L.A. Mass Spectrometry Imaging, Laser Capture Microdissection, and LC-MS/MS of the Same Tissue Section. J. Proteome Res. 2017, 16, 2993–3001. [Google Scholar] [CrossRef]
- Stiehl, D.P.; Tritto, E.; Chibout, S.-D.; Cordier, A.; Moulin, P. The Utility of Gene Expression Profiling from Tissue Samples to Support Drug Safety Assessments. ILAR J. 2017, 58, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Hansen, H.T.; Janfelt, C. Aspects of Quantitation in Mass Spectrometry Imaging Investigated on Cryo-Sections of Spiked Tissue Homogenates. Anal. Chem. 2016, 88, 11513–11520. [Google Scholar] [CrossRef]
- Pirman, D.A.; Kiss, A.; Heeren, R.M.A.; Yost, R.A. Identifying Tissue-Specific Signal Variation in MALDI Mass Spectrometric Imaging by Use of an Internal Standard. Anal. Chem. 2013, 85, 1090–1096. [Google Scholar] [CrossRef]
- Dannhorn, A.; Kazanc, E.; Ling, S.; Nikula, C.; Karali, E.; Serra, M.P.; Vorng, J.-L.; Inglese, P.; Maglennon, G.; Hamm, G.; et al. Universal Sample Preparation Unlocking Multimodal Molecular Tissue Imaging. Anal. Chem. 2020, 92, 11080–11088. [Google Scholar] [CrossRef] [PubMed]
- McEwen, A.B.; Henson, C.M.; Wood, S.G. Quantitative whole-body autoradiography, LC–MS/MS and MALDI for drug-distribution studies in biological samples: The ultimate matrix trilogy. Bioanalysis 2014, 6, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, K.; Maxwell, B.; Grossman, S.J.; Tremaine, L.M.; Humphreys, W.G.; Zhang, D. Tissue Distribution and Elimination of [14C]Apixaban in Rats. Drug Metab. Dispos. 2011, 39, 256. [Google Scholar] [CrossRef] [PubMed]
- Panee, J. Potential Medicinal Application and Toxicity Evaluation of Extracts from Bamboo Plants. J. Med. Plant Res. 2015, 9, 681–692. [Google Scholar] [PubMed] [Green Version]
- Deininger, S.-O.; Ebert, M.P.; Fütterer, A.; Gerhard, M.; Röcken, C. MALDI Imaging Combined with Hierarchical Clustering as a New Tool for the Interpretation of Complex Human Cancers. J. Proteome Res. 2008, 7, 5230–5236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, T.; Becker, M.; Deininger, S.-O.; Ernst, G.; Wehder, L.; Grasmair, M.; von Eggeling, F.; Thiele, H.; Maass, P. Spatial Segmentation of Imaging Mass Spectrometry Data with Edge-Preserving Image Denoising and Clustering. J. Proteome Res. 2010, 9, 6535–6546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Campeneere, D.; Baurain, R.; Slachmuylder-Otte, C.; Trouet, A. Immunological evaluation of blood contamination in tissue distribution studies. Pharm. Res. 1989, 21, 19–26. [Google Scholar] [CrossRef]
- Neubert, H.; Fountain, S.; King, L.; Clark, T.; Weng, Y.; O’Hara, D.M.; Li, W.; Leung, S.; Ray, C.; Palandra, J.; et al. Tissue bioanalysis of biotherapeutics and drug targets to support PK/PD. Bioanalysis 2012, 4, 2589–2604. [Google Scholar] [CrossRef]
- Swales, J.G.; Dexter, A.; Hamm, G.; Nilsson, A.; Strittmatter, N.; Michopoulos, F.; Hardy, C.; Morentin-Gutierrez, P.; Mellor, M.; Andren, P.E.; et al. Quantitation of Endogenous Metabolites in Mouse Tumors Using Mass-Spectrometry Imaging. Anal. Chem. 2018, 90, 6051–6058. [Google Scholar] [CrossRef] [Green Version]
- Takats, Z.; Wiseman, J.M.; Gologan, B.; Cooks, R.G. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 2004, 306, 471–473. [Google Scholar] [CrossRef] [Green Version]
- Adusumilli, R.; Mallick, P. Data Conversion with ProteoWizard msConvert. Methods Mol. Biol. 2017, 1550, 339–368. [Google Scholar] [CrossRef] [PubMed]
- Race, A.M.; Styles, I.B.; Bunch, J. Inclusive sharing of mass spectrometry imaging data requires a converter for all. J. Proteom. 2012, 75, 5111–5112. [Google Scholar] [CrossRef] [PubMed]
- Swales, J.G.; Tucker, J.W.; Strittmatter, N.; Nilsson, A.; Cobice, D.; Clench, M.R.; Mackay, C.L.; Andren, P.E.; Takats, Z.; Webborn, P.J.; et al. Mass spectrometry imaging of cassette-dosed drugs for higher throughput pharmacokinetic and biodistribution analysis. Anal. Chem. 2014, 86, 8473–8480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norris, J.L.; Cornett, D.S.; Mobley, J.A.; Andersson, M.; Seeley, E.H.; Chaurand, P.; Caprioli, R.M. Processing MALDI Mass Spectra to Improve Mass Spectral Direct Tissue Analysis. Int. J. Mass Spectrom. 2007, 260, 212–221. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Absorbed Drug Fraction [%] | |||
|---|---|---|---|
| Drug | Mean ± SD | Min | Max |
| Terfenadine | 56 ± 23 | 8 | 89 |
| Losartan | 66 ± 24 | 12 | 93 |
| Dextromethorphan | 57 ± 22 | 10 | 92 |
| Diphenhydramine | 64 ± 21 | 18 | 93 |
| Quantifier Transition | Qualifier Transition | |
|---|---|---|
| Diphenhydramine | 256.3 > 167.0 | 256.3 > 165.0 |
| Diphenhydramine-d3 | 259.3 > 167.0 | 259.3 > 165.1 |
| Dextromethorphan | 272.4 > 147.0 | 272.4 > 215.1 |
| Dextromethorphan-d3 | 275.4 > 215.1 | 275.4 > 147.0 |
| Losartan | 423.5 > 207.0 | 423.5 > 179.9 |
| Losartan-d4 | 427.5 > 211.1 | 427.5 > 184.0 |
| Terfenadine | 472.1 > 91.0 | 472.7 > 436.3 |
| Terfenadine-d3 | 475.7 > 91.0 | 475.7 > 438.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dannhorn, A.; Kazanc, E.; Hamm, G.; Swales, J.G.; Strittmatter, N.; Maglennon, G.; Goodwin, R.J.A.; Takats, Z. Correlating Mass Spectrometry Imaging and Liquid Chromatography-Tandem Mass Spectrometry for Tissue-Based Pharmacokinetic Studies. Metabolites 2022, 12, 261. https://doi.org/10.3390/metabo12030261
Dannhorn A, Kazanc E, Hamm G, Swales JG, Strittmatter N, Maglennon G, Goodwin RJA, Takats Z. Correlating Mass Spectrometry Imaging and Liquid Chromatography-Tandem Mass Spectrometry for Tissue-Based Pharmacokinetic Studies. Metabolites. 2022; 12(3):261. https://doi.org/10.3390/metabo12030261
Chicago/Turabian StyleDannhorn, Andreas, Emine Kazanc, Gregory Hamm, John G. Swales, Nicole Strittmatter, Gareth Maglennon, Richard J. A. Goodwin, and Zoltan Takats. 2022. "Correlating Mass Spectrometry Imaging and Liquid Chromatography-Tandem Mass Spectrometry for Tissue-Based Pharmacokinetic Studies" Metabolites 12, no. 3: 261. https://doi.org/10.3390/metabo12030261
APA StyleDannhorn, A., Kazanc, E., Hamm, G., Swales, J. G., Strittmatter, N., Maglennon, G., Goodwin, R. J. A., & Takats, Z. (2022). Correlating Mass Spectrometry Imaging and Liquid Chromatography-Tandem Mass Spectrometry for Tissue-Based Pharmacokinetic Studies. Metabolites, 12(3), 261. https://doi.org/10.3390/metabo12030261