
Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology

Abstract
1. Introduction
2. Results
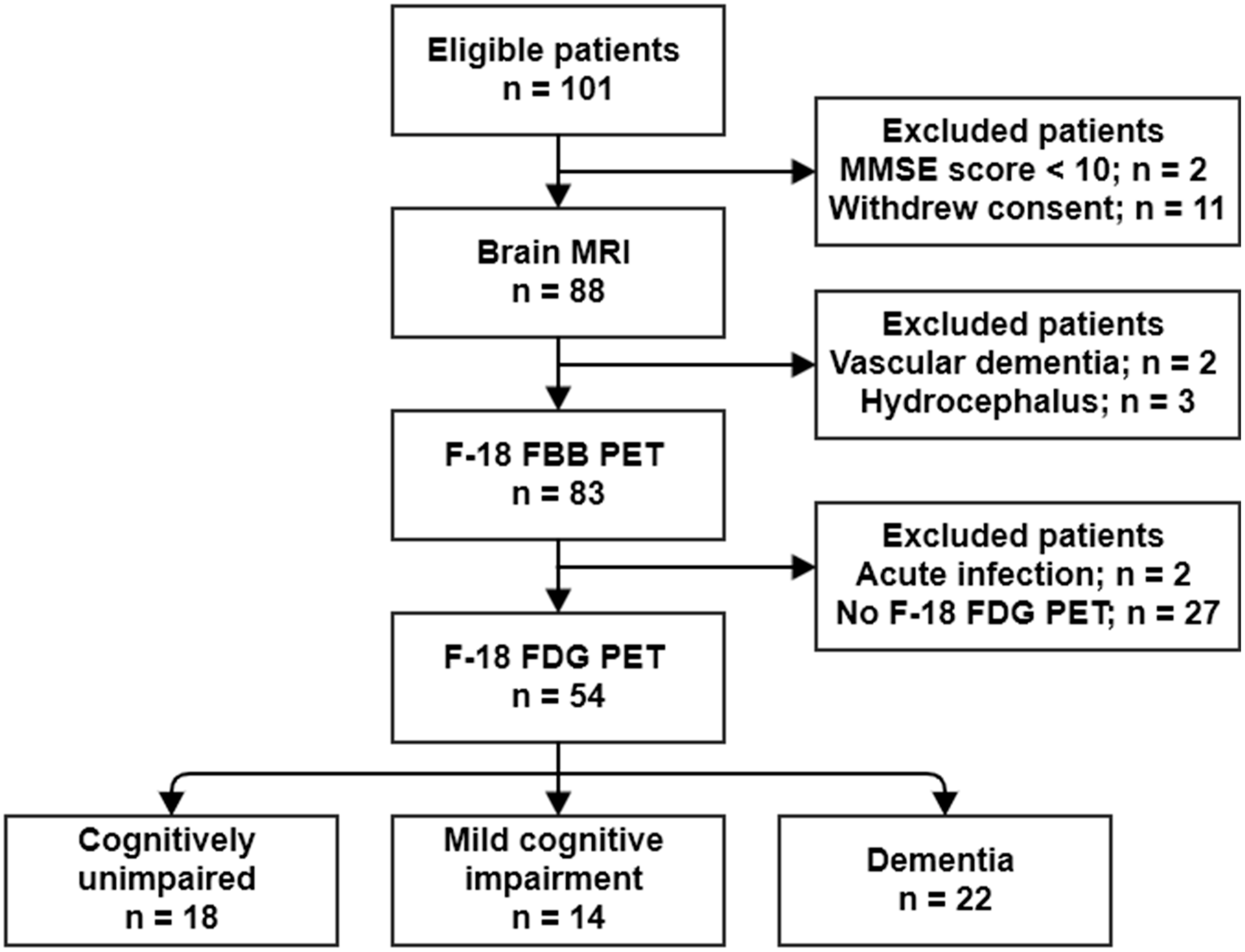
2.1. Population Characteristics
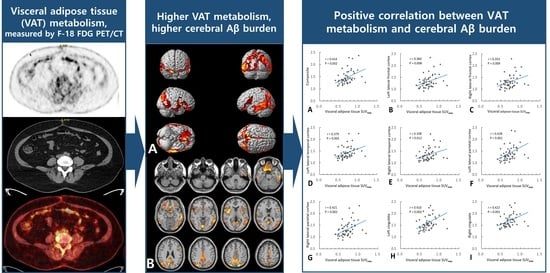
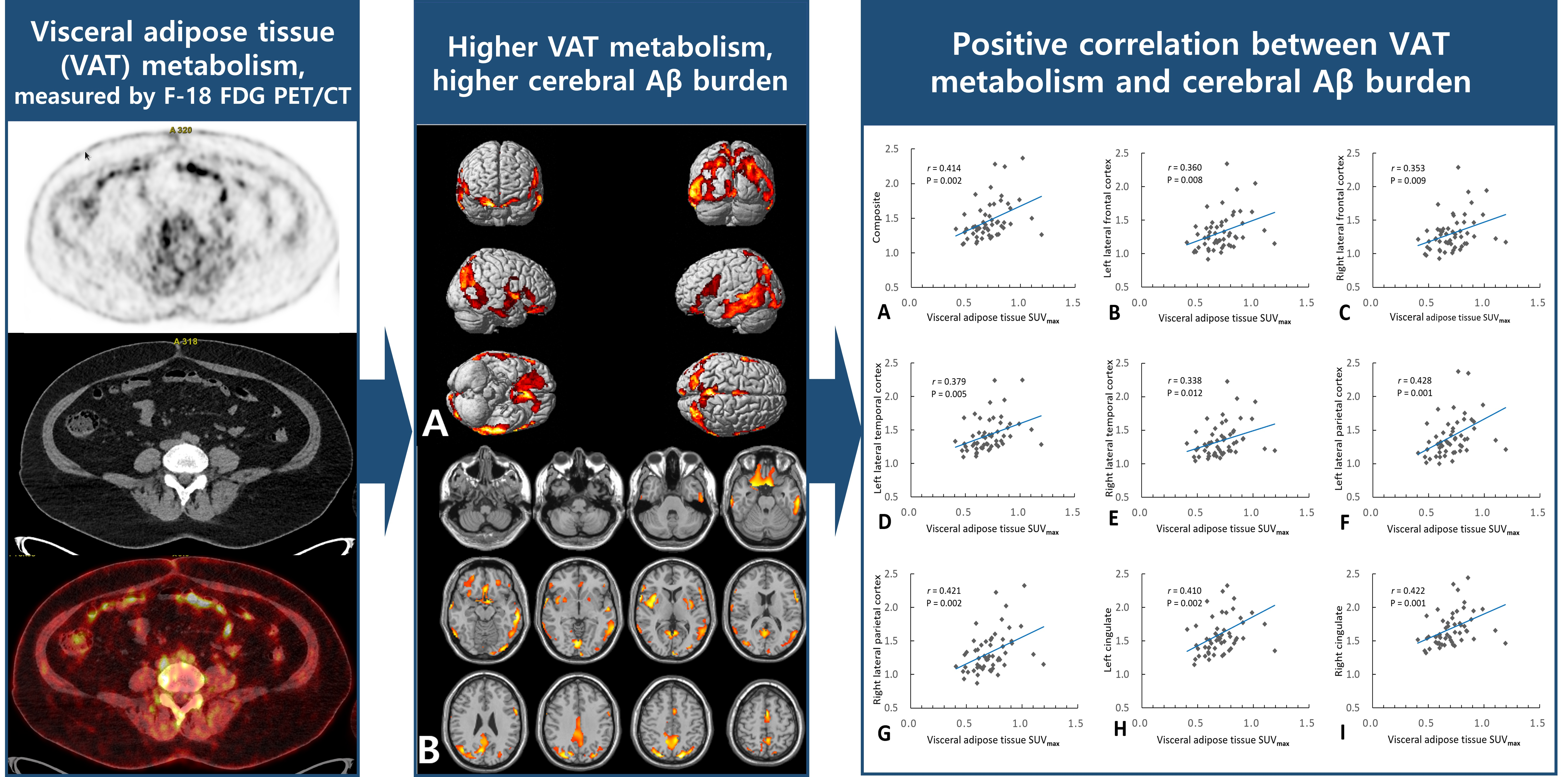
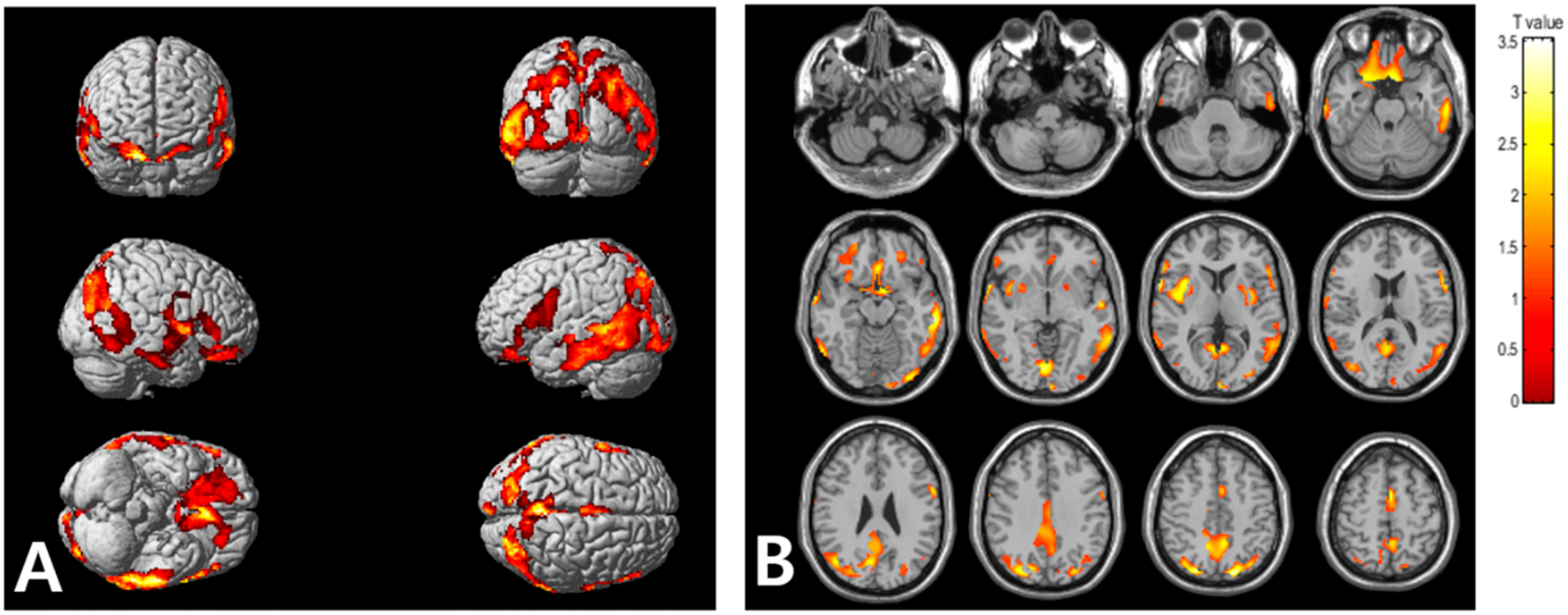
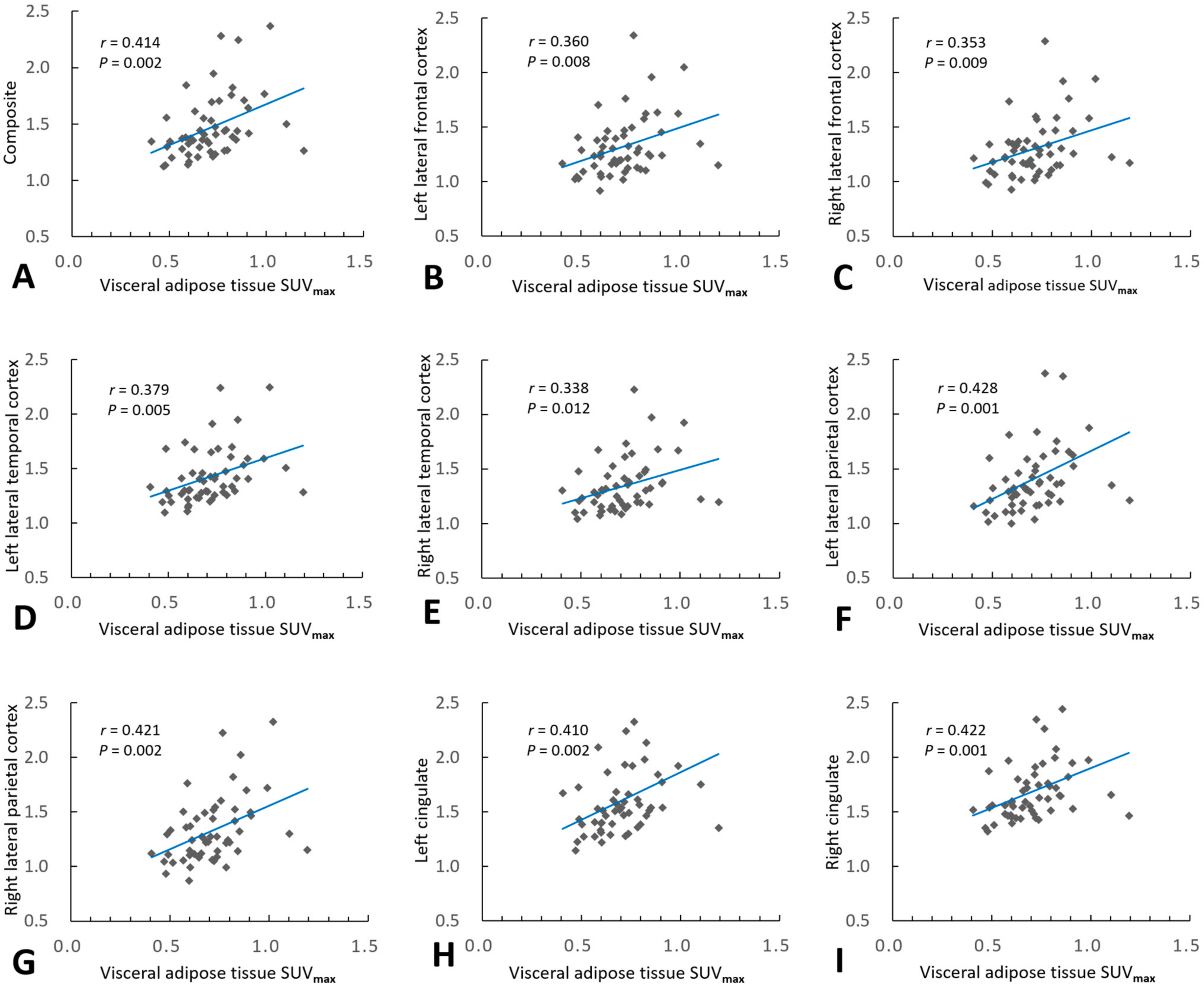
2.2. Association of VAT Metabolism with AD Pathology
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Brain MRI
4.3. 18F-FDG PET
4.4. 18F-FBB PET
4.5. Voxel-Based Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Kiliaan, A.J.; Arnoldussen, I.A.; Gustafson, D.R. Adipokines: A link between obesity and dementia? Lancet Neurol. 2014, 13, 913–923. [Google Scholar] [CrossRef]
- Misiak, B.; Leszek, J.; Kiejna, A. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease—The emerging role of systemic low-grade inflammation and adiposity. Brain Res. Bull. 2012, 89, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Pichiah, P.B.T.; Sankarganesh, D.; Arunachalam, S.; Achiraman, S. Adipose-Derived Molecules-Untouched Horizons in Alzheimer’s Disease Biology. Front. Aging Neurosci. 2020, 12, 17. [Google Scholar] [CrossRef]
- Guo, D.H.; Yamamoto, M.; Hernandez, C.M.; Khodadadi, H.; Baban, B.; Stranahan, A.M. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J. Clin. Investig. 2020, 130, 1961–1976. [Google Scholar] [CrossRef]
- Nazeri, A.; Crandall, J.P.; Fraum, T.J.; Wahl, R.L. Repeatability of Radiomic Features of Brown Adipose Tissue. J. Nucl. Med. 2021, 62, 700–706. [Google Scholar] [CrossRef]
- Reijrink, M.; de Boer, S.A.; Antunes, I.F.; Spoor, D.S.; Heerspink, H.J.; Lodewijk, M.E.; Mastik, M.F.; Boellaard, R.; Greuter, M.J.; Benjamens, S. [18 F] FDG Uptake in Adipose Tissue Is Not Related to Inflammation in Type 2 Diabetes Mellitus. Mol. Imaging Biol. 2021, 23, 117–126. [Google Scholar] [CrossRef]
- Pahk, K.; Kim, E.J.; Lee, Y.J.; Kim, S.; Seo, H.S. Characterization of glucose uptake metabolism in visceral fat by 18 F-FDG PET/CT reflects inflammatory status in metabolic syndrome. PLoS ONE 2020, 15, e0228602. [Google Scholar] [CrossRef]
- Bucerius, J.; Mani, V.; Wong, S.; Moncrieff, C.; Izquierdo-Garcia, D.; Machac, J.; Fuster, V.; Farkouh, M.E.; Rudd, J.H.; Fayad, Z.A. Arterial and fat tissue inflammation are highly correlated: A prospective 18F-FDG PET/CT study. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 934–945. [Google Scholar] [CrossRef]
- Trayhurn, P.; Beattie, J.H. Physiological role of adipose tissue: White adipose tissue as an endocrine and secretory organ. Proc. Nutr. Soc. 2001, 60, 329–339. [Google Scholar] [CrossRef]
- Ishii, M.; Iadecola, C. Adipocyte-derived factors in age-related dementia and their contribution to vascular and Alzheimer pathology. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 966–974. [Google Scholar] [CrossRef]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 2019, 129, 3990–4000. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Naderali, E.K.; Ratcliffe, S.H.; Dale, M.C. Obesity and Alzheimer’s disease: A link between body weight and cognitive function in old age. Am. J. Alzheimers Dis. Other Dement. 2009, 24, 445–449. [Google Scholar] [CrossRef]
- Tziomalos, K.; Dimitroula, H.V.; Katsiki, N.; Savopoulos, C.; Hatzitolios, A.I. Effects of lifestyle measures, antiobesity agents, and bariatric surgery on serological markers of inflammation in obese patients. Mediat. Inflamm. 2010, 2010, 364957. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, Z.-T.; Sun, F.-R.; Shen, X.-N.; Xu, W.; Ma, Y.-H.; Dong, Q.; Tan, L.; Yu, J.-T.; Alzheimer’s Disease Neuroimaging Initiative. Late-life obesity is a protective factor for prodromal Alzheimer’s disease: A longitudinal study. Aging 2020, 12, 2005. [Google Scholar] [CrossRef]
- Yang, F.; Wang, G.; Wang, Z.; Sun, M.; Cao, M.; Zhu, Z.; Fu, Q.; Mao, J.; Shi, Y.; Yang, T. Visceral adiposity index may be a surrogate marker for the assessment of the effects of obesity on arterial stiffness. PLoS ONE 2014, 9, e104365. [Google Scholar] [CrossRef]
- Diehl-Wiesenecker, E.; von Armin, C.A.; Dupuis, L.; Muller, H.P.; Ludolph, A.C.; Kassubek, J. Adipose Tissue Distribution in Patients with Alzheimer’s Disease: A Whole Body MRI Case-Control Study. J. Alzheimers Dis. 2015, 48, 825–832. [Google Scholar] [CrossRef]
- Letra, L.; Matafome, P.; Rodrigues, T.; Duro, D.; Lemos, R.; Baldeiras, I.; Patrício, M.; Castelo-Branco, M.; Caetano, G.; Seiça, R. Association between adipokines and biomarkers of Alzheimer’s disease: A cross-sectional study. J. Alzheimers Dis. 2019, 67, 725–735. [Google Scholar] [CrossRef]
- Puig, K.L.; Floden, A.M.; Adhikari, R.; Golovko, M.Y.; Combs, C.K. Amyloid precursor protein and proinflammatory changes are regulated in brain and adipose tissue in a murine model of high fat diet-induced obesity. PLoS ONE 2012, 7, e30378. [Google Scholar] [CrossRef] [PubMed]
- An, Y.A.; Crewe, C.; Asterholm, I.W.; Sun, K.; Chen, S.; Zhang, F.; Shao, M.; Funcke, J.B.; Zhang, Z.; Straub, L.; et al. Dysregulation of Amyloid Precursor Protein Impairs Adipose Tissue Mitochondrial Function and Promotes Obesity. Nat. Metab. 2019, 1, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Jiang, H.; Xu, X.; Duan, W.; Mattson, M.P. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 2008, 283, 1754–1763. [Google Scholar] [CrossRef]
- Fewlass, D.C.; Noboa, K.; Pi-Sunyer, F.X.; Johnston, J.M.; Yan, S.D.; Tezapsidis, N. Obesity-related leptin regulates Alzheimer’s Abeta. FASEB J. 2004, 18, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Paz-Filho, G.; Wong, M.L.; Licinio, J. The procognitive effects of leptin in the brain and their clinical implications. Int. J. Clin. Pract. 2010, 64, 1808–1812. [Google Scholar] [CrossRef]
- Mangge, H.; Almer, G.; Haj-Yahya, S.; Grandits, N.; Gasser, R.; Pilz, S.; Moller, R.; Horejsi, R. Nuchal thickness of subcutaneous adipose tissue is tightly associated with an increased LMW/total adiponectin ratio in obese juveniles. Atherosclerosis 2009, 203, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Mangge, H.; Almer, G.; Truschnig-Wilders, M.; Schmidt, A.; Gasser, R.; Fuchs, D. Inflammation, adiponectin, obesity and cardiovascular risk. Curr. Med. Chem. 2010, 17, 4511–4520. [Google Scholar] [CrossRef]
- Haase, J.; Weyer, U.; Immig, K.; Klöting, N.; Blüher, M.; Eilers, J.; Bechmann, I.; Gericke, M. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 2014, 57, 562–571. [Google Scholar] [CrossRef]
- Monteiro, R.; Azevedo, I. Chronic inflammation in obesity and the metabolic syndrome. Mediat. Inflamm. 2010, 2010. [Google Scholar] [CrossRef]
- Banks, W.A. Blood-brain barrier transport of cytokines: A mechanism for neuropathology. Curr. Pharm. Des. 2005, 11, 973–984. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Nakandakari, S.C.B.R.; Munoz, V.R.; Kuga, G.K.; Gaspar, R.C.; Sant’Ana, M.R.; Pavan, I.C.B.; da Silva, L.G.S.; Morelli, A.P.; Simabuco, F.M.; da Silva, A.S.R. Short-term high-fat diet modulates several inflammatory, ER stress, and apoptosis markers in the hippocampus of young mice. Brain Behav. Immun. 2019, 79, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Van Leijsen, E.M.C.; Bergkamp, M.I.; van Uden, I.W.M.; Ghafoorian, M.; van der Holst, H.M.; Norris, D.G.; Platel, B.; Tuladhar, A.M.; de Leeuw, F.E. Progression of White Matter Hyperintensities Preceded by Heterogeneous Decline of Microstructural Integrity. Stroke 2018, 49, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- No, H.J.; Yi, H.A.; Won, K.S.; Chang, H.W.; Kim, H.W. Association between white matter lesions and the cerebral glucose metabolism in patients with cognitive impairment. Rev. Esp. Med. Nucl. Imagen Mol. 2019, 38, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Tzourio-Mazoyer, N.; Landeau, B.; Papathanassiou, D.; Crivello, F.; Etard, O.; Delcroix, N.; Mazoyer, B.; Joliot, M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage 2002, 15, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Bullich, S.; Seibyl, J.; Catafau, A.M.; Jovalekic, A.; Koglin, N.; Barthel, H.; Sabri, O.; De Santi, S. Optimized classification of (18)F-Florbetaben PET scans as positive and negative using an SUVR quantitative approach and comparison to visual assessment. Neuroimage Clin. 2017, 15, 325–332. [Google Scholar] [CrossRef]
- Barthel, H.; Gertz, H.J.; Dresel, S.; Peters, O.; Bartenstein, P.; Buerger, K.; Hiemeyer, F.; Wittemer-Rump, S.M.; Seibyl, J.; Reininger, C.; et al. Cerebral amyloid-beta PET with florbetaben (18F) in patients with Alzheimer’s disease and healthy controls: A multicentre phase 2 diagnostic study. Lancet Neurol. 2011, 10, 424–435. [Google Scholar] [CrossRef]
- Talairach, J.; Tournoux, P. Co-Planar Stereotaxic Atlas of the Human Brain: Three-Dimensional Proportional System; Thieme Medical: New York, NY, USA, 1988. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total (n = 54) | CU (n = 18) | MCI (n = 14) | Dementia (n = 22) | p |
|---|---|---|---|---|---|
| Age, years (SD) | 66.4 (8.4) | 62.7 (5.6) | 63.9 (8.9) | 71.1 (8.2) | 0.002 1 |
| Sex, female, n (%) | 34 (63.0) | 11 (61.1) | 7 (50.0) | 16 (72.7) | 0.380 |
| Body mass index (SD) | 23.3 (3.4) | 24.3 (4.4) | 22.6 (1.0) | 22.9 (3.5) | 0.277 |
| Education, years (SD) | 11.5 (6.1) | 14.3 (3.5) | 13.8 (6.4) | 7.6 (5.6) | <0.001 2 |
| Diabetes, n (%) | 8 (14.8) | 1 (5.6%) | 2 (14.3) | 5 (22.7) | 0.314 |
| Hypertension, n (%) | 16 (29.6) | 1 (7.1) | 4 (28.6) | 11 (50.0) | 0.0261 |
| Cardiovascular disease, n (%) | 6 (11.1) | 1 (7.1) | 2 (15.4) | 3 (14.3) | 0.768 |
| Hyperlipidemia, n (%) | 9 (16.7) | 1 (8.3) | 3 (21.4) | 5 (22.7) | 0.563 |
| WMH volume (SD) | 3.4 (5.1) | 1.1 (2.2) | 1.6 (2.0) | 6.4 (6.5) | 0.001 1 |
| MMSE (SD) | 24.6 (5.3) | 28.9 (1.2) | 25.9 (2.9) | 20.3 (5.3) | <0.001 2 |
| K-BNT (SD) | 42.4 (13.5) | 52.0 (4.0) | 47.8 (10.1) | 31.1 (12.2) | <0.001 2 |
| Aβ positivity, n (%) | 26 (48.1) | 5 (27.8) | 7 (50.0) | 14 (63.6) | 0.077 |
| Composite SUVRFBB | 1.47 (0.28) | 1.33 (0.11) | 1.48 (0.22) | 1.57 (0.36) | 0.017 1 |
| VAT SUVmax (SD) | 0.71 (0.16) | 0.69 (0.17) | 0.67 (0.11) | 0.76 (0.17) | 0.200 |
| VAT SUVmean (SD) | 0.44 (0.11) | 0.41 (0.11) | 0.41 (0.08) | 0.48 (0.12) | 0.067 |
| Variables | Total (n = 54) | Low VAT Metabolism Group (n = 31) | High VAT Metabolism Group (n = 23) | p |
|---|---|---|---|---|
| Age, years (SD) | 66.4 (8.4) | 65.3 (8.3) | 67.9 (8.5) | 0.269 |
| Sex, female, n (%) | 34 (63.0) | 18 (58.1) | 16 (69.6) | 0.412 |
| Body mass index (SD) | 23.3 (3.4) | 23.8 (3.0) | 22.5 (3.9) | 0.133 |
| Education, years (SD) | 11.5 (6.1) | 12.1 (5.9) | 10.7 (6.3) | 0.403 |
| Diabetes, n (%) | 8 (14.8) | 4 (12.9) | 4 (17.4) | 0.711 |
| Hypertension, n (%) | 16 (29.6) | 10 (37.0) | 6 (26.1) | 0.546 |
| Cardiovascular disease, n (%) | 6 (11.1) | 4 (14.8) | 2 (9.5) | 0.683 |
| Hyperlipidemia, n (%) | 9 (16.7) | 6 (23.1) | 3 (13.6) | 0.478 |
| WMH volume (SD) | 3.4 (5.08) | 2.7 (4.0) | 4.3 (6.2) | 0.245 |
| Cognitive stage | ||||
| CU, n (%) | 18 (33.3) | 13 (41.9) | 5 (21.7) | 0.234 |
| MCI, n (%) | 14 (26.0) | 8 (25.8) | 6 (26.1) | |
| Dementia, n (%) | 22 (40.7) | 10 (32.3) | 12 (52.2) | |
| MMSE (SD) | 24.6 (5.3) | 25.8 (3.8) | 23.0 (6.6) | 0.245 |
| K-BNT (SD) | 42.4 (13.5) | 45.0 (10.8) | 38.9 (15.9) | 0.107 |
| Aβ positivity, n (%) | 26 (48.1%) | 8 (25.8) | 18 (78.3) | <0.001 |
| VAT SUVmax (SD) | 0.71 (0.16) | 0.61 (0.09) | 0.85 (0.12) | <0.001 |
| VAT SUVmean (SD) | 0.44 (0.11) | 0.37 (0.06) | 0.54 (0.83) | <0.001 |
| Regions | Brodmann Area | Size | MNI Coordinates | T Value | p | ||
|---|---|---|---|---|---|---|---|
| X | Y | Z | |||||
| Right occipital lobe, lingual gyrus | BA 18 | 5896 | 2 | −84 | −8 | 3.56 | <0.001 |
| Right parietal lobe, precuneus | BA 19 | 26 | −80 | 42 | 3.52 | <0.001 | |
| Right parietal lobe, precuneus | BA 31 | 8 | −68 | 24 | 3.23 | 0.001 | |
| Right frontal lobe, precentral gyrus | BA 44 | 1315 | 62 | 8 | 4 | 3.55 | <0.001 |
| Right temporal lobe, middle temporal gyrus | BA 21 | 64 | 0 | −8 | 3.35 | <0.001 | |
| Right insula | BA 13 | 36 | 10 | 4 | 3.31 | <0.001 | |
| Left parietal lobe, precuneus | BA 7 | 4022 | −18 | −78 | 48 | 3.54 | <0.001 |
| Left temporal lobe, inferior temporal gyrus | BA 20 | −62 | −28 | −16 | 3.44 | <0.001 | |
| Left occipital lobe, inferior occipital gyrus | BA 18 | −34 | −90 | −14 | 3.2 | 0.001 | |
| Left frontal lobe, rectal gyrus | BA 11 | 2258 | −8 | 10 | −24 | 3.37 | <0.001 |
| Right frontal lobe, inferior frontal gyrus | BA 47 | 26 | 12 | −22 | 3.28 | <0.001 | |
| Left frontal lobe, medial frontal gyrus | BA 25 | −6 | 6 | −16 | 3.26 | <0.001 | |
| Left frontal lobe, superior frontal gyrus | BA 6 | 348 | −2 | 4 | 54 | 3.3 | <0.001 |
| Left frontal lobe, medial frontal gyrus | BA 6 | −4 | −8 | 58 | 3.02 | 0.002 | |
| Left frontal lobe, inferior frontal gyrus | BA 44 | 450 | −60 | 8 | 18 | 3.17 | 0.001 |
| Left cerebrum, frontal lobe, precentral gyrus | BA 6 | −60 | 6 | 30 | 3.17 | 0.001 | |
| Left frontal lobe, inferior frontal gyrus | BA 45 | −56 | 20 | 12 | 3.07 | 0.002 | |
| Regions | Univariable Model | Multivariable Model | |||
|---|---|---|---|---|---|
| r | p | Adjusted R2 | Standardized β 2 | p | |
| Composite 1 | 0.414 | 0.002 | 0.195 | 0.359 | 0.007 |
| Left lateral frontal cortex | 0.360 | 0.008 | 0.113 | 0.360 | 0.008 |
| Right lateral frontal cortex | 0.353 | 0.009 | 0.140 | 0.302 | 0.025 |
| Left lateral temporal cortex | 0.379 | 0.005 | 0.127 | 0.379 | 0.005 |
| Right lateral temporal cortex | 0.338 | 0.012 | 0.147 | 0.278 | 0.038 |
| Left lateral parietal cortex | 0.428 | 0.001 | 0.218 | 0.369 | 0.005 |
| Right lateral parietal cortex | 0.421 | 0.002 | 0.201 | 0.366 | 0.005 |
| Left cingulate | 0.410 | 0.002 | 0.199 | 0.352 | 0.008 |
| Right cingulate | 0.422 | 0.001 | 0.162 | 0.422 | 0.001 |
| Regions | Univariable Model | Multivariable Model | |||
|---|---|---|---|---|---|
| r | p | Adjusted R2 | Standardized β 2 | p | |
| Composite 1 | 0.367 | 0.006 | 0.150 | 0.295 | 0.032 |
| Left lateral frontal cortex | 0.319 | 0.019 | 0.085 | 0.319 | 0.019 |
| Right lateral frontal cortex | 0.318 | 0.019 | 0.084 | 0.318 | 0.019 |
| Left lateral temporal cortex | 0.340 | 0.012 | 0.098 | 0.340 | 0.012 |
| Right lateral temporal cortex | 0.304 | 0.025 | 0.118 | 0.224 | 0.106 |
| Left lateral parietal cortex | 0.380 | 0.005 | 0.169 | 0.302 | 0.026 |
| Right lateral parietal cortex | 0.379 | 0.005 | 0.158 | 0.308 | 0.024 |
| Left cingulate | 0.360 | 0.008 | 0.152 | 0.282 | 0.039 |
| Right cingulate | 0.369 | 0.006 | 0.120 | 0369 | 0.006 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Yi, H.-A.; Won, K.S.; Lee, J.S.; Kim, H.W. Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology. Metabolites 2022, 12, 258. https://doi.org/10.3390/metabo12030258
Kim S, Yi H-A, Won KS, Lee JS, Kim HW. Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology. Metabolites. 2022; 12(3):258. https://doi.org/10.3390/metabo12030258
Chicago/Turabian StyleKim, Shin, Hyon-Ah Yi, Kyoung Sook Won, Ji Soo Lee, and Hae Won Kim. 2022. "Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology" Metabolites 12, no. 3: 258. https://doi.org/10.3390/metabo12030258
APA StyleKim, S., Yi, H.-A., Won, K. S., Lee, J. S., & Kim, H. W. (2022). Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology. Metabolites, 12(3), 258. https://doi.org/10.3390/metabo12030258