
High-Throughput UHPLC-MS to Screen Metabolites in Feces for Gut Metabolic Health

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
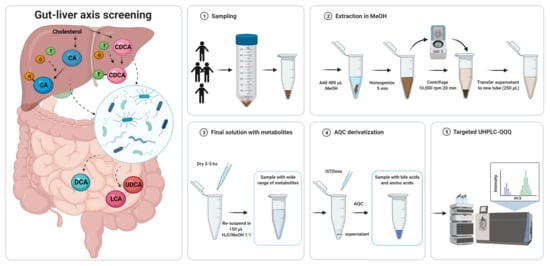
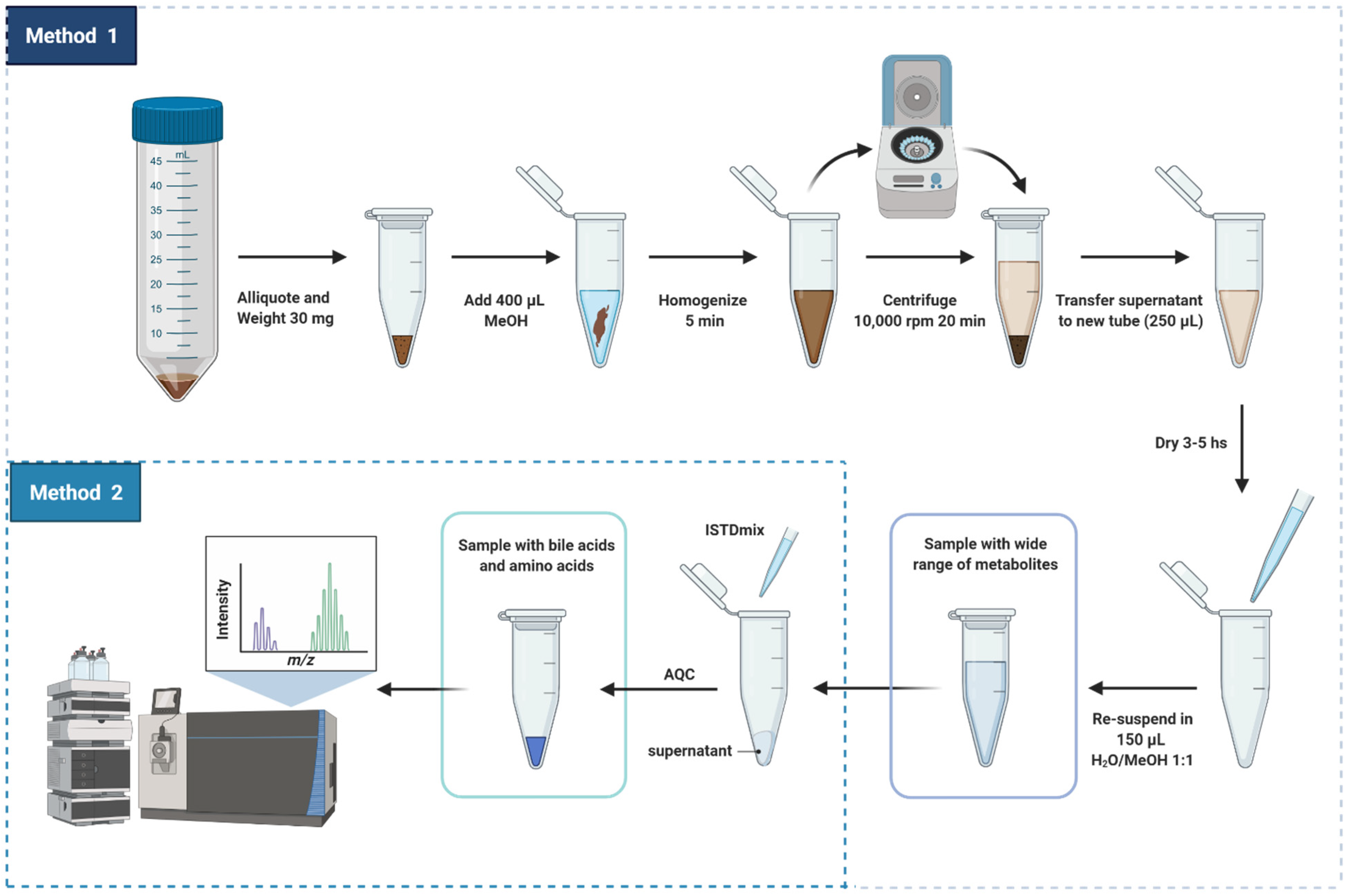
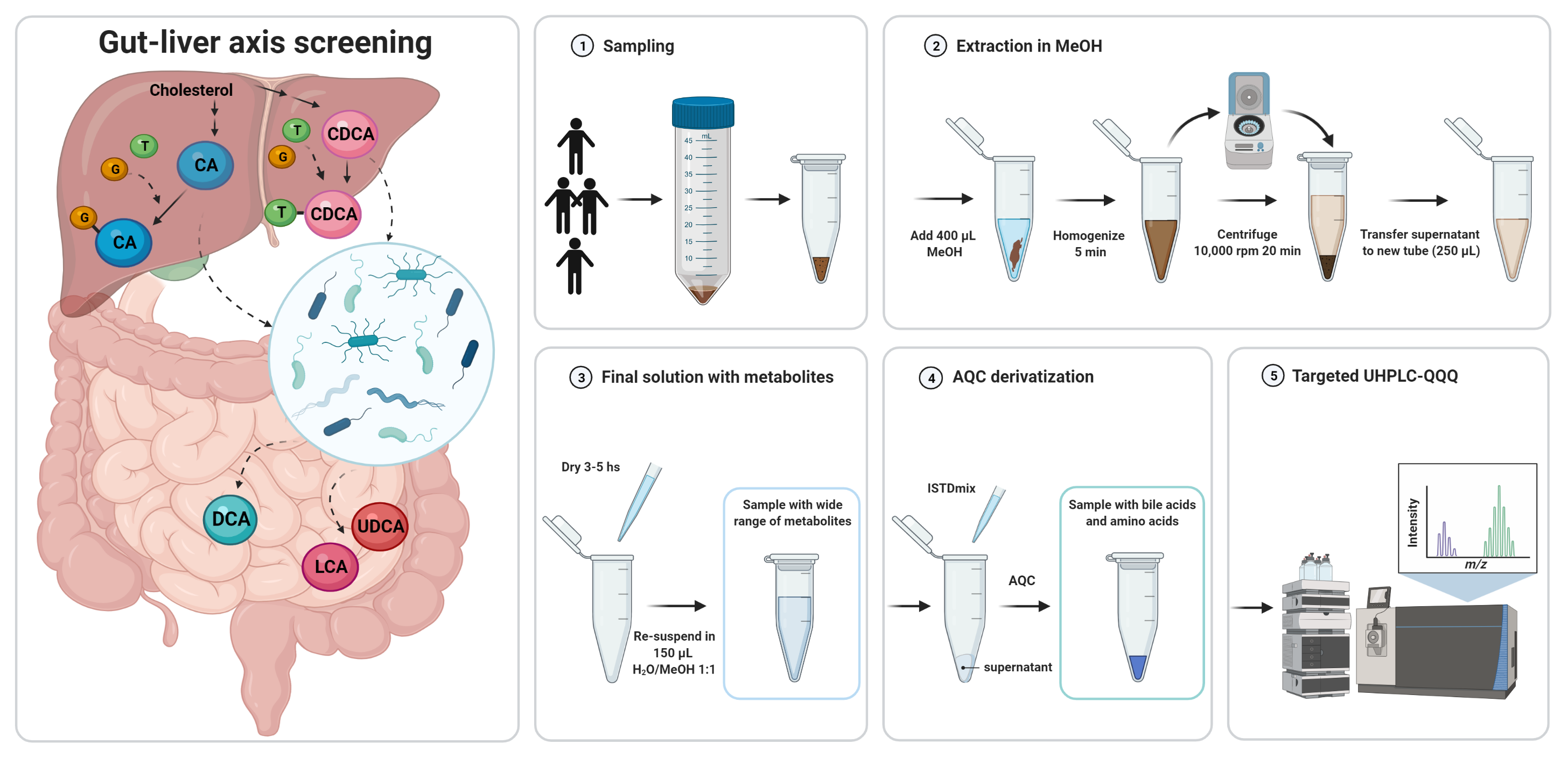
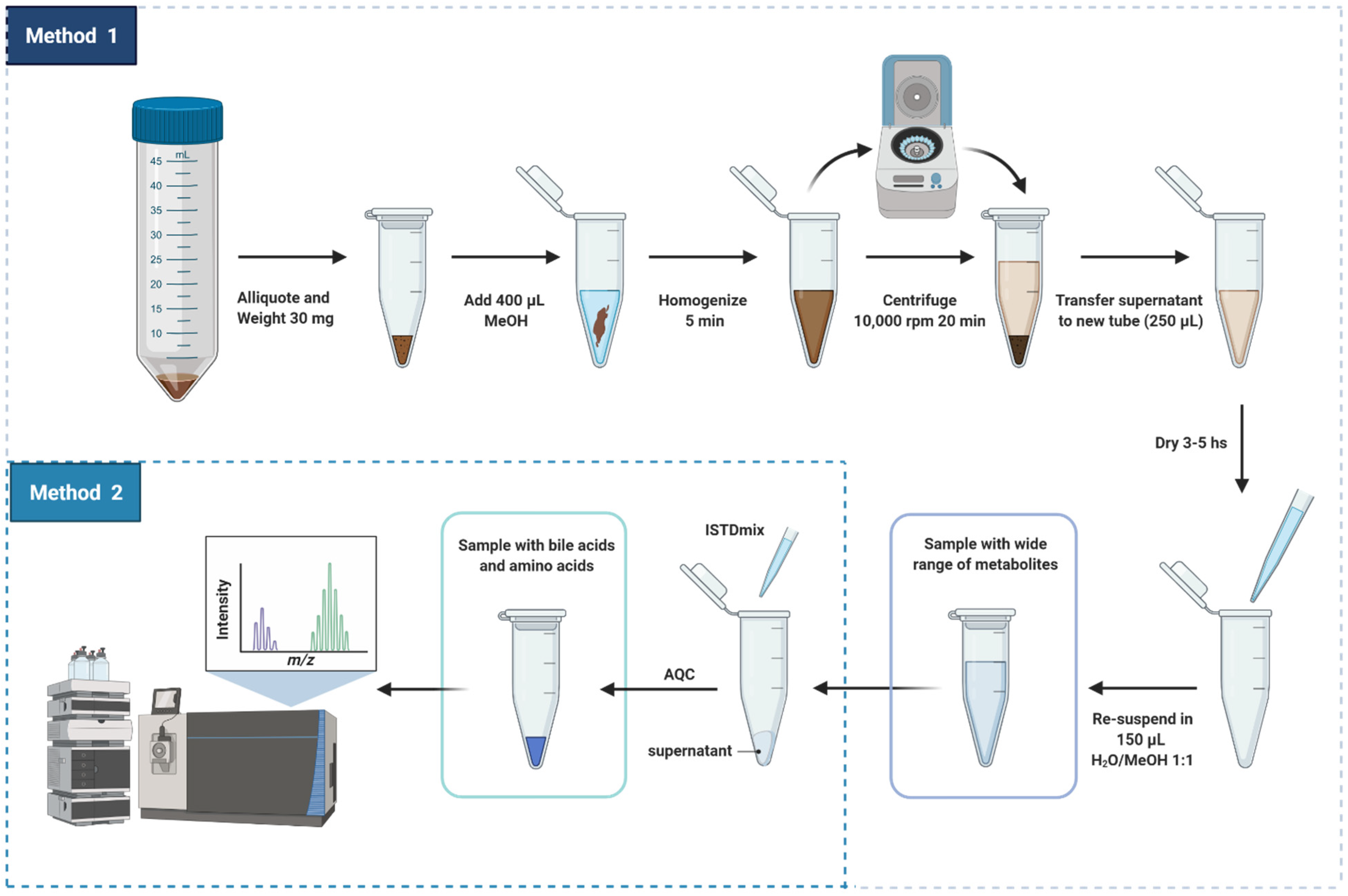
2.1. Sample Preparation
- (i)
- Addition of internal reference standards
- (ii)
- Derivatization of amino acids with AQC reagent
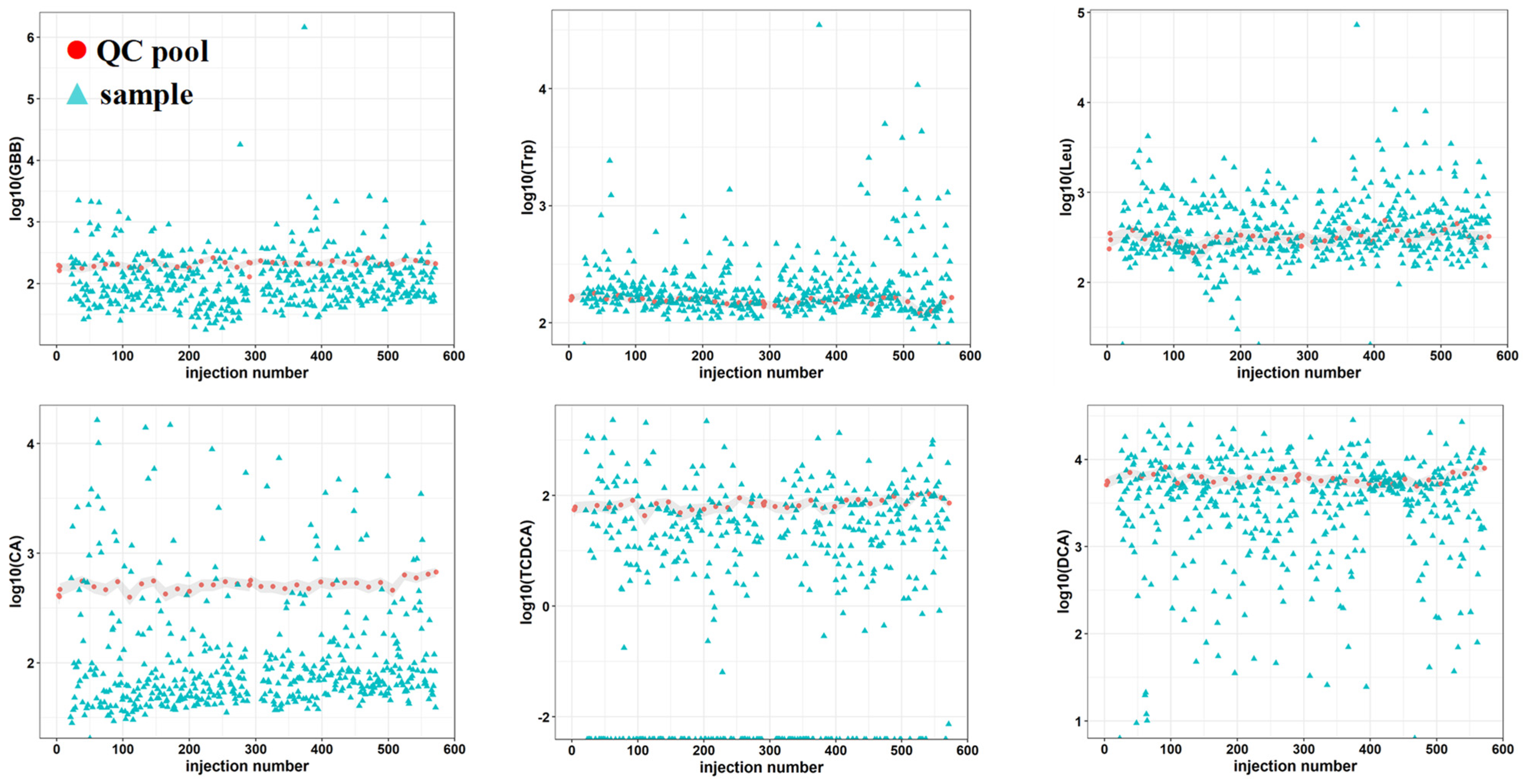
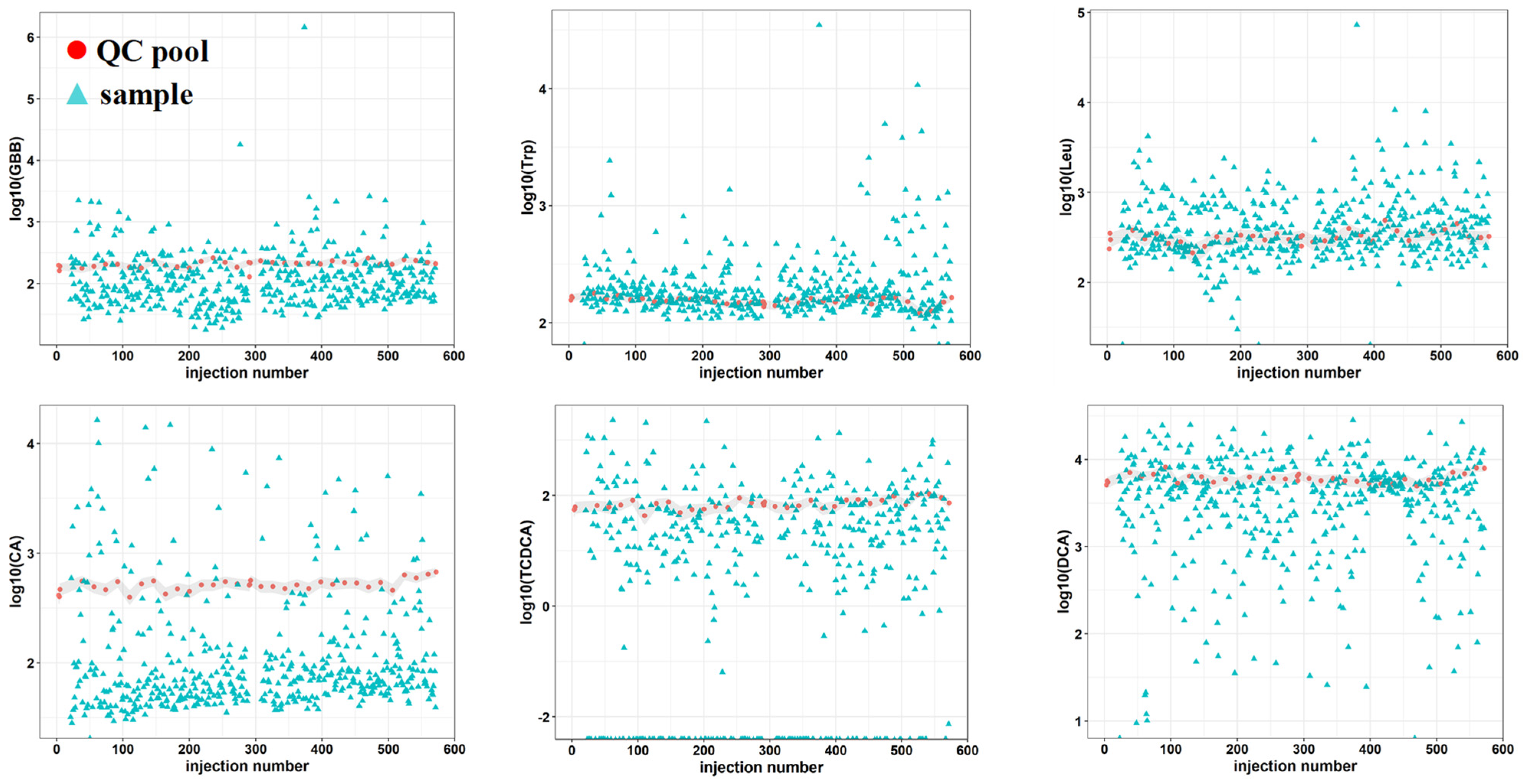
2.2. Method Optimization
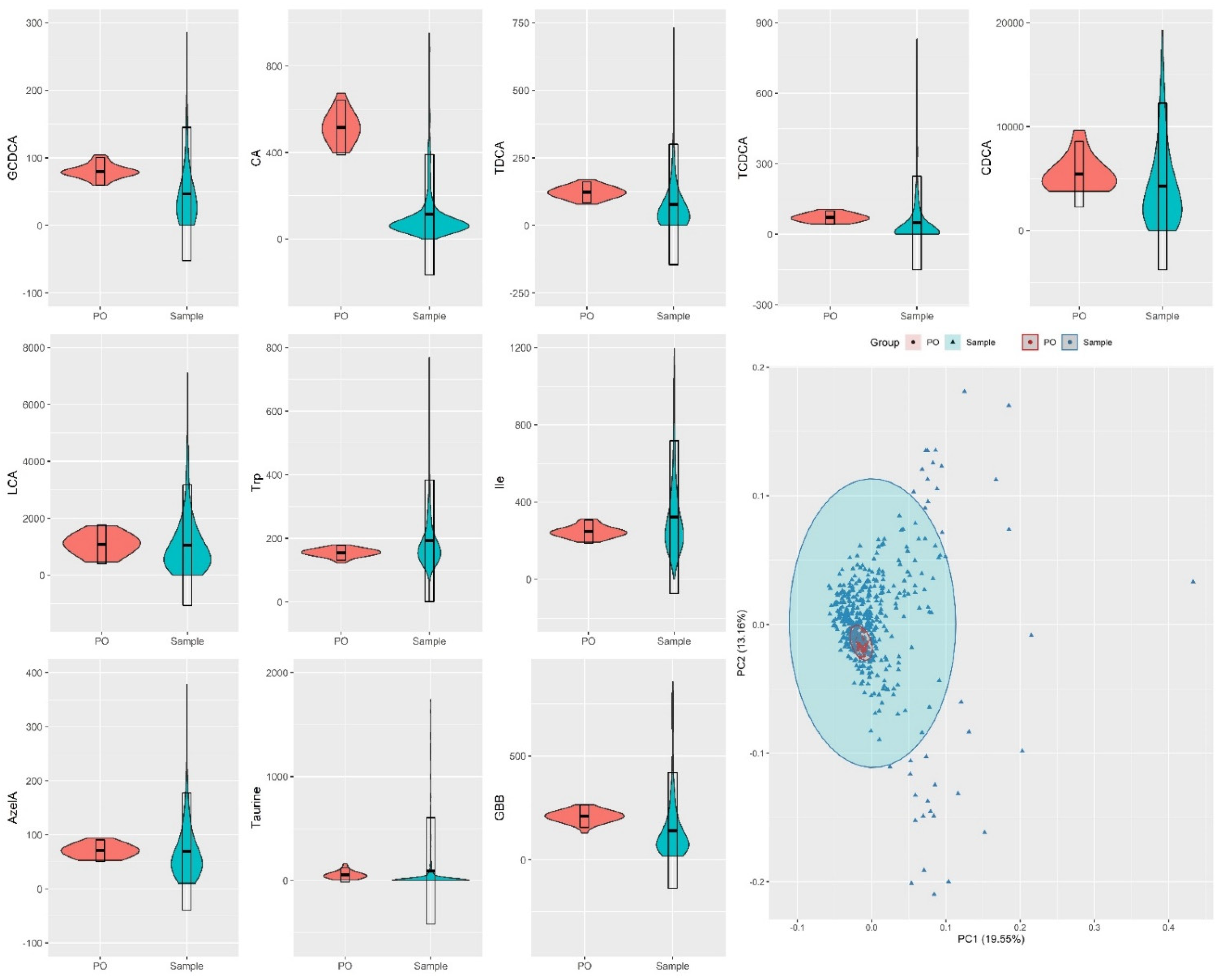
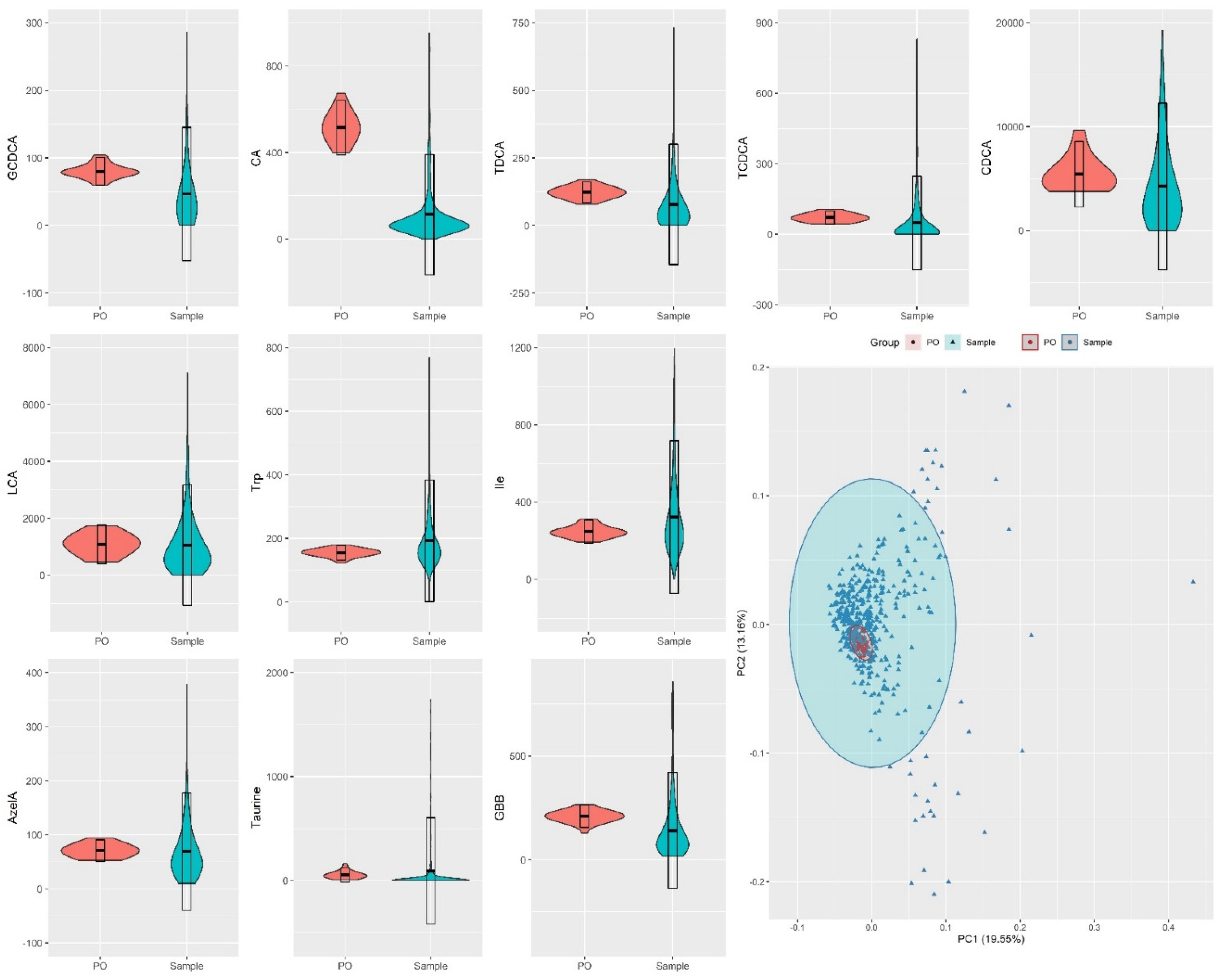
2.3. Method Feasibility in Large/Scale Cohorts
3. Materials and Methods
3.1. Patients
3.2. Sample Collection
3.3. Preparation of Pooled Samples
3.4. Study Design for Evaluation of Analytical Performance and Suitability
3.5. Chemicals
3.6. Preparation of Standards and Calibration Standards
3.7. Instrumentation
3.8. Sample Preparation
3.9. Study Design for Evaluation of Analytical Performance, Suitability and Recovery Efficiency
3.10. Data Analysis and Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef] [PubMed]
- Karu, N.; Deng, L.; Slae, M.; Guo, A.C.; Sajed, T.; Huynh, H.; Wine, E.; Wishart, D.S. A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal. Chim. Acta 2018, 1030, 1–24. [Google Scholar] [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [Green Version]
- Theodoridis, G.A.; Gika, H.G.; Want, E.J.; Wilson, I.D. Liquid chromatography-mass spectrometry based global metabolite profiling: A review. Anal. Chim. Acta 2012, 711, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Trošt, K.; Ahonen, L.; Suvitaival, T.; Christiansen, N.; Nielsen, T.; Thiele, M.; Jacobsen, S.; Krag, A.; Rossing, P.; Hansen, T.; et al. Describing the fecal metabolome in cryogenically collected samples from healthy participants. Sci. Rep. 2020, 10, 885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseinkhani, F.; Dubbelman, A.-C.; Karu, N.; Harms, A.C.; Hankemeier, T. Towards Standards for Human Fecal Sample Preparation in Targeted and Untargeted LC-HRMS Studies. Metabolites 2021, 11, 364. [Google Scholar] [CrossRef]
- Shafaei, A.; Rees, J.; Christophersen, C.T.; Devine, A.; Broadhurst, D.; Boyce, M.C. Extraction and quantitative determination of bile acids in feces. Anal. Chim. Acta 2021, 1150, 338224. [Google Scholar] [CrossRef]
- Chen, M.X.; Wang, S.Y.; Kuo, C.H.; Tsai, I.L. Metabolome analysis for investigating host-gut microbiota interactions. J. Formos. Med. Assoc. 2019, 118, S10–S22. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Fan, J. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2020, 19, 77–94. [Google Scholar] [CrossRef]
- Deda, O.; Virgiliou, C.; Orfanidis, A.; Gika, H.G. Study of fecal and urinary metabolite perturbations induced by chronic ethanol treatment in mice by UHPLC-MS/MS targeted profiling. Metabolites 2019, 9, 232. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caffaratti, C.; Plazy, C.; Mery, G.; Tidjani, A.-R.; Fiorini, F.; Thiroux, S.; Toussaint, B.; Hannani, D.; Le Gouellec, A. What We Know So Far about the Metabolite-Mediated Microbiota-Intestinal Immunity Dialogue and How to Hear the Sound of This Crosstalk. Metabolites 2021, 11, 406. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sokol, H. Gut microbiota- derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Salic, K.; Kleemann, R.; Wilkins-Port, C.; McNulty, J.; Verschuren, L.; Palmer, M. Apical sodium-dependent bile acid transporter inhibition with volixibat improves metabolic aspects and components of nonalcoholic steatohepatitis in Ldlr-/-.Leiden mice. PLoS ONE 2019, 14, e0218459. [Google Scholar] [CrossRef]
- Ahonen, L.; Jäntti, S.; Suvitaival, T.; Theilade, S.; Risz, C.; Kostiainen, R.; Rossing, P.; Orešič, M.; Hyötyläinen, T. Targeted clinical metabolite profiling platform for the stratification of diabetic patients. Metabolites 2019, 9, 184. [Google Scholar] [CrossRef] [Green Version]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.G.; Reily, M.D.; Lehman-Mckeeman, L.D.; Vaillancourt, R.R.; Cherrington, N.J. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids 2015, 47, 603–615. [Google Scholar] [CrossRef]
- Cheng, S.; Wiklund, P.; Autio, R.; Borra, R.; Ojanen, X.; Xu, L.; Törmäkangas, T.; Alen, M. Adipose tissue dysfunction and altered systemic amino acid metabolism are associated with non-alcoholic fatty liver disease. PLoS ONE 2015, 10, e0138889. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Anesi, A.; Rubert, J.; Oluwagbemigun, K.; Orozco-Ruiz, X.; Nöthlings, U.; Breteler, M.M.B.; Mattivi, F. Metabolic profiling of human plasma and urine, targeting tryptophan, tyrosine and branched chain amino acid pathways. Metabolites 2019, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Clària, J.; Moreau, R.; Fenaille, F.; Amorós, A.; Junot, C.; Gronbaek, H.; Coenraad, M.J.; Pruvost, A.; Ghettas, A.; Chu-Van, E.; et al. Orchestration of Tryptophan-Kynurenine Pathway, Acute Decompensation, and Acute-on-Chronic Liver Failure in Cirrhosis. Hepatology 2019, 69, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Muthulakshmi, S.; Saravanan, R. Efficacy of azelaic acid on hepatic key enzymes of carbohydrate metabolism in high fat diet induced type 2 diabetic mice. Biochimie 2013, 95, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Reiter, S.; Dunkel, A.; Metwaly, A.; Panes, J.; Haller, D.; Hofmann, T.; Berg, W. Development of a highly-sensitive UHPLC—MS/MS quantitation method for fecal bile acids and application on Crohn ’s Disease studies. J. Agric. Food Chem. 2021, 69, 5238–5251. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Muto, A.; Takei, H.; Nittono, H.; Murai, T.; Kurosawa, T.; Hofmann, A.F.; Pandak, W.M.; Bajaj, J.S. A simple and accurate HPLC method for fecal bile acid profile in healthy and cirrhotic subjects: Validation by GC-MS and LC-MS. J. Lipid Res. 2014, 55, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northfield, T.C.; McColl, I. Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 1973, 14, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Wang, L.; Chen, T.; Zhou, K.; Zhang, Z.; Li, J.; Sun, B.; Guo, Y.; Wang, X.; Wang, Y.; et al. A Metabolite Array Technology for Precision Medicine. Anal. Chem. 2021, 93, 5709–5717. [Google Scholar] [CrossRef]
- Katsidzira, L.; Ocvirk, S.; Wilson, A.; Li, J.; Mahachi, C.B.; Soni, D.; DeLany, J.; Nicholson, J.K.; Zoetendal, E.G.; O’Keefe, S.J.D. Differences in Fecal Gut Microbiota, Short-Chain Fatty Acids and Bile Acids Link Colorectal Cancer Risk to Dietary Changes Associated with Urbanization Among Zimbabweans. Nutr. Cancer 2019, 71, 1313–1324. [Google Scholar] [CrossRef]
- Zheng, X.; Qiu, Y.; Zhong, W.; Baxter, S.; Su, M.; Li, Q.; Xie, G.; Ore, B.M.; Qiao, S.; Spencer, M.D.; et al. A targeted metabolomic protocol for short-chain fatty acids and branched-chain amino acids. Metabolomics 2013, 9, 818–827. [Google Scholar] [CrossRef] [Green Version]
- Røst, L.M.; Brekke Thorfinnsdottir, L.; Kumar, K.; Fuchino, K.; Eide Langørgen, I.; Bartosova, Z.; Kristiansen, K.A.; Bruheim, P. Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems. Metabolites 2020, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Guo, P.; Hai, D.; Xu, L.; Shu, J.; Zhang, W.; Khan, M.I.; Kurland, I.J.; Qiu, Y.; Liu, Y. Optimization of GC/TOF MS analysis conditions for assessing host-gut microbiota metabolic interactions: Chinese rhubarb alters fecal aromatic amino acids and phenol metabolism. Anal. Chim. Acta 2017, 995, 21–33. [Google Scholar] [CrossRef]
- Van Eijk, H.M.H.; Bloemen, J.G.; Dejong, C.H.C. Application of liquid chromatography—Mass spectrometry to measure short chain fatty acids in blood. J. Chromatogr. B 2009, 877, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Cao, H. Fast quantification of short chain fatty acids and ketone bodies by liquid chromatography-tandem mass spectrometry after facile derivatization coupled with liquid-liquid extraction. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1083, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Orešič, M.; Anderson, G.; Mattila, I.; Manoucheri, M.; Soininen, H.; Hyötyläinen, T.B.C. Targeted Serum Metabolite Profiling Identifies Metabolic Signatures in Patients with Alzheimer ’s Disease, Normal Pressure Hydrocephalus and Brain Tumor. Front. Neurosci. 2018, 11, 747. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Coira, J.; Delgado-Dolset, M.I.; Obeso, D.; Dolores-Hernández, M.; Quintás, G.; Angulo, S.; Barber, D.; Carrillo, T.; Escribese, M.M.; Villaseñor, A. Troubleshooting in large-scale LC-ToF-MS metabolomics analysis: Solving complex issues in big cohorts. Metabolites 2019, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Krautbauer, S.; Buüchler, C.L.G. Relevance in the use of appropriate internal standards for accurate quantification using LC–MS/MS: Tauro-conjugated bile acids as an example. Anal. Chem. 2016, 88, 10957–10961. [Google Scholar] [CrossRef]
- Bajad, S.U.; Lu, W.; Kimball, E.H.; Yuan, J.; Peterson, C.; Rabinowitz, J.D. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography tandem mass spectrometry. J. Chromatogr. A 2006, 1125, 76–88. [Google Scholar] [CrossRef]
- Xu, W.; Zhong, C.; Zou, C.; Wang, B.; Zhang, N. Analytical methods for amino acid determination in organisms. Amino Acids 2020, 52, 1071–1088. [Google Scholar] [CrossRef]
- Cohen, S.A.; Michaud, D.P. Synthesis of a Fluorescent Derivatizing Reagent, 6-Aminoquinolyl-N-Hydroxysuccinimidyl Carbamate, and Its Application for the Analysis of Hydrolysate Amino Acids via High-Performance Liquid Chromato. Anal. Biochem. 1993, 211, 279–287. [Google Scholar] [CrossRef]
- Song, Y.; Xu, C.; Kuroki, H.; Liao, Y.; Tsunoda, M. Recent trends in analytical methods for the determination of amino acids in biological samples. J. Pharm. Biomed. Anal. 2018, 147, 35–49. [Google Scholar] [CrossRef]
- Lajczak-McGinley, N.K.; Porru, E.; Fallon, C.M.; Smyth, J.; Curley, C.; McCarron, P.A.; Tambuwala, M.M.; Roda, A.; Keely, S.J. The secondary bile acids, ursodeoxycholic acid and lithocholic acid, protect against intestinal inflammation by inhibition of epithelial apoptosis. Physiol. Rep. 2020, 8, e14456. [Google Scholar] [CrossRef]
- Kakiyama, G.; Pandak, W.M.; Gilleve, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the Fecal Bile Acid Profile by Gut Microbiota in Cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godzien, J.; Alonso-Herranz, V.; Barbas, C.; Armitage, E.G. Controlling the quality of metabolomics data: New strategies to get the best out of the QC sample. Metabolomics 2015, 11, 518–528. [Google Scholar] [CrossRef]
- Gika, H.G.; Theodoridis, G.A.; Earll, M.; Wilson, I.D. A QC approach to the determination of day-to-day reproducibility and robustness of LC-MS methods for global metabolite profiling in metabonomics/metabolomics. Bioanalysis 2012, 4, 2239–2247. [Google Scholar] [CrossRef]
- Szabo, G. Gut-Liver Axis in Alcoholic Liver Disease. Gastroenterology 2015, 148, 10–36. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Wang, C.; Li, B.; Zhang, A. Metabolomics of Alcoholic Liver Disease: A Clinical Discovery Study. RSC Adv. 2015, 5, 80381–80387. [Google Scholar] [CrossRef]
- Ismail, R.; Lee, H.Y.; Mahyudin, N.A.; Abu Bakar, F. Linearity study on detection and quantification limits for the determination of avermectins using linear regression. J. Food Drug Anal. 2014, 22, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot 2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; pp. 10–1007. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bile Acids | |||||
| Primary | Secondary | ||||
| Concentration | Normal Range | Concentration | Normal Range | ||
| CA | 0.26 ± 0.35 | 0.07–2.50 | LCA | 2.95 ± 3.02 | 0.08–19.0 |
| CDCA | 8.99 ± 8.98 | 0.22–62.0 | DCA | 10.16 ± 10.04 | 0.05–52.0 |
| TCA | 0.11 ± 0.05 | 0.06–0.45 | GLCA | 0.17 ± 0.44 | 0.10–0.40 |
| TCDCA | 0.21 ± 0.34 | 0–3.20 | TDCA | 0.13 ± 0.21 | 0–1.50 |
| GCA | 0.11 ± 0.12 | 0–0.85 | TUDCA | 0.10 ± 0.09 | 0.02–0.60 |
| GCDCA | 0.16 ± 0.33 | 0–2.30 | GDCA | 0.14 ± 0.23 | 0–2.00 |
| UDCA | BLOQ * | ||||
| GUDCA | BLOQ * | ||||
| Ratio secondary and primary bile acids | |||||
| DCA/CA | 38.59 | ||||
| LCA/CDCA | 0.33 | ||||
| TUDCA/CDCA | 0.01 | ||||
| Total ratio | 1.40 | ||||
| Amino Acids | |||||
| Concentration | Normal Range | Concentration | Normal Range | ||
| Alanine | 6.49 ± 8.71 | 0.2–58.0 | Kynurenine | 0.60 ± 0.16 | 0.36–1.10 |
| ADMA | BLOQ * | Leucine | 4.15 ± 3.82 | 0.80–26.0 | |
| Citruline | 1.31 ± 1.12 | 0.5–9.0 | Isoleucine | 2.92 ± 2.21 | 1.0–15.0 |
| Glutamine | BLOQ * | Phenylalanine | 2.57 ± 2.51 | 0.8–19.0 | |
| Glutamate | 1.21 v 2.71 | 0.2–28.0 | Taurine | 0.81 ± 2.45 | 0–20.0 |
| Glycine | BLOQ * | Tryptophan | 1.05 ± 0.85 | 0.50–6.50 | |
| Homocitruline | BLOQ * | Tyrosine | 0.17 ± 0.03 | 0.10–0.33 | |
| Other Compounds | |||||
| Concentration | Normal Range | ||||
| AADA | BLOQ * | ||||
| Azelaic acid | 0.50 ± 0.37 | 0.10–2.30 | |||
| β-OHB | 73.58 ± 280.51 | 17.0–355.0 | |||
| GBB | 0.80 ± 0.89 | 0.13–6.20 | |||
| IndS | BLOQ * | ||||
| N-MNA | BLOQ * | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zawadzki, A.d.; Thiele, M.; Suvitaival, T.; Wretlind, A.; Kim, M.; Ali, M.; Bjerre, A.F.; Stahr, K.; Mattila, I.; Hansen, T.; et al. High-Throughput UHPLC-MS to Screen Metabolites in Feces for Gut Metabolic Health. Metabolites 2022, 12, 211. https://doi.org/10.3390/metabo12030211
Zawadzki Ad, Thiele M, Suvitaival T, Wretlind A, Kim M, Ali M, Bjerre AF, Stahr K, Mattila I, Hansen T, et al. High-Throughput UHPLC-MS to Screen Metabolites in Feces for Gut Metabolic Health. Metabolites. 2022; 12(3):211. https://doi.org/10.3390/metabo12030211
Chicago/Turabian StyleZawadzki, Andressa de, Maja Thiele, Tommi Suvitaival, Asger Wretlind, Min Kim, Mina Ali, Annette F. Bjerre, Karin Stahr, Ismo Mattila, Torben Hansen, and et al. 2022. "High-Throughput UHPLC-MS to Screen Metabolites in Feces for Gut Metabolic Health" Metabolites 12, no. 3: 211. https://doi.org/10.3390/metabo12030211
APA StyleZawadzki, A. d., Thiele, M., Suvitaival, T., Wretlind, A., Kim, M., Ali, M., Bjerre, A. F., Stahr, K., Mattila, I., Hansen, T., Krag, A., & Legido-Quigley, C. (2022). High-Throughput UHPLC-MS to Screen Metabolites in Feces for Gut Metabolic Health. Metabolites, 12(3), 211. https://doi.org/10.3390/metabo12030211