
Mangosteen Metabolites as Promising Alpha-Amylase Inhibitor Candidates: In Silico and In Vitro Evaluations

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Protein
2.2. Ligand Preparation
2.3. Receptor Grid Generation and Molecular Docking
2.4. Molecular Dynamic Simulation (MDS)
2.5. Prediction of ADMET Properties
2.6. Alpha-Amylase Inhibition Assay
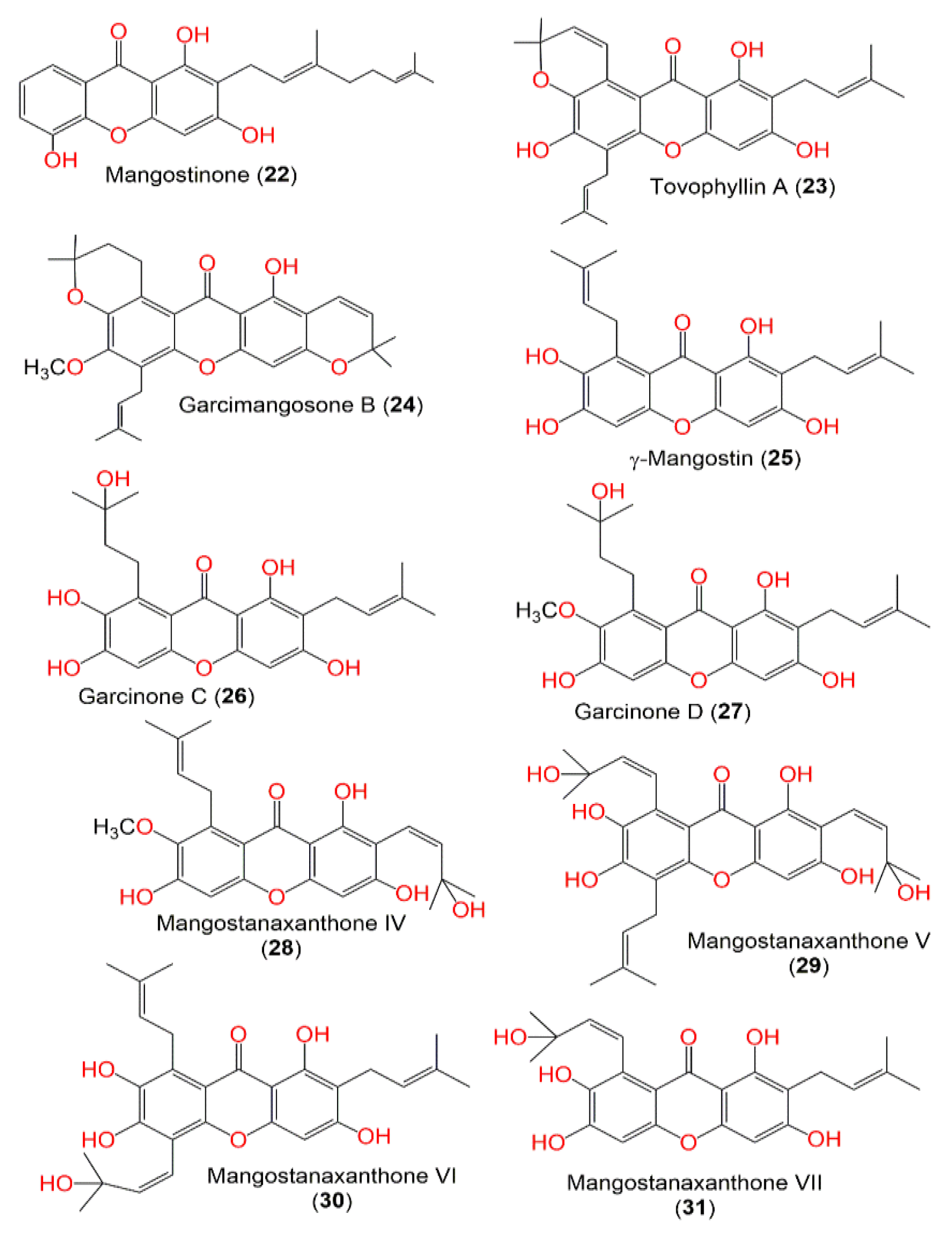
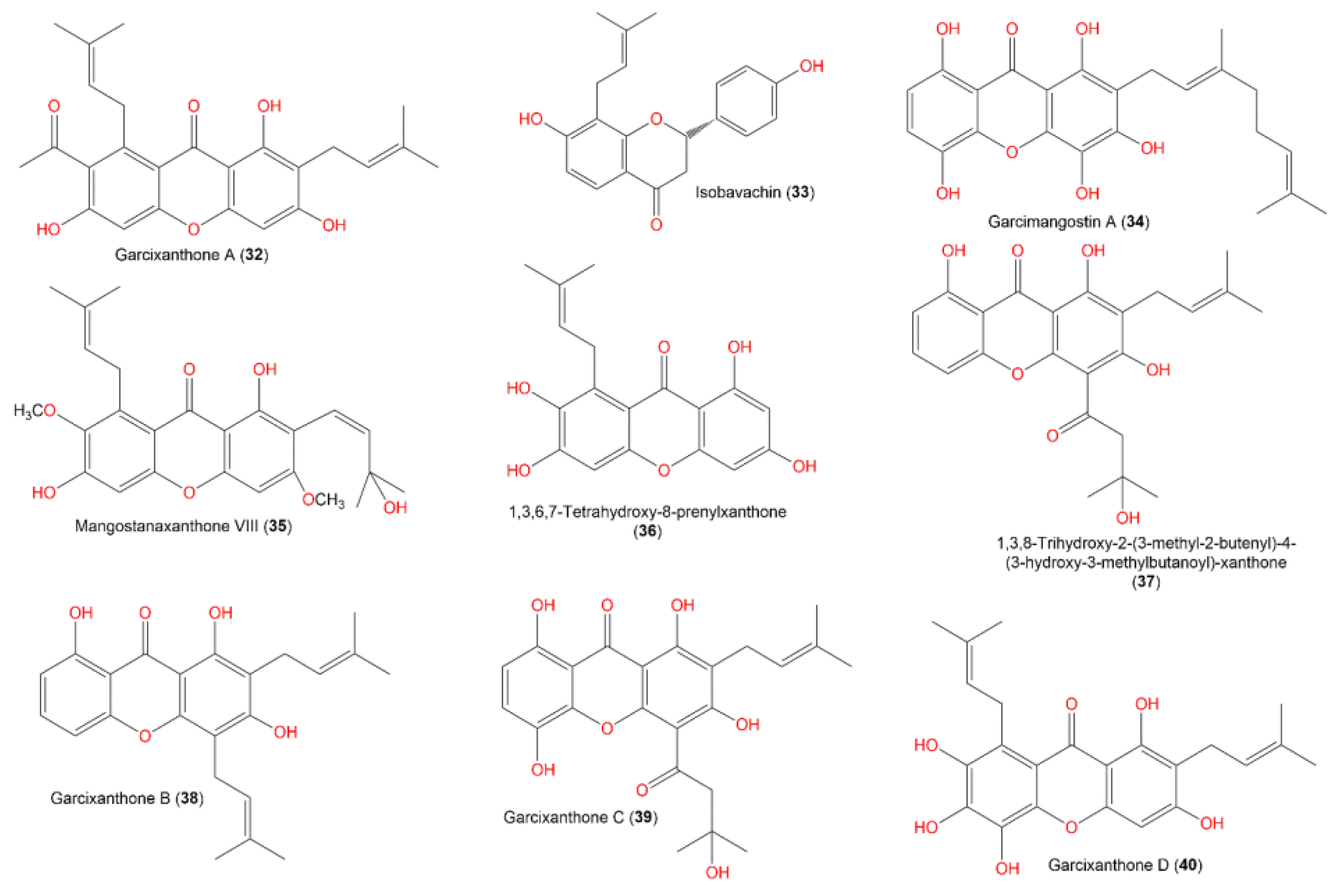
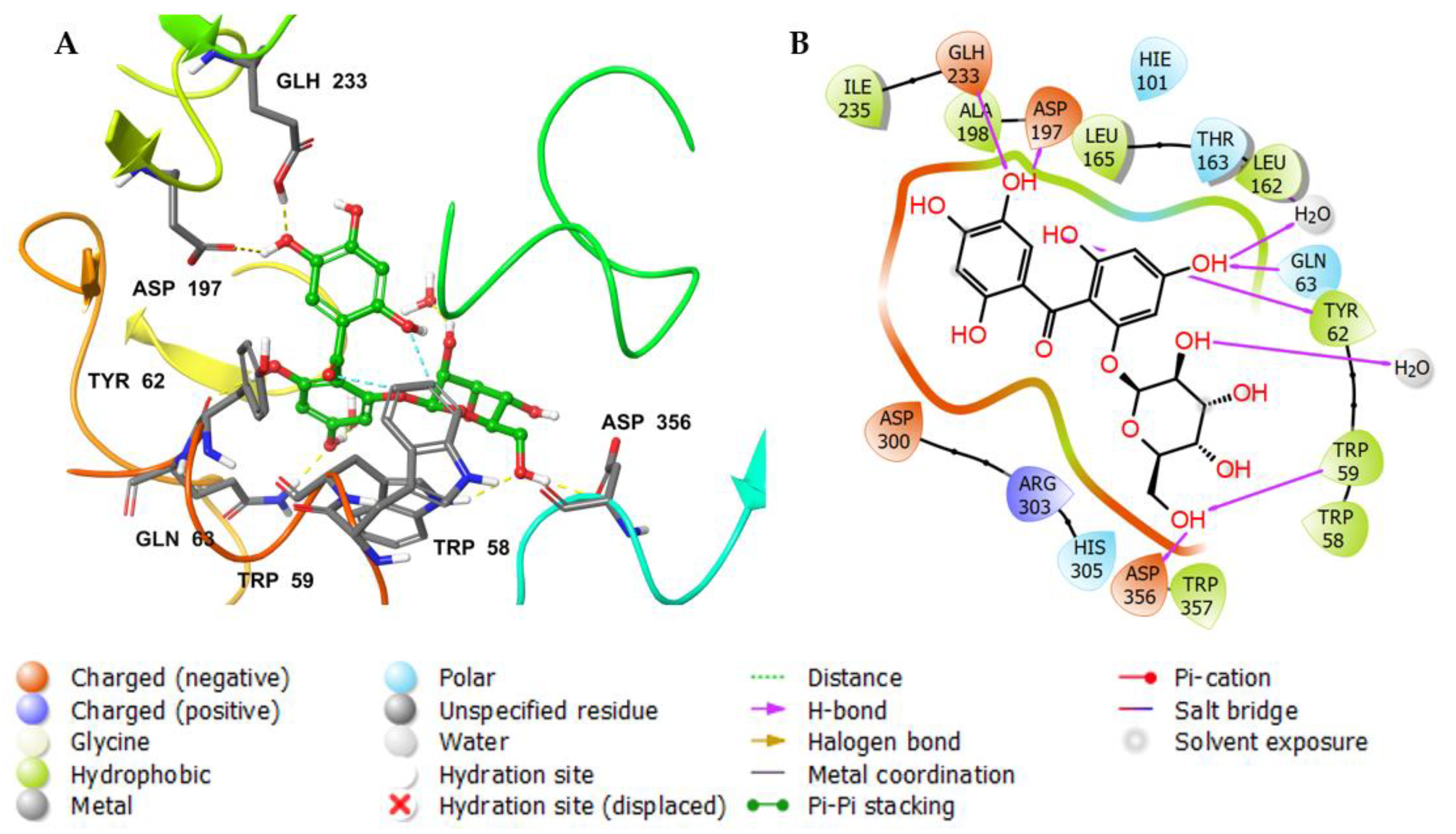
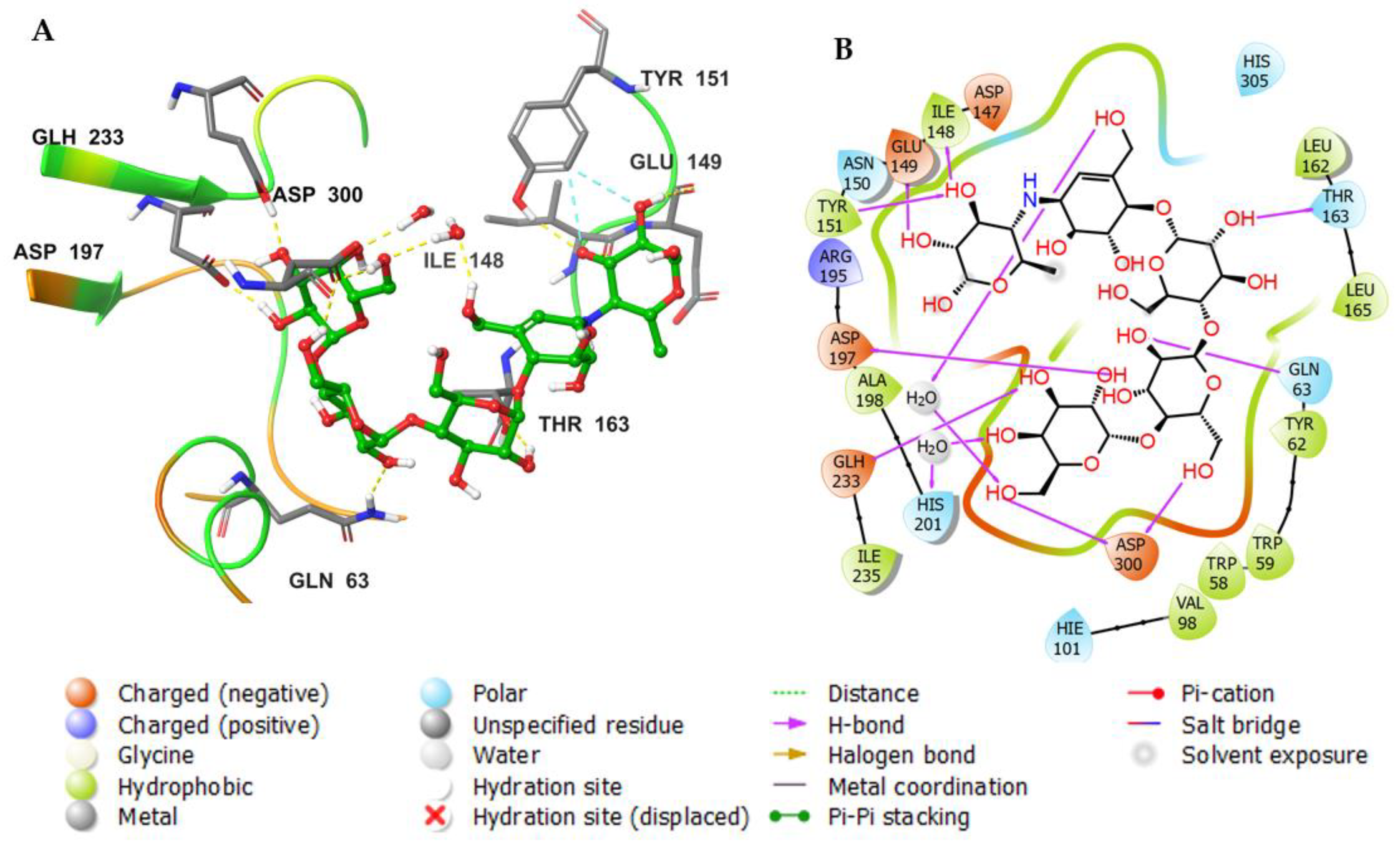
3. Results and Discussion
3.1. In Silico ADME Properties of Selected Ligands
3.2. Protein and Ligand Preparation
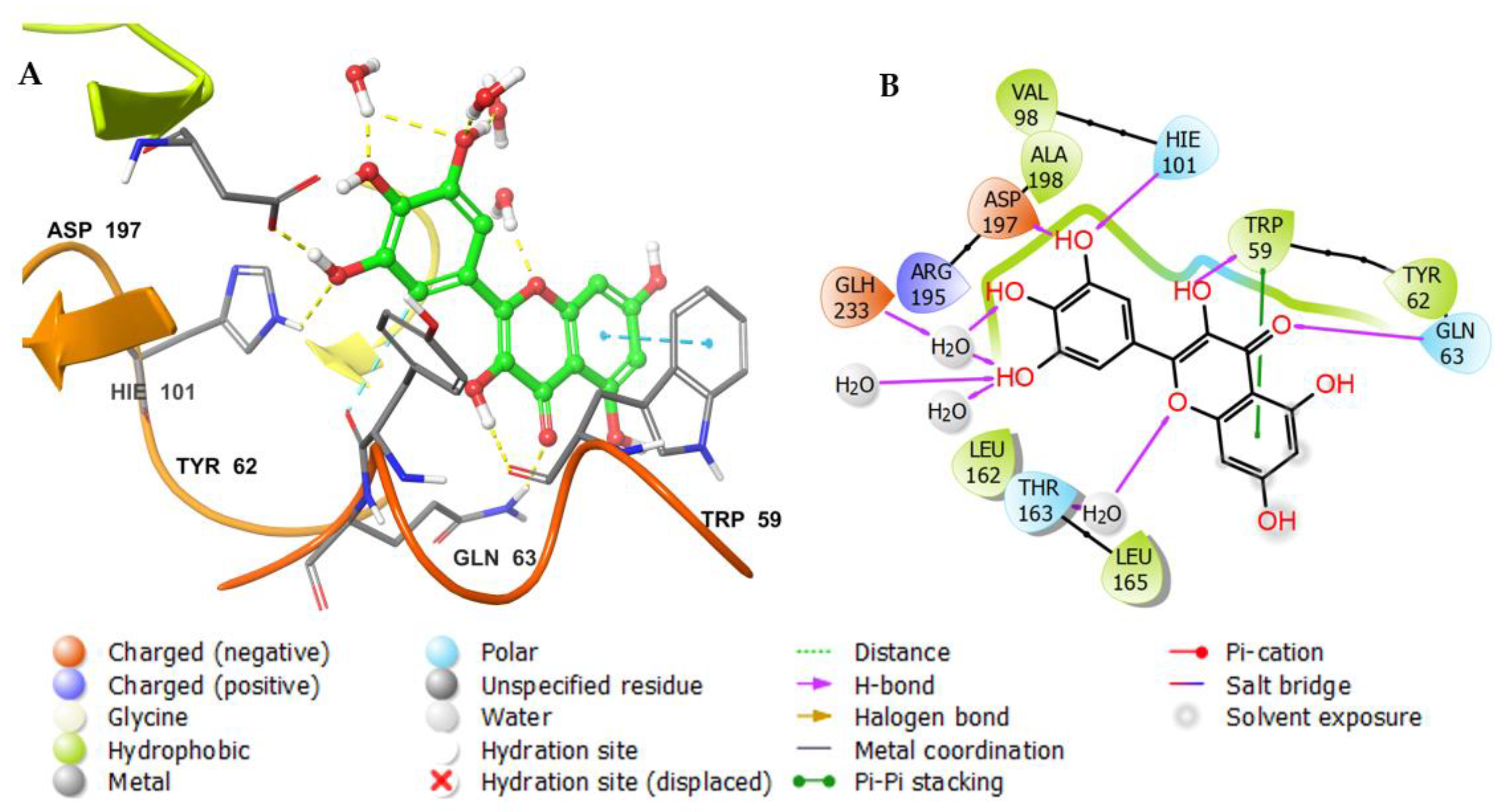
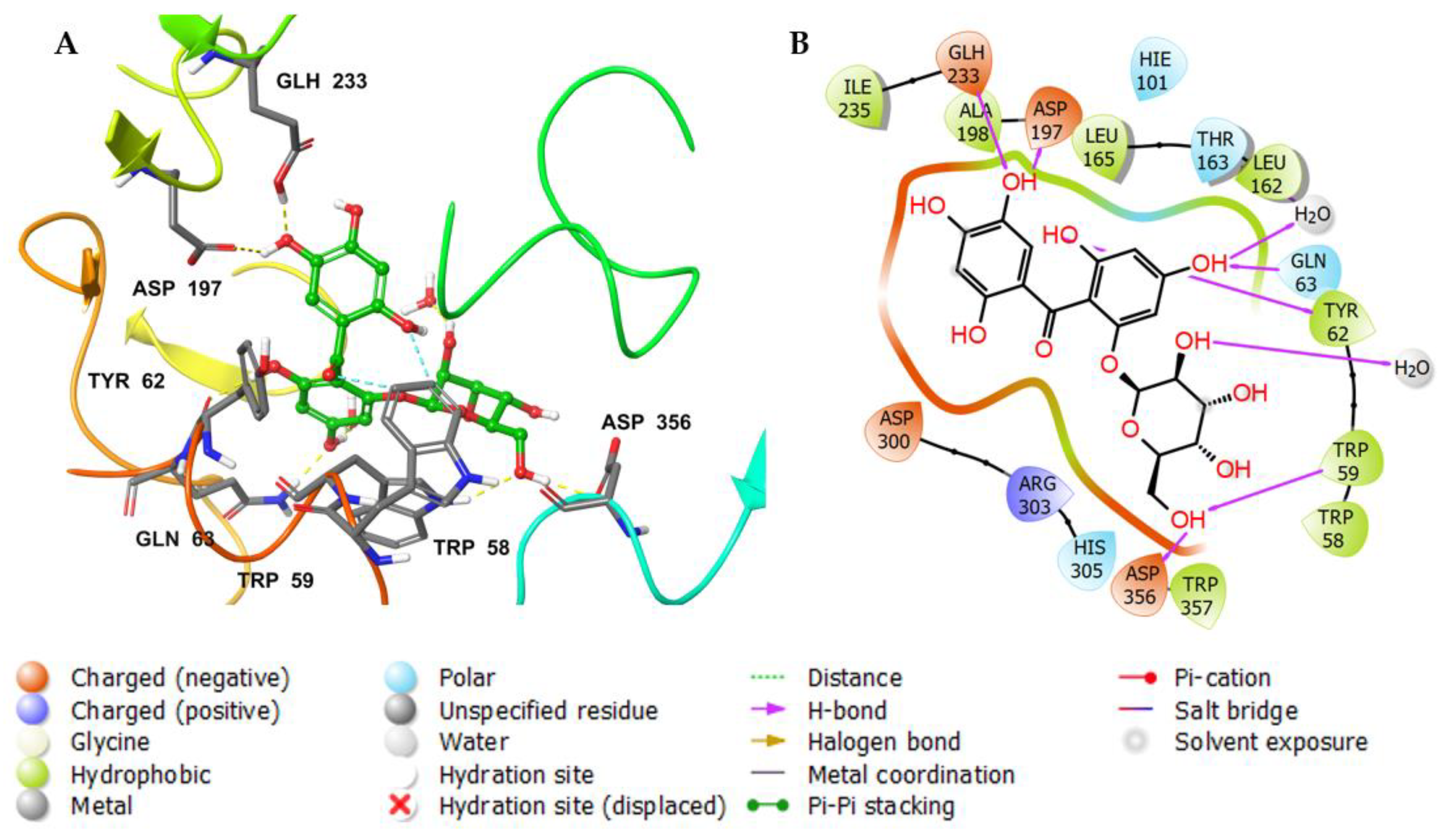
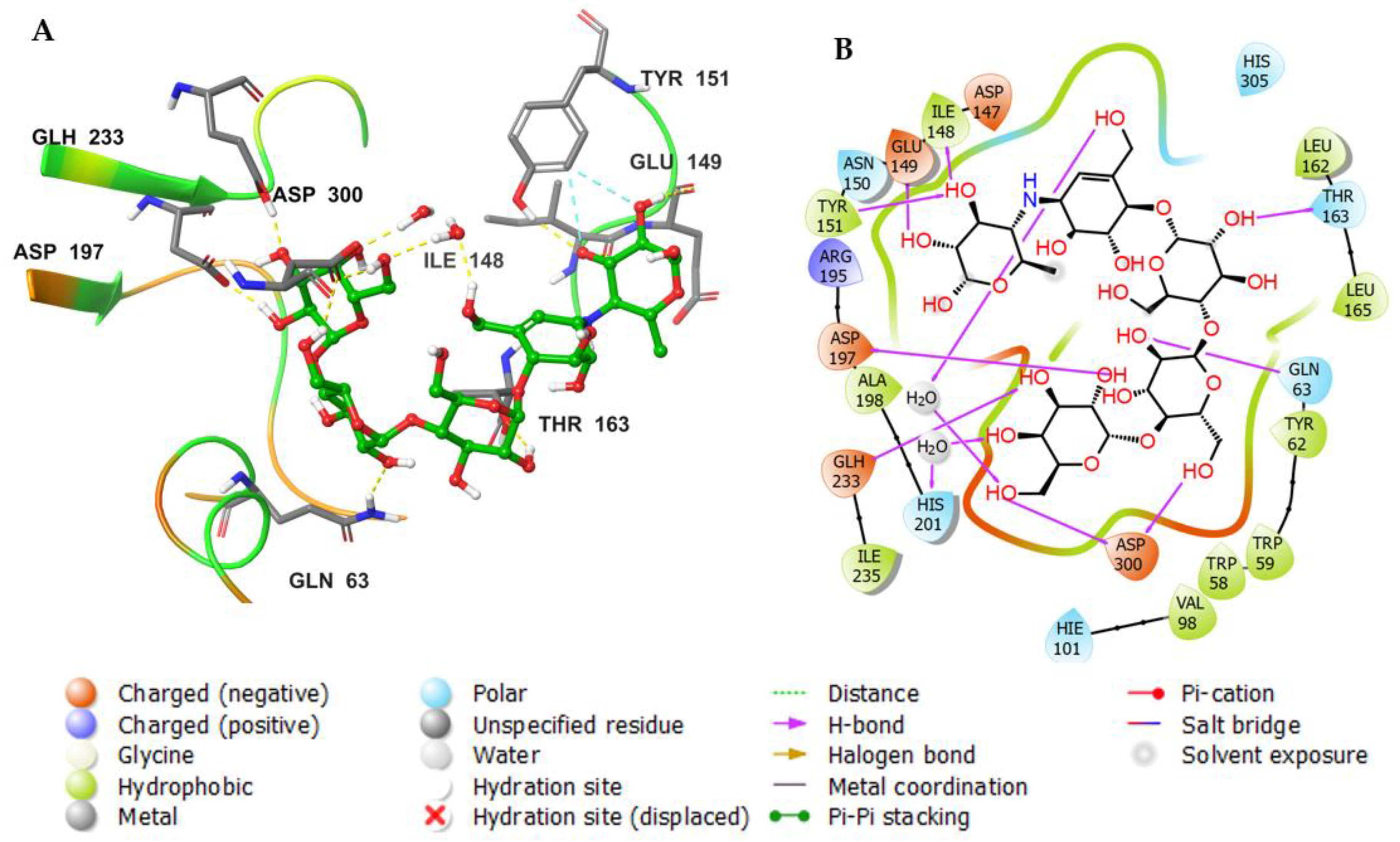
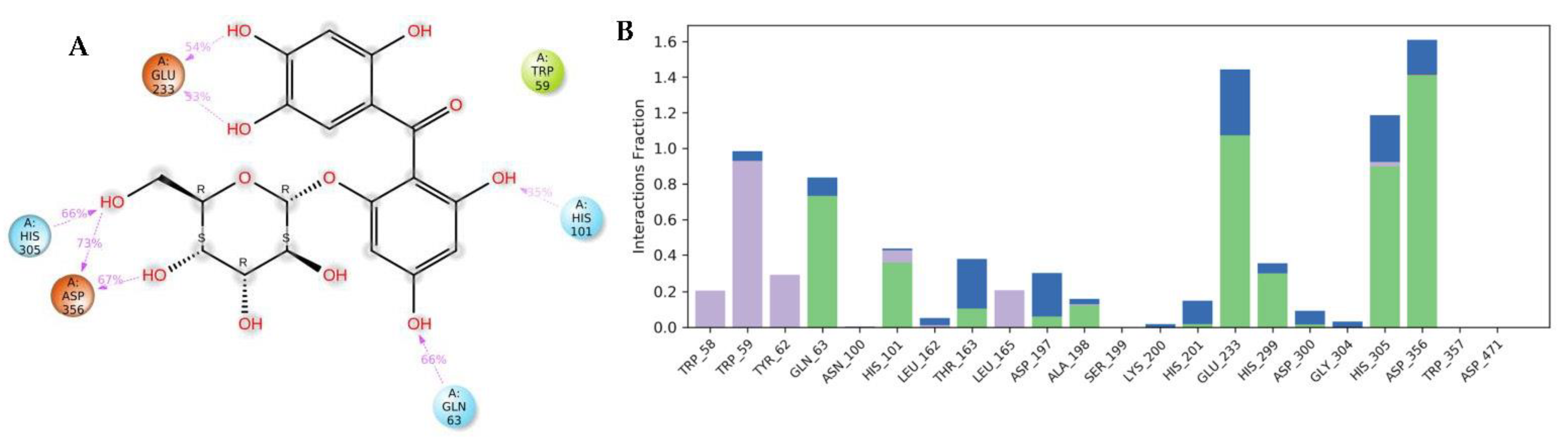
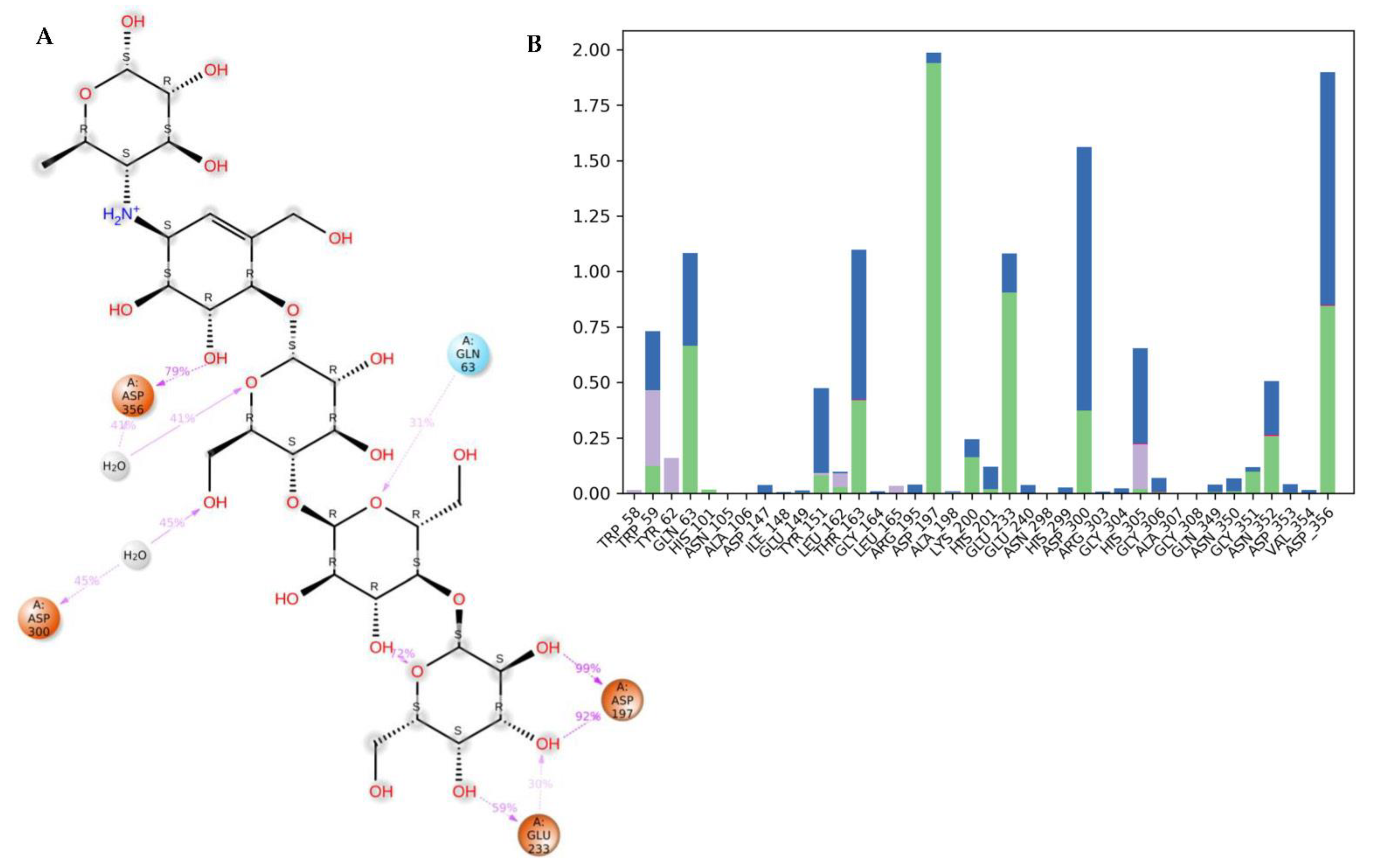
3.3. Molecular Docking Studies
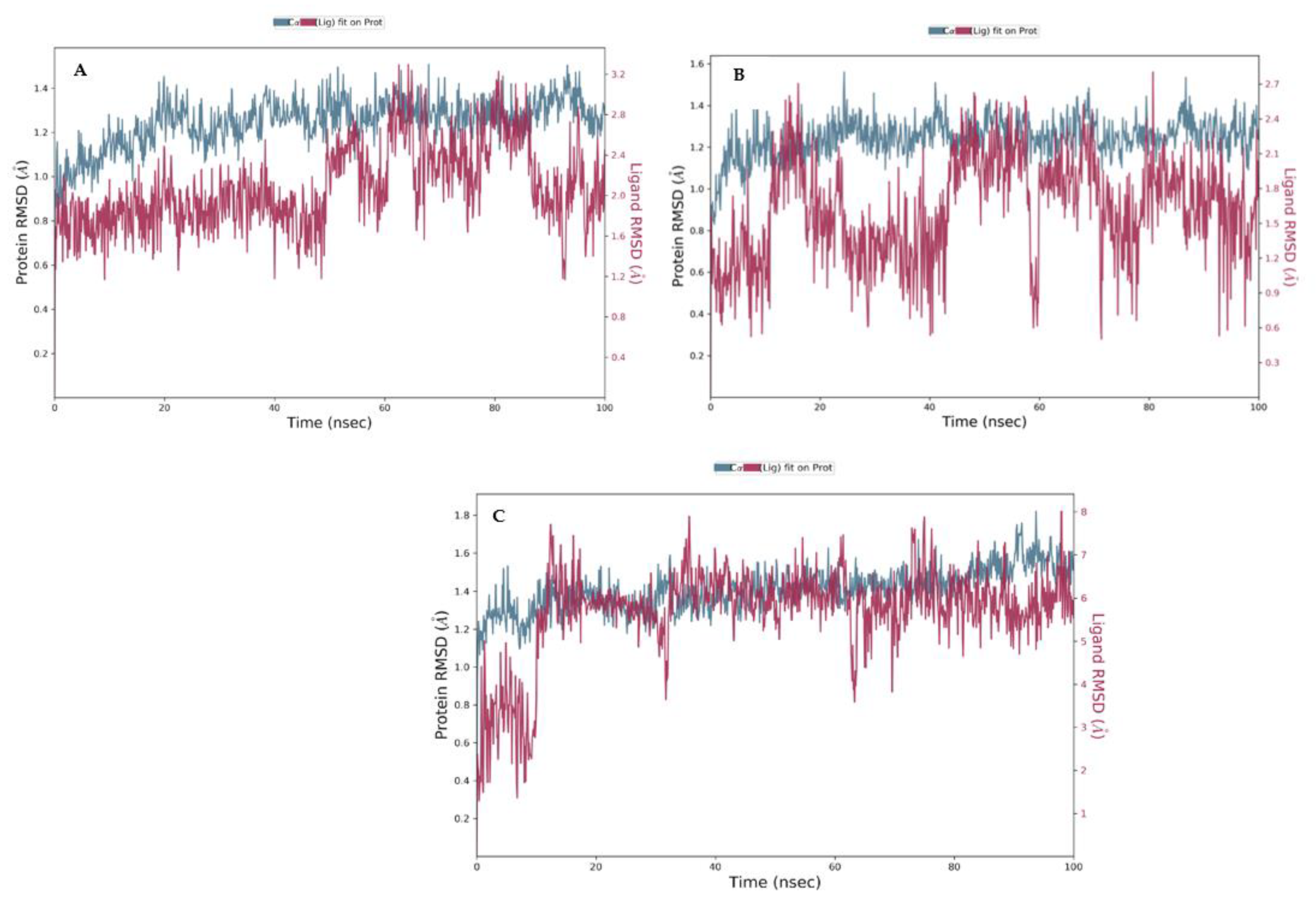
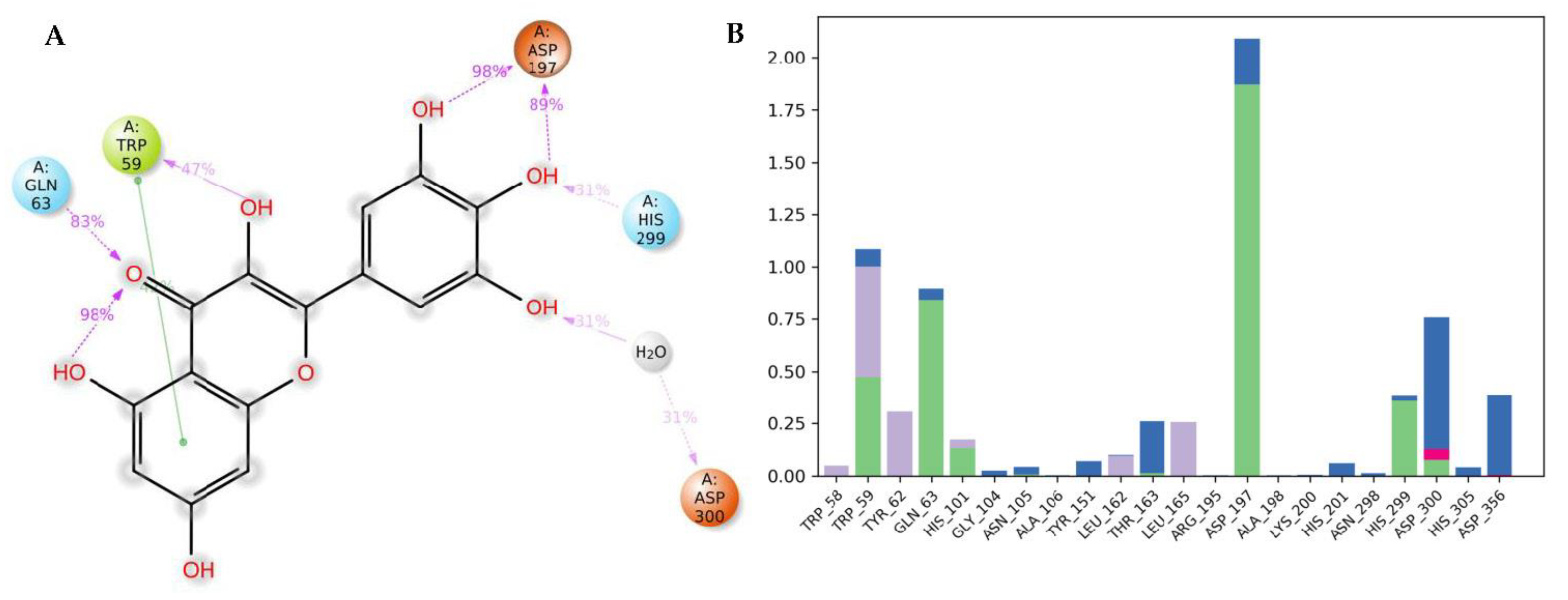
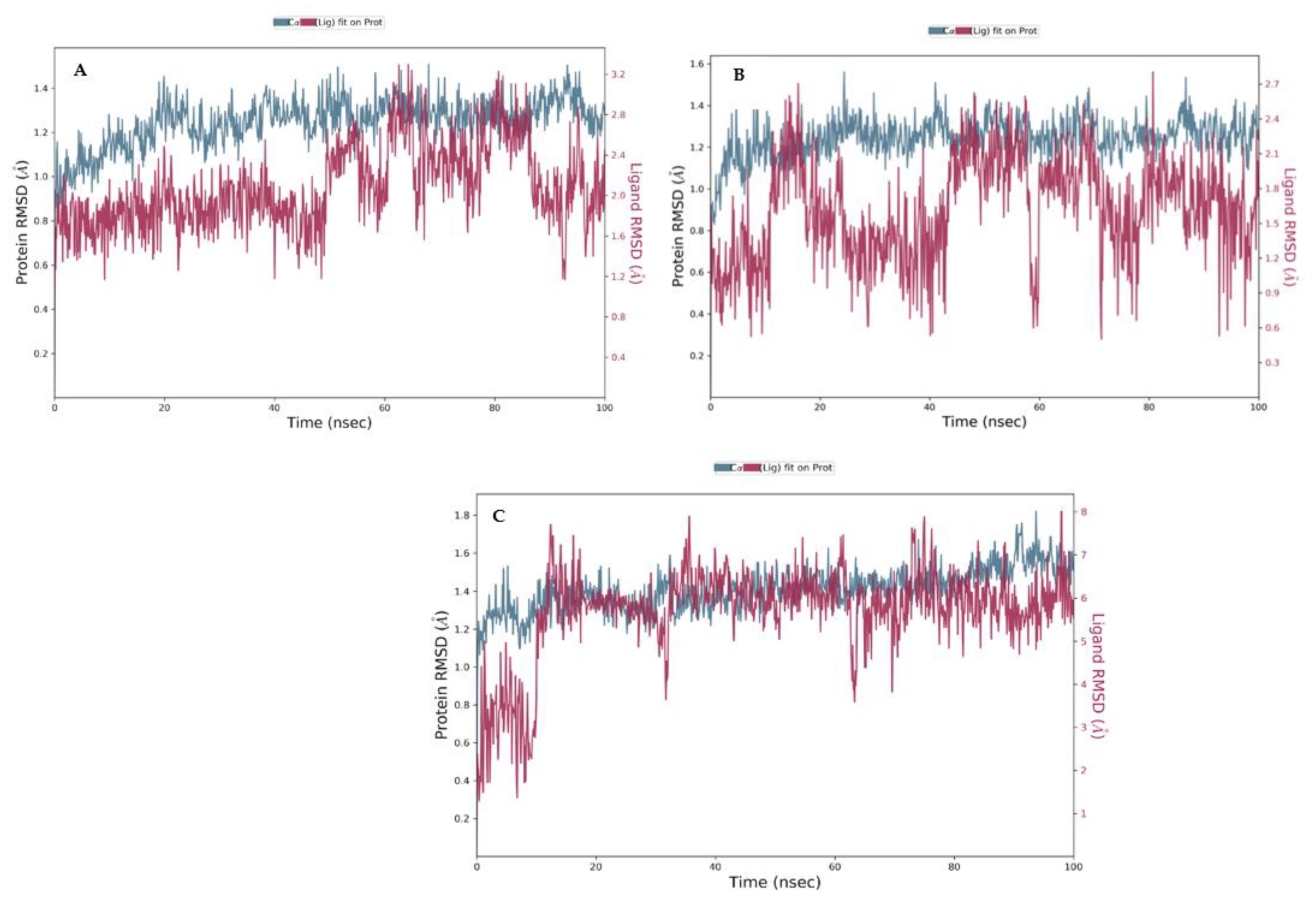
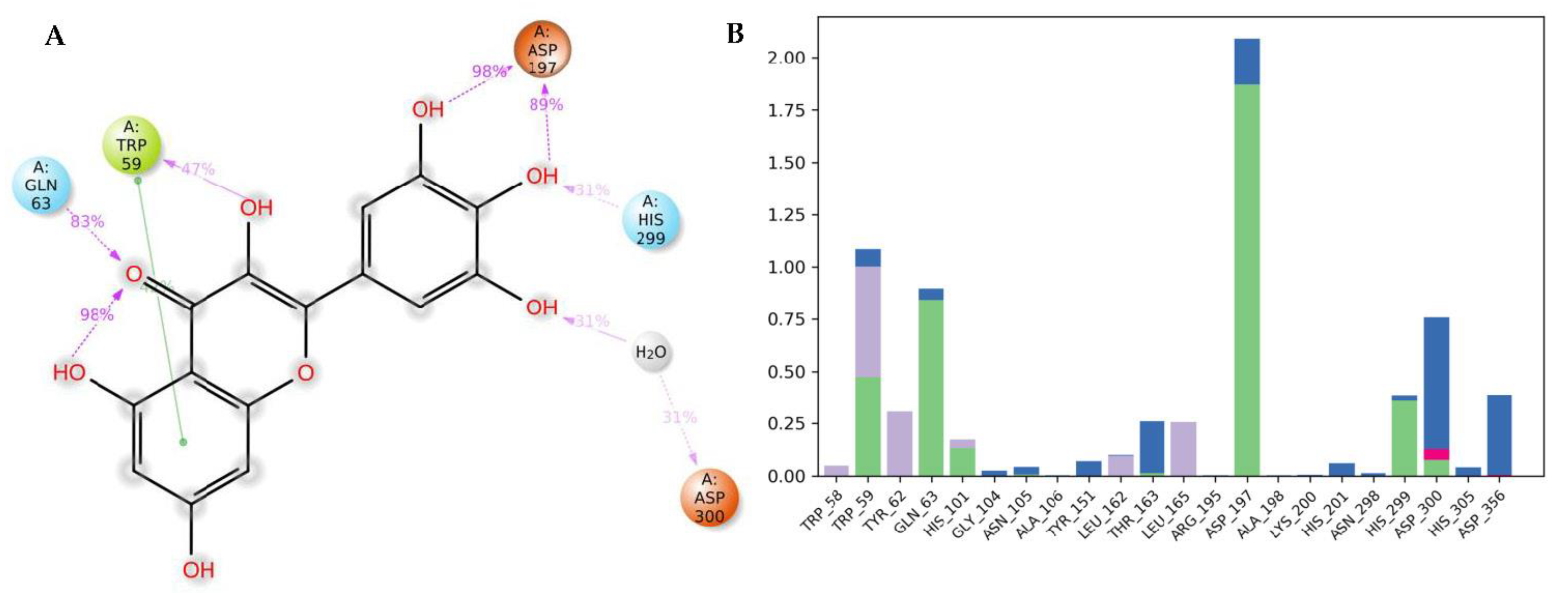
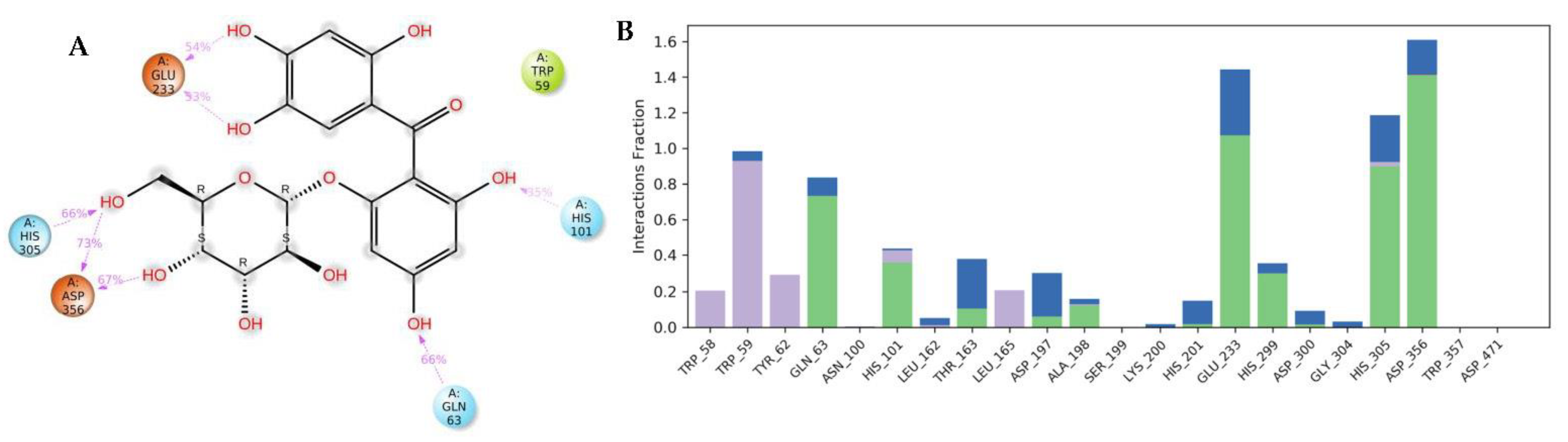
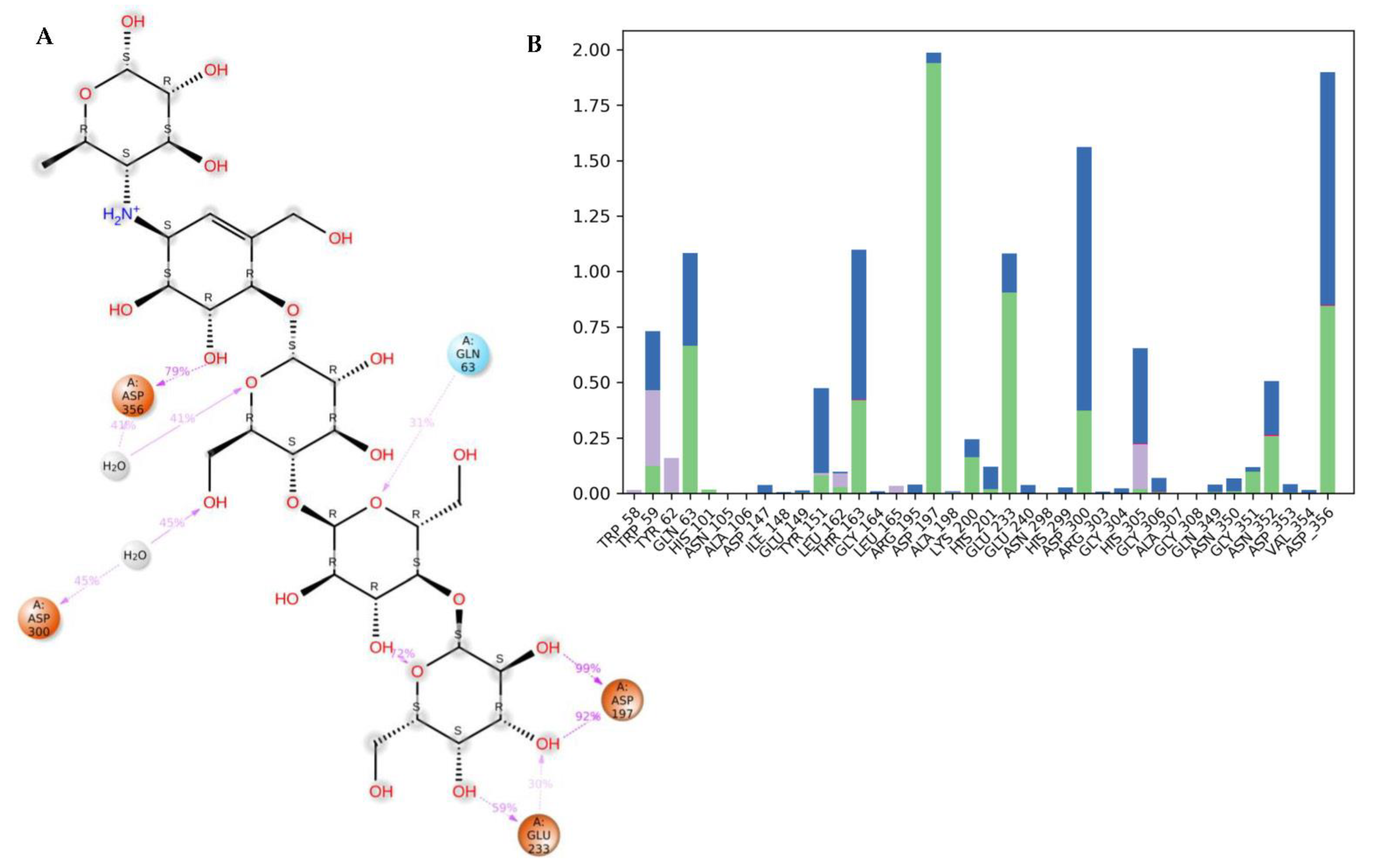
3.4. Molecular Dynamics (MDs)
3.5. In Vitro Alpha-Amylase Inhibitory Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dandekar, P.D.; Kotmale, A.S.; Chavan, S.R.; Kadlag, P.P.; Sawant, S.V.; Dhavale, D.D.; RaviKumar, A. Insights into the Inhibition Mechanism of Human Pancreatic α-Amylase, a Type 2 Diabetes Target, by Dehydrodieugenol B Isolated from Ocimum tenuiflorum. ACS Omega 2021, 6, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Diabetes: Key Facts—World Health Organization. Available online: http://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 15 October 2018).
- Al Dawish, M.A.; Robert, A.A.; Braham, R.; Al Hayek, A.A.; Al Saeed, A.; Ahmed, R.A.; Al Sabaan, F.S. Diabetes Mellitus in Saudi Arabia: A Review of the Recent Literature. Curr. Diabetes Rev. 2016, 12, 359–368. [Google Scholar] [CrossRef]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 2015, 24, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef]
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef]
- Mattio, L.M.; Marengo, M.; Parravicini, C.; Eberini, I.; Dallavalle, S.; Bonomi, F.; Iametti, S.; Pinto, A. Inhibition of Pancreatic α-amylase by Resveratrol Derivatives: Biological Activity and Molecular Modelling Evidence for Cooperativity between Viniferin Enantiomers. Molecules 2019, 24, 3225. [Google Scholar] [CrossRef] [Green Version]
- Chiasson, J.-L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M.; STOP-NIDDM Trail Research Group. Acarbose for prevention of type 2 diabetes mellitus: The STOP-NIDDM randomised trial. Lancet 2002, 359, 2072–2077. [Google Scholar] [CrossRef]
- Riccardi, G.; Giacco, R.; Parillo, M.; Turco, S.; Rivellese, A.A.; Ventura, M.R.; Contadini, S.; Marra, G.; Monteduro, M.; Santeusanio, F.; et al. Efficacy and safety of acarbose in the treatment of Type 1 diabetes mellitus: A placebo-controlled, double-blind, multicentre study. Diabet. Med. 1999, 16, 228–232. [Google Scholar] [CrossRef]
- Henrissat, B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991, 280, 309–316. [Google Scholar] [CrossRef]
- Qin, X.; Ren, L.; Yang, X.; Bai, F.; Wang, L.; Geng, P.; Bai, G.; Shen, Y. Structures of human pancreatic α-amylase in complex with acarviostatins: Implications for drug design against type II diabetes. J. Struct. Biol. 2011, 174, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Akinfemiwa, O.; Muniraj, T. “Amylase”, StatPearls. 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557738/ (accessed on 15 February 2022).
- Ramasubbu, N.; Paloth, V.; Luo, Y.; Brayer, G.D.; Levine, M.J. Structure of Human Salivary α-Amylase at 1.6 Å Resolution: Implications for its Role in the Oral Cavity. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Brás, N.F.; Fernandes, P.A.; Ramos, M.J. QM/MM Studies on the β-Galactosidase Catalytic Mechanism: Hydrolysis and Transglycosylation Reactions. J. Chem. Theory Comput. 2010, 6, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Brayer, G.D.; Sidhu, G.; Maurus, R.; Rydberg, E.H.; Braun, C.; Wang, Y.; Nguyen, N.T.; Overall, C.M.; Withers, S.G. Subsite Mapping of the Human Pancreatic α-Amylase Active Site through Structural, Kinetic, and Mutagenesis Techniques. Biochemistry 2000, 39, 4778–4791. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, M. The Role of Acarbose in the Treatment of Non–Insulin-Dependent Diabetes Mellitus. J. Diabetes its Complicat. 1998, 12, 228–237. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- Anadón, A.; Martínez, M.A.; Castellano, V.; Martínez-Larrañaga, M.R. The role of in vitro methods as alternatives to animals in toxicity testing. Expert Opin. Drug Metab. Toxicol. 2013, 10, 67–79. [Google Scholar] [CrossRef]
- Yeung, A.W.K.; Tzvetkov, N.T.; Durazzo, A.; Lucarini, M.; Souto, E.B.; Santini, A.; Gan, R.-Y.; Jozwik, A.; Grzybek, W.; Horbańczuk, J.O.; et al. Natural products in diabetes research: Quantitative literature analysis. Nat. Prod. Res. 2020, 35, 5813–5827. [Google Scholar] [CrossRef]
- Choudhury, H.; Pandey, M.; Hua, C.K.; Mun, C.S.; Jing, J.K.; Kong, L.; Ern, L.Y.; Ashraf, N.A.; Kit, S.W.; Yee, T.S.; et al. An update on natural compounds in the remedy of diabetes mellitus: A systematic review. J. Tradit. Complement. Med. 2017, 8, 361–376. [Google Scholar] [CrossRef]
- Jugran, A.K.; Rawat, S.; Devkota, H.P.; Bhatt, I.D.; Rawal, R.S. Diabetes and plant-derived natural products: From ethnopharmacological approaches to their potential for modern drug discovery and development. Phytotherapy Res. 2020, 35, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Alhakamy, N.A.; Mohamed, G.A.; Fahmy, U.A.; Eid, B.G.; Ahmed, O.A.A.; Al-Rabia, M.W.; Khedr, A.I.M.; Nasrullah, M.Z.; Ibrahim, S.R.M. New Alpha-Amylase Inhibitory Metabolites from Pericarps of Garcinia mangostana. Life 2022, 12, 384. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Mohamed, G.A.A.; Khayat, M.T.A.; Ahmed, S.; Abo-Haded, H. Garcixanthone D, a New Xanthone, and Other Xanthone Derivatives from Garcinia mangostana Pericarps: Their α-Amylase Inhibitory Potential and Molecular Docking Studies. Starch 2019, 71, 1800354. [Google Scholar] [CrossRef]
- Ibrahim, S.R.; Abdallah, H.M.; El-Halawany, A.M.; Radwan, M.F.; Shehata, I.A.; Al-Harshany, E.M.; Zayed, M.F.; Mohamed, G.A. Garcixanthones B and C, new xanthones from the pericarps of Garcinia mangostana and their cytotoxic activity. Phytochem. Lett. 2018, 25, 12–16. [Google Scholar] [CrossRef]
- Ibrahim, S.R.; Mohamed, G.A.; Elfaky, M.A.; Zayed, M.F.; El-Kholy, A.A.; Abdelmageed, O.H.; Ross, S.A. Mangostanaxanthone VII, a new cytotoxic xanthone from Garcinia mangostana. Z. Naturforsch. C. J. Biosci. 2017, 73, 185–189. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Abdallah, H.M.; El-Halawany, A.M.; Nafady, A.M.; Mohamed, G.A. Mangostanaxanthone VIII, a new xanthone from Garcinia mangostana and its cytotoxic activity. Nat. Prod. Res. 2019, 33, 258–265. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Ibrahim, S.R.; Shaaban, M.I.; Ross, S.A. Mangostanaxanthones I and II, new xanthones from the pericarp of Garcinia mangostana. Fitoterapia 2014, 98, 215–221. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Al-Abd, A.M.; El-Halawany, A.M.; Abdallah, H.; Ibrahim, S.R.M. New xanthones and cytotoxic constituents from Garcinia mangostana fruit hulls against human hepatocellular, breast, and colorectal cancer cell lines. J. Ethnopharmacol. 2017, 198, 302–312. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Ibrahim, S.R. New benzophenones and a dihydroflavanonol from Garcinia mangostana pericarps and their antioxidant and cytotoxic activities. Phytochem. Lett. 2020, 39, 43–48. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Chin, Y.-W. Structural Characterization, Biological Effects, and Synthetic Studies on Xanthones from Mangosteen (Garcinia mangostana), a Popular Botanical Dietary Supplement. Mini-Reviews Org. Chem. 2008, 5, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauman, M.C.; Johnson, J.J. The purple mangosteen (Garcinia mangostana): Defining the anticancer potential of selected xanthones. Pharmacol. Res. 2021, 175, 106032. [Google Scholar] [CrossRef] [PubMed]
- Panda, K.; Alagarasu, K.; Patil, P.; Agrawal, M.; More, A.; Kumar, N.; Mainkar, P.; Parashar, D.; Cherian, S. In Vitro Antiviral Activity of α-Mangostin against Dengue Virus Serotype-2 (DENV-2). Molecules 2021, 26, 3016. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.-Q.; Hou, B.; Yang, L.; Zi, C.-T.; Lv, Y.-F.; Li, J.-Y.; Ren, F.-C.; Yuan, M.-Y.; Hu, J.-M.; Zhou, J. Design, synthesis and cholinesterase inhibitory activity of α-mangostin derivatives. Nat. Prod. Res. 2018, 34, 1380–1388. [Google Scholar] [CrossRef]
- Jiang, M.; Huang, S.; Duan, W.; Liu, Q.; Lei, M. Alpha-mangostin improves endothelial dysfunction in db/db mice through inhibition of aSMase/ceramide pathway. J. Cell. Mol. Med. 2021, 25, 3601–3609. [Google Scholar] [CrossRef]
- Jiang, T.-T.; Ji, C.-F.; Cheng, X.-P.; Gu, S.-F.; Wang, R.; Li, Y.; Zuo, J.; Han, J. α-Mangostin Alleviated HIF-1α-Mediated Angiogenesis in Rats With Adjuvant-Induced Arthritis by Suppressing Aerobic Glycolysis. Front. Pharmacol. 2021, 12, 785586. [Google Scholar] [CrossRef]
- Mahmudah, R.; Adnyana, I.K.; Sukandar, E.Y. Molecular docking studies of α-mangostin, γ-mangostin, and xanthone on peroxisome proliferator-activated receptor gamma diphenyl peptidase-4 enzyme, and aldose reductase enzyme as an antidiabetic drug candidate. J. Adv. Pharm. Technol. Res. 2021, 12, 196–208. [Google Scholar] [CrossRef]
- Usman, F.; Shah, H.S.; Zaib, S.; Manee, S.; Mudassir, J.; Khan, A.; Batiha, G.E.-S.; Abualnaja, K.M.; Alhashmialameer, D.; Khan, I. Fabrication and Biological Assessment of Antidiabetic α-Mangostin Loaded Nanosponges: In Vitro, In Vivo, and In Silico Studies. Molecules 2021, 26, 6633. [Google Scholar] [CrossRef]
- Chen, S.-P.; Lin, S.-R.; Chen, T.-H.; Ng, H.-S.; Yim, H.-S.; Leong, M.K.; Weng, C.-F. Mangosteen xanthone γ-mangostin exerts lowering blood glucose effect with potentiating insulin sensitivity through the mediation of AMPK/PPARγ. Biomed. Pharmacother. 2021, 144, 112333. [Google Scholar] [CrossRef]
- Yeong, K.Y.; Khaw, K.Y.; Takahashi, Y.; Itoh, Y.; Murugaiyah, V.; Suzuki, T. Discovery of gamma-mangostin from Garcinia mangostana as a potent and selective natural SIRT2 inhibitor. Bioorganic Chem. 2019, 94, 103403. [Google Scholar] [CrossRef]
- Akawa, O.B.; Subair, T.I.; Soremekun, O.S.; Olotu, F.A.; Soliman, M.E.S. Structural alterations in the catalytic core of hSIRT2 enzyme predict therapeutic benefits of Garcinia mangostana derivatives in Alzheimer’s disease: Molecular dynamics simulation study. RSC Adv. 2021, 11, 8003–8018. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Jia, L.; Jia, J. γ-mangostin attenuates amyloid-β42-induced neuroinflammation and oxidative stress in microglia-like BV2 cells via the mitogen-activated protein kinases signaling pathway. Eur. J. Pharmacol. 2022, 917, 174744. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Z.; Quan, X.F.; Tian, W.X.; Hu, J.M.; Wang, P.C.; Huang, S.Z.; Cheng, Z.Q.; Liang, W.J.; Zhou, J.; Ma, X.F.; et al. Fatty acid synthase inhibitors of phenolic constituents isolated from Garcinia mangostana. Bioorganic Med. Chem. Lett. 2010, 20, 6045–6047. [Google Scholar] [CrossRef]
- Abdallah, H.M.; El-Bassossy, H.; Mohamed, G.A.; El-Halawany, A.M.; Alshali, K.Z.; Banjar, Z.M. Phenolics from Garcinia mangostana Inhibit Advanced Glycation Endproducts Formation: Effect on Amadori Products, Cross-Linked Structures and Protein Thiols. Molecules 2016, 21, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, L.L.C. Schrödinger Release 2021-4:Ligprerp; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger, L.L.C. Schrödinger Release 2021-4: Glide; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger, L.L.C. Schrödinger Release 2021-4: Desmond Molecular Dynamics System, D.E.; Shaw Research: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger, L.L.C. Schrödinger Release 2021-4: QikProp; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Adcock, S.A.; McCammon, J.A. Molecular Dynamics: Survey of Methods for Simulating the Activity of Proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | # Stars | # rtvFG | CNS | mol_MW | SASA | donorHB | accptHB | QPlogPo/w | QPlogHERG | QPPCaco | QPlogBB | # Metab | QPlogKhsa | PercentHumanOralAbsorption |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recommended range | (0.0–5.0) | (0–2) | (−2 inactive) (+2 active) | (130–725) | (300–1000) | (0–6) | (2.0–20.0) | (−2–6.5) | concern below −5 | <25 poor, >500 great | (−3–1.2) | (1–8) | (−1.5–1.5) | (<25% poor; >80% high) |
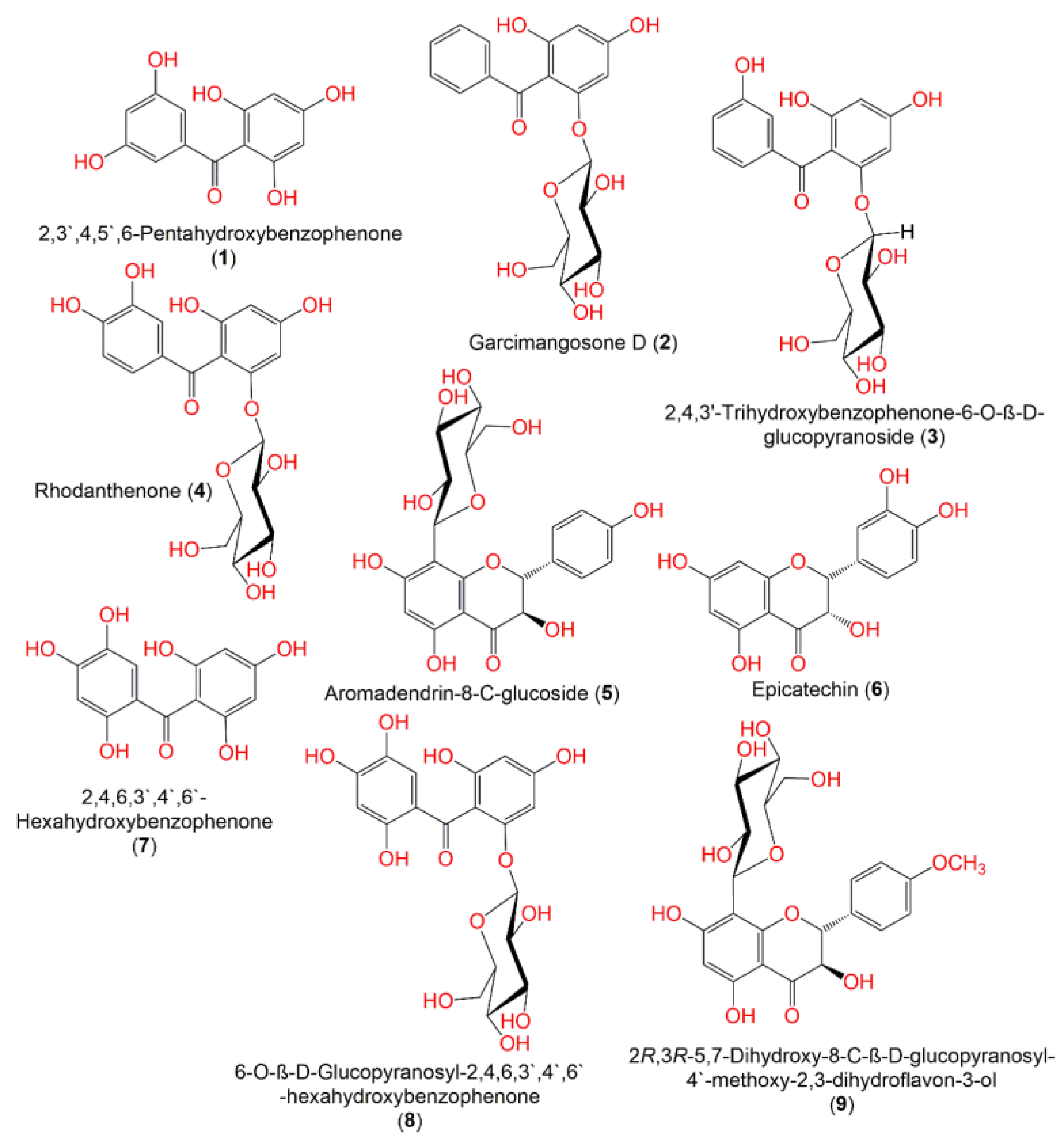
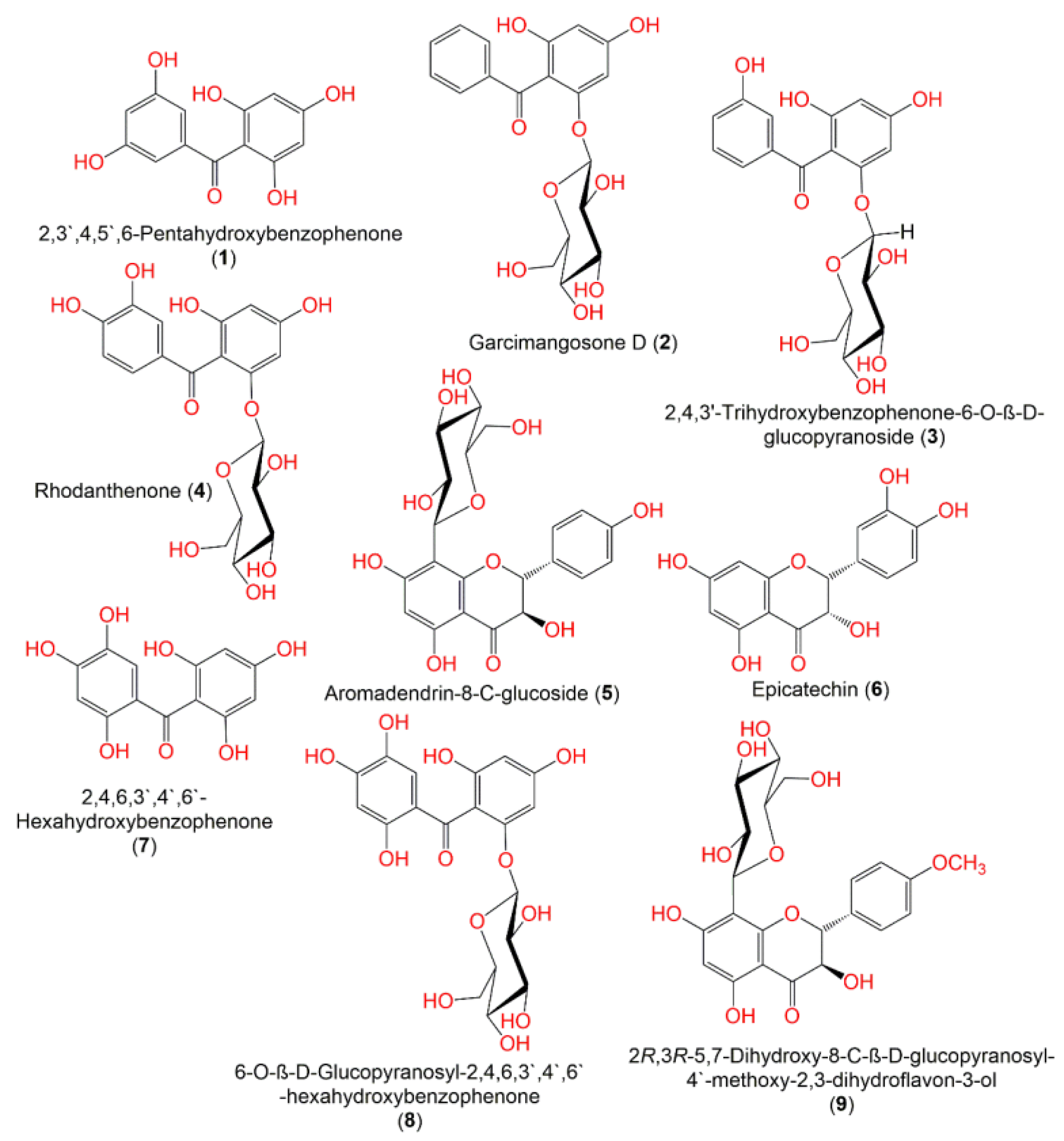
| 2,3′,4,5′,6-Pentahydroxybenzophenone (1) | 0 | 0 | −2 | 262.218 | 470.594 | 3 | 4 | 0.554 | −4.527 | 21.754 | −2.322 | 5 | −0.373 | 54.131 |
| Garcimangosone D (2) | 0 | 1 | −2 | 392.362 | 602.925 | 5 | 12 | −0.474 | −4.889 | 20.469 | −2.663 | 6 | −0.814 | 34.68 |
| 2,4,3’-Trihydroxy benzophenone-6-O-β-D-glucopyranoside (3) | 1 | 1 | −2 | 408.361 | 613.179 | 6 | 13 | −1.068 | −4.798 | 7.724 | −3.154 | 7 | −0.905 | 23.626 |
| Rhodanthenone (4) | 4 | 1 | −2 | 424.36 | 616.186 | 7 | 13 | −1.664 | −4.537 | 2.94 | −3.583 | 8 | −0.982 | 0 |
| Aromadendrin-8-C-glucoside (5) | 5 | 0 | −2 | 450.398 | 681.978 | 7 | 14 | −1.532 | −5.347 | 2.688 | −3.702 | 11 | −0.852 | 0 |
| Epicatechin (6) | 0 | 0 | −2 | 290.08 | 519.049 | 4 | 6 | 0.098 | −4.869 | 21.252 | −2.3 | 6 | −0.426 | 51.276 |
| 2,4,6,3′,4′,6′-hexahydroxybenzophenone (7) | 0 | 0 | −2 | 278.218 | 474.191 | 3 | 4 | 0.561 | −4.376 | 13.745 | −2.542 | 6 | −0.35 | 37.642 |
| 6-O-β-D-Glucopyranosyl-2,4,6,3′,4′,6′-hexahydroxybenzophenone (8) | 5 | 1 | −2 | 440.36 | 663.47 | 7 | 13 | −1.65 | −5.119 | 1.514 | −4.259 | 9 | −1.028 | 0 |
| 2R,3R-5,7-Dihydroxy-8-C-β-D-glucopyranosyl-4-methoxy-2,3-dihydroflavon-3-ol (9) | 2 | 0 | −2 | 464.425 | 708.735 | 6 | 14 | −0.782 | −5.418 | 8.732 | −3.231 | 11 | −0.769 | 13.292 |
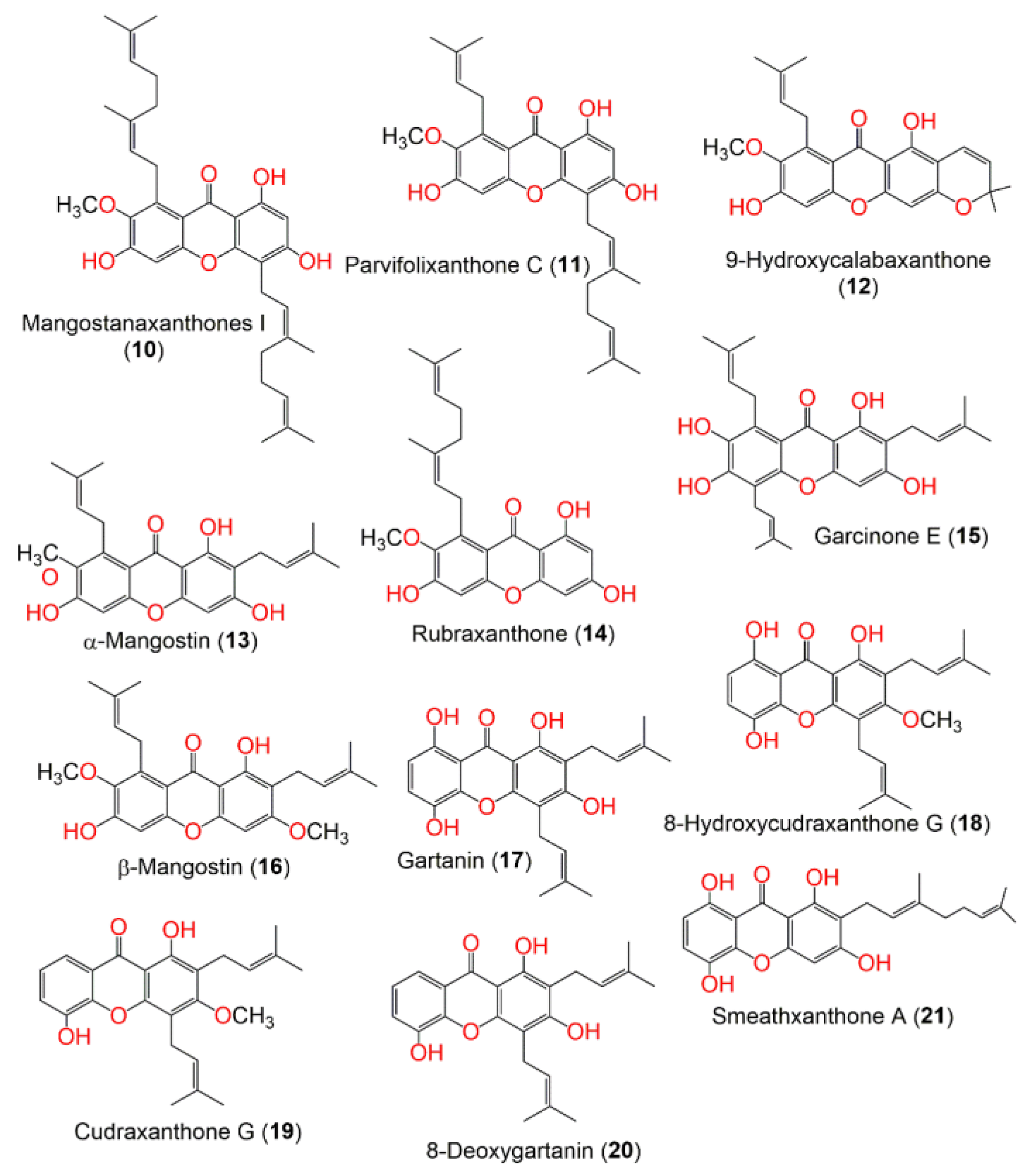
| Mangostanaxanthone I (10) | 4 | 0 | −2 | 546.702 | 951.529 | 2 | 5 | 7.657 | −6.17 | 366.708 | −2.059 | 16 | 1.967 | 91.76 |
| Parvifolixanthone C (11) | 2 | 0 | −2 | 478.584 | 815.402 | 2 | 5 | 5.954 | −5.517 | 369.062 | −1.714 | 13 | 1.369 | 94.798 |
| 9-Hydroxycalabaxanthone (12) | 0 | 0 | 0 | 408.45 | 671.549 | 1 | 5 | 4.793 | −5.212 | 1503.476 | −0.575 | 6 | 0.896 | 100 |
| α-Mangostin (13) | 1 | 0 | −2 | 410.466 | 693.506 | 2 | 5 | 4.655 | −5.125 | 882.919 | −1.013 | 10 | 0.813 | 100 |
| Rubraxanthone (14) | 1 | 0 | −2 | 410.466 | 676.001 | 2 | 5 | 4.469 | −5.042 | 596.126 | −1.209 | 10 | 0.733 | 100 |
| Garcinone E (15) | 2 | 0 | −2 | 464.557 | 796.788 | 3 | 5 | 5.513 | −5.427 | 485.861 | −1.511 | 13 | 1.178 | 94.349 |
| β-Mangostin (16) | 2 | 0 | −1 | 424.493 | 748.032 | 1 | 5 | 5.511 | −5.493 | 1511.156 | −0.829 | 10 | 1.103 | 100 |
| Gartanin (17) | 1 | 0 | −2 | 396.439 | 687.965 | 2 | 4 | 4.512 | −5.282 | 304.523 | −1.524 | 10 | 0.9 | 100 |
| 8-Hydroxycudraxanthone G (18) | 1 | 0 | −2 | 410.466 | 696.392 | 1 | 4 | 5.004 | −5.163 | 369.475 | −1.415 | 10 | 1.095 | 89.241 |
| Cudraxanthone G (19) | 2 | 0 | −1 | 394.466 | 706.416 | 1 | 4 | 5.273 | −5.489 | 1075.829 | −0.897 | 9 | 1.085 | 100 |
| 8-Deoxygartanin (20) | 1 | 0 | −1 | 380.44 | 667.452 | 2 | 4 | 4.546 | −5.262 | 782.505 | −1 | 9 | 0.813 | 100 |
| Smeathxanthone A (21) | 1 | 0 | −2 | 396.439 | 710.514 | 2 | 4 | 4.445 | −5.747 | 146.974 | −2.007 | 10 | 0.905 | 91.759 |
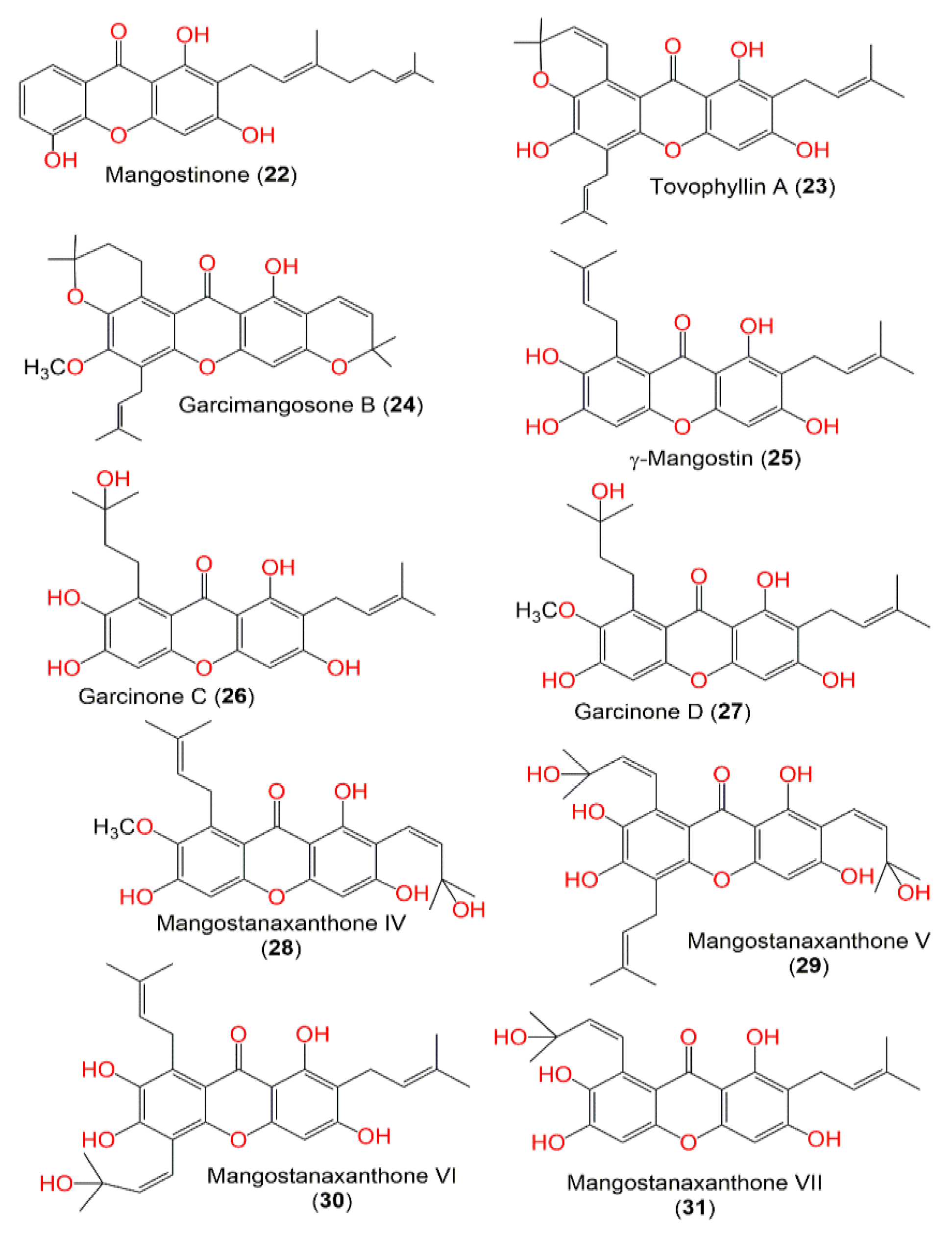
| Mangostinone (22) | 1 | 0 | −2 | 380.44 | 697.748 | 2 | 4 | 4.513 | −5.815 | 352.029 | −1.507 | 9 | 0.841 | 100 |
| Tovophyllin A (23) | 2 | 0 | −2 | 462.541 | 784.186 | 2 | 5 | 5.712 | −5.601 | 755.786 | −1.111 | 9 | 1.323 | 100 |
| Garcimangosone B (24) | 2 | 0 | 0 | 476.568 | 784.948 | 0 | 5 | 6.434 | −5.468 | 2337.985 | −0.369 | 6 | 1.526 | 100 |
| γ-Mangostin (25) | 1 | 0 | −2 | 396.439 | 675.17 | 3 | 5 | 3.803 | −5.104 | 289.754 | −1.511 | 10 | 0.595 | 93.281 |
| Garcinone C (26) | 1 | 0 | −2 | 414.454 | 700.302 | 4 | 5 | 3.29 | −5.268 | 123.883 | −2.072 | 9 | 0.397 | 83.673 |
| GarcinoneD (27) | 1 | 0 | −2 | 428.481 | 671.867 | 3 | 5.25 | 3.932 | −4.683 | 433.902 | −1.372 | 9 | 0.524 | 100 |
| Mangostanaxanthone IV (28) | 0 | 0 | −2 | 426.465 | 691.651 | 3 | 5 | 4.098 | −4.918 | 588.906 | −1.233 | 8 | 0.594 | 100 |
| Mangostanaxanthone V (29) | 1 | 0 | −2 | 496.556 | 788.467 | 5 | 6 | 4.363 | −4.975 | 185.916 | −2.009 | 9 | 0.724 | 80.147 |
| Mangostanaxanthone VI (30) | 2 | 0 | −2 | 480.557 | 798.713 | 4 | 5 | 4.955 | −5.285 | 295.03 | −1.792 | 11 | 0.954 | 100 |
| Mangostanaxanthone VI (30) | 1 | 0 | −2 | 480.557 | 794.228 | 4 | 5 | 4.842 | −5.262 | 275.224 | −1.827 | 11 | 0.918 | 100 |
| Mangostanaxanthone VII (31) | 0 | 0 | −2 | 412.438 | 689.283 | 4 | 5 | 3.301 | −5.171 | 184.105 | −1.805 | 8 | 0.396 | 86.814 |
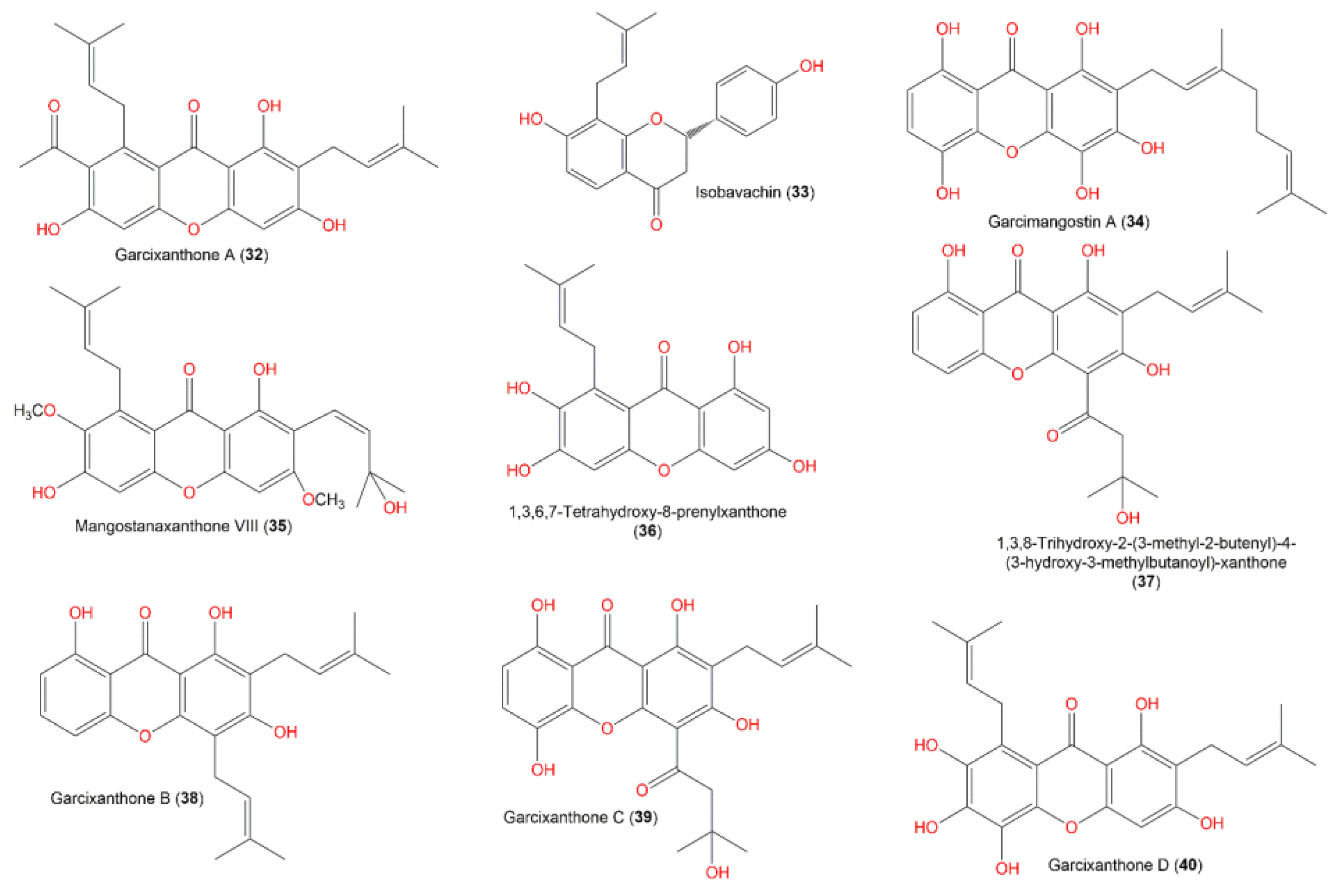
| Garcixanthone A (32) | 1 | 0 | −2 | 422.477 | 716.018 | 1 | 5 | 4.725 | −5.209 | 396.428 | −1.407 | 9 | 0.942 | 100 |
| Isobavachin (33) | 0 | 0 | −1 | 324.376 | 562.774 | 2 | 4 | 3.082 | −4.496 | 389.48 | −0.954 | 7 | 0.412 | 91.356 |
| Garcimangostin A (34) | 1 | 0 | −2 | 412.438 | 710.47 | 3 | 4 | 3.756 | −5.504 | 69.776 | −2.389 | 11 | 0.667 | 81.934 |
| Mangostanaxanthone VIII (35) | 0 | 0 | −1 | 440.492 | 724.392 | 2 | 5 | 4.855 | −5.094 | 1113.098 | −0.982 | 8 | 0.827 | 100 |
| 1,3,6,7-tetrahydroxy-8-prenylxanthone (36) | 0 | 0 | −2 | 328.321 | 550.081 | 3 | 5 | 2.08 | −4.565 | 147.3 | −1.514 | 7 | 0.055 | 77.93 |
| 1,3,8-Trihydroxy-2-(3-methyl-2-butenyl)-4-(3-hydroxy-3-methylbutanoyl)-xanthone (37) | 0 | 0 | −2 | 412.438 | 677.829 | 0 | 4 | 4.66 | −5.009 | 139.018 | −1.856 | 8 | 0.962 | 92.589 |
| Garcixanthone B (38) | 1 | 0 | −2 | 380.44 | 657.216 | 1 | 3 | 5.031 | −4.999 | 605.344 | −1.083 | 9 | 1.088 | 93.235 |
| Garcixanthone C (39) | 1 | 0 | −2 | 428.438 | 704.892 | 1 | 4 | 4.215 | −5.258 | 75.781 | −2.296 | 9 | 0.871 | 85.268 |
| GarcixanthoneD (40) | 1 | 0 | −2 | 412.438 | 706.392 | 4 | 5.25 | 3.144 | −5.245 | 86.596 | −2.214 | 11 | 0.436 | 80.031 |
| Compound No. | XP G.Score | Glide G.Score | Docking Score (Kcal/mol) | Glide Emodel | Prime Energy | MMGBSA dG Bind | IC50 (µM) |
|---|---|---|---|---|---|---|---|
| 6-O-β-D-Glucopyranosyl-2,4,6,3′,4′,6′-hexahydroxybenzophenone (8) | −12.425 | −12.425 | −12.227 | −73.234 | −21,238.5 | −24.48 | 7.1 |
| Myricetin | −12.319 | −12.319 | −12.319 | −68.813 | −21,398.5 | −51.23 | - |
| Acarbose | −12.201 | −12.201 | −12.201 | −75.139 | −20,979.6 | −15.9 | 6.7 |
| Aromadendrin-8-C-glucoside (5) | −11.855 | −11.855 | −11.823 | −58.225 | −21,216.7 | −36.82 | 8.3 |
| Epicatechin (6) | −11.135 | −11.135 | −11.108 | −61.064 | −21,253.4 | −43.73 | 8.9 |
| Rhodanthenone (4) | −11.048 | −11.048 | −10.887 | −65.449 | −21,207.1 | −44.87 | 10.4 |
| Garcixanthone D (40) | −10.989 | −10.989 | −10.764 | −53.919 | −21,260.7 | −41.35 | 11.1 |
| Mangostanaxanthone V (29) | −10.254 | −10.254 | −10.088 | −70.702 | −21,346.8 | −38.65 | 11.9 |
| 2R,3R-5,7-Dihydroxy-8-C-β-D-glucopyranosyl-4-methoxy-2,3-dihydroflavon-3-ol (9) | −10.107 | −10.107 | −10.094 | −52.556 | −21,192.9 | −26.05 | 12.8 |
| 1,3,8-Trihydroxy-2-(3-methyl-2-butenyl)-4-(3-hydroxy-3-methylbutanoyl)-xanthone (37) | −9.99 | −9.99 | −8.574 | −59.986 | −21,411.4 | −33.11 | 14.2 |
| 2,4,3’-Trihydroxy benzophenone-6-O-β-D-glucopyranoside (3) | −9.77 | −9.77 | −9.615 | −64.384 | −21,203.7 | −19.85 | 15.8 |
| Garcimangosone D (2) | −9.696 | −9.696 | −9.541 | −56.996 | −21,194.9 | −23.33 | - |
| γ-Mangostin (25) | −9.518 | −9.518 | −7.058 | −54.095 | −21,255.6 | −2.33 | - |
| Isobavachin (33) | −8.79 | −8.79 | −8.79 | −59.656 | −21,270.1 | −35.87 | - |
| 2,4,6,3′,4′,6′-Hexahydroxybenzophenone (7) | −8.647 | −8.647 | −8.11 | −43.182 | −21,308.1 | 3.74 | - |
| Garcixanthone C (39) | −8.617 | −8.617 | −6.406 | −55.483 | −21,348.2 | −3.66 | 17.1 |
| Garcimangostin A (34) | −8.367 | −8.367 | −8.248 | −60.875 | −21,243.7 | −29.37 | - |
| Garcinone E (15) | −8.245 | −8.245 | −8.114 | −59.531 | −21,285.8 | −33.75 | 18.8 |
| 1,3,6,7-Tetrahydroxy-8-prenylxanthone (36) | −8.213 | −8.213 | −7.915 | −50.339 | −21,298.3 | −33.28 | - |
| Garcixanthone B (38) | −7.978 | −7.978 | −7.904 | −58.072 | −21,314.5 | −25.91 | - |
| Smeathxanthone A (21) | −7.976 | −7.976 | −7.864 | −56.159 | −21,321.2 | −39.59 | - |
| Mangostanaxanthone VII (31) | −7.871 | −7.871 | −6.501 | −61.697 | −21,260.9 | −4.38 | 19.5 |
| Mangostinone (22) | −7.843 | −7.843 | −6.507 | −47.68 | −21,267.2 | −13.3 | - |
| Tovophyllin A (23) | −7.646 | −7.646 | −6.164 | −46.506 | −21,311.1 | −35.81 | 20.4 |
| Gartanin (17) | −7.635 | −7.635 | −5.417 | −52.868 | −21,268 | −11.2 | 21.2 |
| Mangostanaxanthone VI (30) | −7.577 | −7.577 | −7.479 | −61.529 | −21,297.6 | −24.81 | 21.9 |
| 2,3′,4,5′,6-Pentahydroxybenzophenone (1) | −7.502 | −7.502 | −7.094 | −49.619 | −21,321.2 | −28.82 | - |
| 8-Deoxygartanin (20) | −7.293 | −7.293 | −4.769 | −53.856 | −21,244.6 | −17.72 | - |
| Cudraxanthone G (19) | −7.12 | −7.12 | −7.12 | −55 | −21,237.8 | −44.68 | - |
| Mangostanaxanthone I (10) | −7.107 | −7.107 | −6.924 | −53.946 | −21,323.1 | −48.36 | 28.3 |
| Garcinone C (26) | −7.098 | −7.098 | −6.962 | −53.746 | −21,319.8 | −37.48 | - |
| 8-Hydroxycudraxanthone G (18) | −6.667 | −6.667 | −6.66 | −49.088 | −21,268.2 | −41.81 | - |
| β-Mangostin (16) | −6.62 | −6.62 | −5.284 | −38.5 | −21,270.8 | −24.24 | - |
| Parvifolixanthone C (11) | −6.566 | −6.566 | −6.381 | −56.272 | −21,326.6 | −39.95 | - |
| Mangostanaxanthone VIII (35) | −6.541 | −6.541 | −6.483 | −45.301 | −21,299.1 | −30.71 | 40.6 |
| α-Mangostin (13) | −6.323 | −6.323 | −5.038 | −41.996 | −21,302.6 | −29.84 | 44.1 |
| Mangostanaxanthone IV (28) | −6.293 | −6.293 | −4.907 | −42.202 | −21,323.6 | −24.42 | 46.5 |
| Garcinone D (27) | −5.823 | −5.823 | −4.457 | −49.712 | −21,335.2 | −34.69 | - |
| Garcixanthone A (32) | −5.082 | −5.082 | −3.764 | −47.142 | −21,352.5 | −37.06 | - |
| Garcimangosone B (24) | −4.978 | −4.978 | −4.976 | −45.586 | −21,289.8 | −39.15 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omar, A.M.; AlKharboush, D.F.; Mohammad, K.A.; Mohamed, G.A.; Abdallah, H.M.; Ibrahim, S.R.M. Mangosteen Metabolites as Promising Alpha-Amylase Inhibitor Candidates: In Silico and In Vitro Evaluations. Metabolites 2022, 12, 1229. https://doi.org/10.3390/metabo12121229
Omar AM, AlKharboush DF, Mohammad KA, Mohamed GA, Abdallah HM, Ibrahim SRM. Mangosteen Metabolites as Promising Alpha-Amylase Inhibitor Candidates: In Silico and In Vitro Evaluations. Metabolites. 2022; 12(12):1229. https://doi.org/10.3390/metabo12121229
Chicago/Turabian StyleOmar, Abdelsattar M., Dana F. AlKharboush, Khadijah A. Mohammad, Gamal A. Mohamed, Hossam M. Abdallah, and Sabrin R. M. Ibrahim. 2022. "Mangosteen Metabolites as Promising Alpha-Amylase Inhibitor Candidates: In Silico and In Vitro Evaluations" Metabolites 12, no. 12: 1229. https://doi.org/10.3390/metabo12121229
APA StyleOmar, A. M., AlKharboush, D. F., Mohammad, K. A., Mohamed, G. A., Abdallah, H. M., & Ibrahim, S. R. M. (2022). Mangosteen Metabolites as Promising Alpha-Amylase Inhibitor Candidates: In Silico and In Vitro Evaluations. Metabolites, 12(12), 1229. https://doi.org/10.3390/metabo12121229