
An Optimised Monophasic Faecal Extraction Method for LC-MS Analysis and Its Application in Gastrointestinal Disease

 , ,
, ,  , and
, and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Faecal Samples
2.3. Chemicals and Reagents
2.4. Extraction Protocol
2.5. Untargeted LC-MS Metabolite Measurement
2.6. Targeted LC-MS Metabolite Measurement
2.7. Method Application
2.8. Mass Spectrometry Data Processing
2.9. Data and Statistical Analysis
2.10. Putative Metabolite Identification
3. Results
3.1. Analysis of Sample Weight
3.2. Analysis of Extraction Solvent
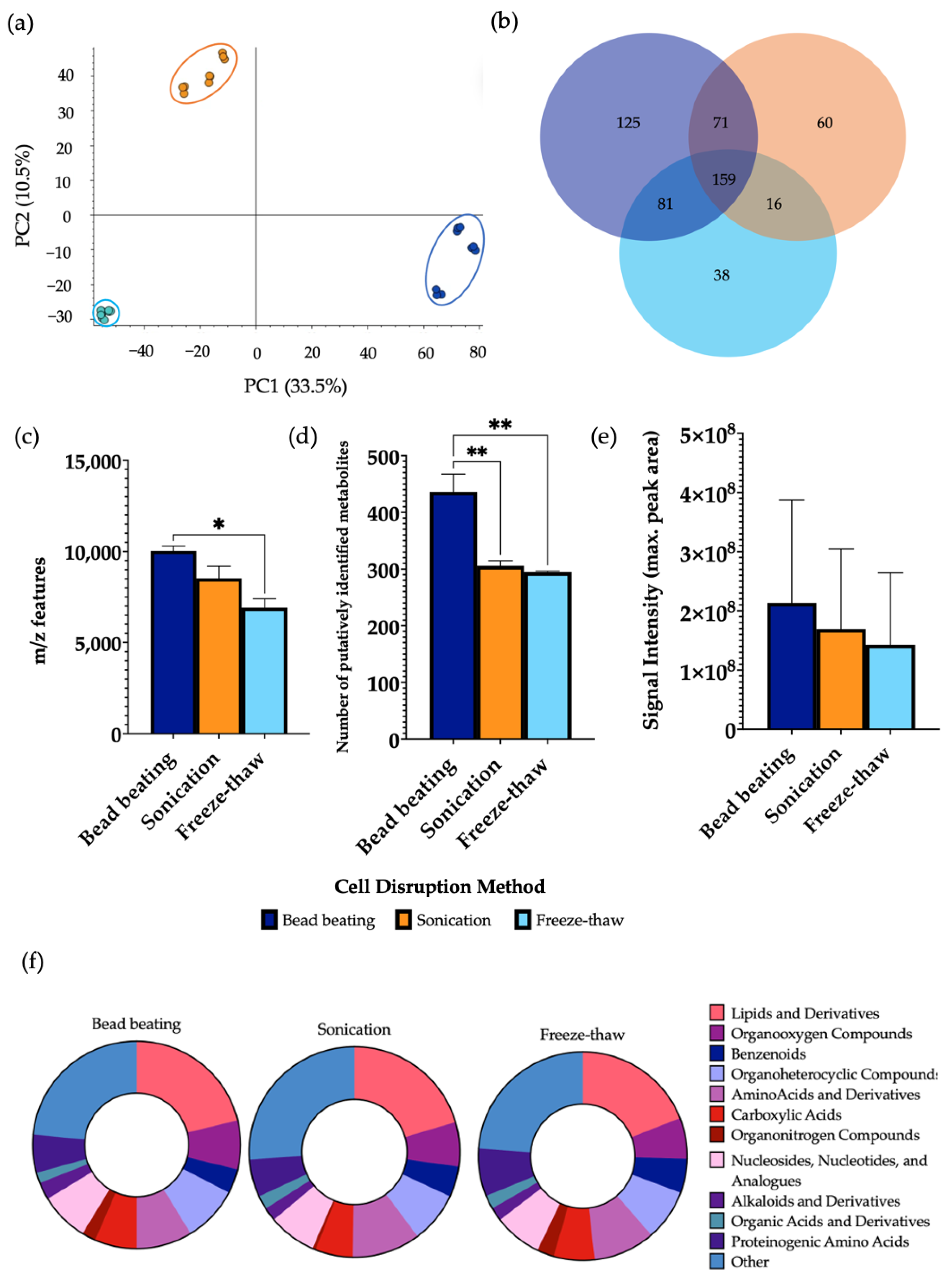
3.3. Analysis of the Cellular Disruption Method
3.4. Analysis of Sample-to Solvent Ratio
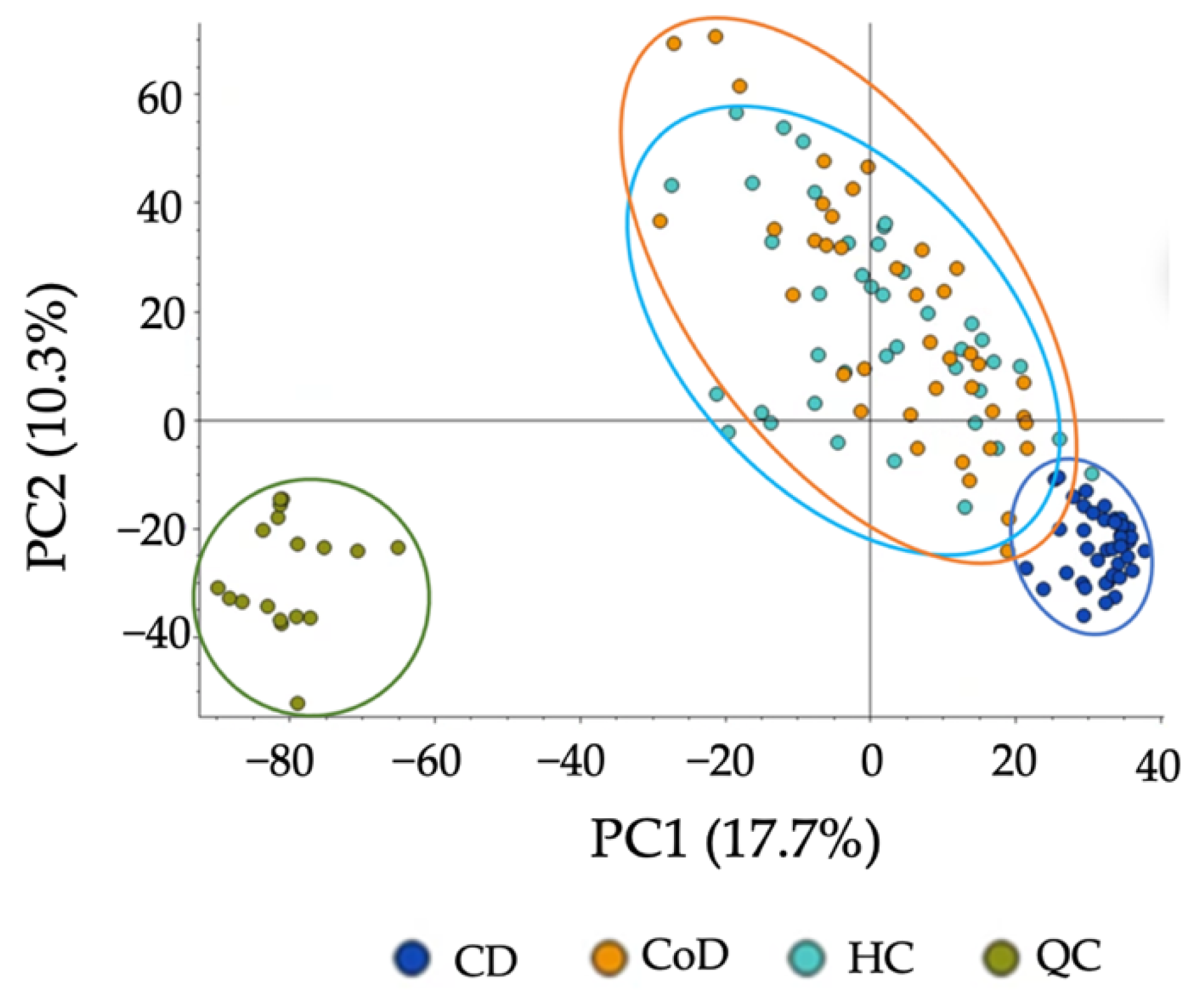
3.5. Applicability of the Method to Patients with Gastrointestinal Disease
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef] [PubMed]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Erben, V.; Poschet, G.; Schrotz-King, P.; Brenner, H. Evaluation of different stool extraction methods for metabolomics measurements in human faecal samples. BMJ NPH 2020, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Coker, O.O.; Liu, C.; Wu, W.K.K.; Wong, S.H.; Jia, W.; Sung, J.J.Y.; Yu, J. Altered gut metabolites and microbiota interactions are implicated in colorectal carcinogenesis and can be non-invasive diagnostic biomarkers. Microbiome 2022, 10, 1–12. [Google Scholar] [CrossRef]
- Yu, D.; Du, J.; Pu, X.; Zheng, L.; Chen, S.; Wang, N.; Li, J.; Chen, S.; Pan, S.; Shen, B. The Gut Microbiome and Metabolites Are Altered and Interrelated in Patients with Rheumatoid Arthritis. Front. Cell. Infect Microbiol. 2022, 11, 763507. [Google Scholar] [CrossRef]
- Fang, Q.; Liu, N.; Zheng, B.; Guo, F.; Zeng, X.; Huang, X.; Ouyang, D. Roles of Gut Microbial Metabolites in Diabetic Kidney Disease. Front. Endocrinol. 2021, 12, 636175. [Google Scholar] [CrossRef]
- Zheng, L.; Wen, X.L.; Duan, S.L. Role of metabolites derived from gut microbiota in inflammatory bowel disease. World J. Clin. Cases 2022, 10, 2660–2677. [Google Scholar] [CrossRef]
- Di Cagno, R.; Rizzello, C.G.; Gagliardi, F.; Ricciuti, P.; Ndagijimana, M.; Francavilla, R.; Guerzoni, M.E.; Crecchio, C.; Gobbetti, M.; De Angelis, M. Different fecal microbiotas and volatile organic compounds in treated and untreated children with celiac disease. Appl. Environ. Microbiol. 2009, 75, 3963–3971. [Google Scholar] [CrossRef]
- Metwaly, A.; Dunkel, A.; Waldschmitt, N.; Raj, A.C.D.; Lagkouvardos, I.; Corraliza, A.M.; Mayorgas, A.; Martinez-Medina, M.; Reiter, S.; Schloter, M.; et al. Integrated microbiota and metabolite profiles link Crohn’s disease to sulfur metabolism. Nat. Commun. 2020, 11, 4322. [Google Scholar] [CrossRef]
- Heinken, A.; Hertel, J.; Thiele, I. Metabolic modelling reveals broad changes in gut microbial metabolism in inflammatory bowel disease patients with dysbiosis. NPJ Syst. Biol. Appl. 2021, 7, 19. [Google Scholar] [CrossRef]
- Khalkhal, E.; Rezaei-Tavirani, M.; Fathi, F.; Nobakht, M.; Gh, B.F.; Taherkhani, A.; Rostami-Nejad, M.; Asri, N.; Haidari, M.H. Screening of Altered Metabolites and Metabolic Pathways in Celiac Disease Using NMR Spectroscopy. Biomed. Res. Int. 2021, 1798783. [Google Scholar] [CrossRef] [PubMed]
- Martín-Masot, R.; Mota-Martorell, N.; Jové, M.; Maldonado, J.; Pamplona, R.; Nestares, T. Alterations in one-carbon metabolism in celiac disease. Nutrients 2020, 12, 3723. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, F.; Dubbelman, A.C.; Karu, N.; Harms, A.C.; Hankemeier, T. Towards standards for human fecal sample preparation in targeted and untargeted lc-hrms studies. Metabolites 2021, 11, 364. [Google Scholar] [CrossRef] [PubMed]
- Moosmang, S.; Pitscheider, M.; Sturm, S.; Seger, C.; Tilg, H.; Halabalaki, M.; Stuppner, H. Metabolomic analysis—Addressing NMR and LC-MS related problems in human feces sample preparation. Clin. Chim. Acta 2019, 489, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Nandania, J.; Peddinti, G.; Pessia, A.; Kokkonen, M.; Velagapudi, V. Validation and automation of a high-throughput multitargeted method for semiquantification of endogenous metabolites from different biological matrices using tandem mass spectrometry. Metabolites 2018, 8, 44. [Google Scholar] [CrossRef]
- Berardi, D.; Hunter, Y.; van den Driest, L.; Farrell, G.; Rattray, N.J.W.; Rattray, Z. The Differential Metabolic Signature of Breast Cancer Cellular Response to Olaparib Treatment. Cancers 2022, 14, 3661. [Google Scholar] [CrossRef]
- Wu, J.; An, Y.; Yao, J.; Wang, Y.; Tang, H. An optimised sample preparation method for NMR-based faecal metabonomic analysis. Analyst 2010, 135, 1023–1030. [Google Scholar] [CrossRef]
- Gholib, G.; Heistermann, M.; Agil, M.; Supriatna, I.; Purwantara, B.; Nugraha, T.P.; Engelhardt, A. Comparison of fecal preservation and extraction methods for steroid hormone metabolite analysis in wild crested macaques. Primates 2018, 59, 281–292. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Z.; Hu, B.; Huang, W.; Yuan, C.; Zou, L. Response of gut microbiota to metabolite changes induced by endurance exercise. Front. Micro. 2018, 9, 765. [Google Scholar] [CrossRef]
- De Zawadzki, A.; Thiele, M.; Suvitaival, T.; Wretlind, A.; Kim, M.; Ali, M.; Bjerre, A.F.; Stahr, K.; Mattila, I.; Hansen, T.; et al. High-Throughput UHPLC-MS to Screen Metabolites in Feces for Gut Metabolic Health. Metabolites 2022, 12, 211. [Google Scholar] [CrossRef]
- Want, E.J.; O’Maille, G.; Smith, C.A.; Brandon, T.R.; Uritboonthai, W.; Qin, C.; Trauger, S.A.; Siuzdak, G. Solvent-dependent metabolite distribution, clustering, and protein extraction for serum profiling with mass spectrometry. Anal. Chem. 2006, 78, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.M.; Betts, E.L.; Yiangou, L.; Roldan, J.O.; Tsaousis, A.D.; Thompson, G.S. Establishing a metabolite extraction method to study the metabolome of blastocystis using nmr. Molecules 2011, 26, 3285. [Google Scholar] [CrossRef] [PubMed]
- Deda, O.; Gika, H.G.; Wilson, I.D.; Theodoridis, G.A. An overview of fecal sample preparation for global metabolic profiling. J. Pharm. Biomedical. 2015, 113, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Cao, H. Fast quantification of short chain fatty acids and ketone bodies by liquid chromatography-tandem massspectrometry after facile derivatization coupled with liquid-liquid extraction. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1083, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography-massspectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef]
- Fu, X.; Calderón, C.; Harm, T.; Gawaz, M.; Lämmerhofer, M. Advanced unified monophasic lipid extraction protocol with wide coverage on the polarity scale optimized for large-scale untargeted clinical lipidomics analysis of platelets. Anal. Chim. Acta 2022, 1221, 340155. [Google Scholar] [CrossRef]
- Cheng, K.; Brunius, C.; Fristedt, R.; Landberg, R. An LC-QToF MS based method for untargeted metabolomics of human fecal samples. Metabolomics 2020, 16, 1–8. [Google Scholar] [CrossRef]
- Macioszek, S.; Dudzik, D.; Jacyna, J.; Wozniak, A.; Schöffski, P.; Markuszewski, M.J. A robust method for sample preparation of gastrointestinal stromal tumour for LC/MS untargeted metabolomics. Metabolites 2021, 11, 554. [Google Scholar] [CrossRef]
- Southam, A.D.; Pursell, H.; Frigerio, G.; Jankevics, A.; Weber, R.J.M.; Dunn, W.B. Characterization of Monophasic Solvent-Based Tissue Extractions for the Detection of Polar Metabolites and Lipids Applying Ultrahigh-Performance Liquid Chromatography-Mass Spectrometry Clinical Metabolic Phenotyping Assays. J. Proteome Res. 2021, 20, 831–840. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Medina, J.; van der Velpen, V.; Teav, T.; Guitton, Y.; Gallart-Ayala, H.; Ivanisevic, J. Single-step extraction coupled with targeted hilic-ms/ms approach for comprehensive analysis of human plasma lipidome and polar metabolome. Metabolites 2020, 10, 495. [Google Scholar] [CrossRef] [PubMed]
- Alshehry, Z.H.; Barlow, C.K.; Weir, J.M.; Zhou, Y.; McConville, M.J.; Meikle, P.J. An efficient single phase method for the extraction of plasma lipids. Metabolites 2015, 5, 389–403. [Google Scholar] [CrossRef]
- Peterson, A.L.; Walker, A.K.; Sloan, E.K.; Creek, D.J. Optimized Method for Untargeted Metabolomics Analysis of MDA-MB-231 Breast Cancer Cells. Metabolites 2016, 6, 30. [Google Scholar] [CrossRef]
- Lydic, T.A.; Busik, J.v; Reid, G.E. A monophasic extraction strategy for the simultaneous lipidome analysis of polar and nonpolar retina lipids. J. Lipid Res. 2014, 55, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H.; Sullards, M.C.; Allegood, J.C.; Kelly, S.; Wang, E. Sphingolipidomics: High-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods 2005, 36, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Southam, A.D.; Haglington, L.D.; Najdekr, L.; Jankevics, A.; Weber, R.J.M.; Dunn, W.B. Assessment of human plasma and urine sample preparation for reproducible and high-throughput UHPLC-MS clinical metabolic phenotyping. Analyst 2020, 145, 6511–6523. [Google Scholar] [CrossRef]
- Joshi, D.R.; Adhikari, N. An Overview on Common Organic Solvents and Their Toxicity. J. Pharm. Res. Int. 2019, 28, 1–18. [Google Scholar] [CrossRef]
- Wong, M.W.K.; Braidy, N.; Pickford, R.; Sachdev, P.S.; Poljak, A. Comparison of single phase and biphasic extraction protocols for lipidomic studies using human plasma. Front. Neurol. 2019, 10, 879. [Google Scholar] [CrossRef]
- Calderón, C.; Sanwald, C.; Schlotterbeck, J.; Drotleff, B.; Lämmerhofer, M. Comparison of simple monophasic versus classical biphasic extraction protocols for comprehensive UHPLC-MS/MS lipidomic analysis of Hela cells. Anal. Chim. Acta 2019, 1048, 66–74. [Google Scholar] [CrossRef]
- Zafeiropoulou, K.; Nichols, B.; Mackinder, M.; Biskou, O.; Rizou, E.; Karanikolou, A.; Clark, C.; Buchanan, E.; Cardigan, T.; Duncan, H.; et al. Alterations in Intestinal Microbiota of Children with Celiac Disease at the Time of Diagnosis and on a Gluten-free Diet. Gastroenterology 2020, 159, 2039–2051.e20. [Google Scholar] [CrossRef]
- Quince, C.; Ijaz, U.Z.; Loman, N.; Eren, A.M.; Saulnier, D.; Russell, J.; Haig, S.; Calus, S.; Quick, J.; Barclay, A.; et al. Extensive Modulation of the Fecal Metagenome in Children with Crohn’s Disease During Exclusive Enteral Nutrition. Am. J. Gastroenterol. 2015, 110, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Yu, J.; Cairns, T.C.; Zhang, L.; Zhang, Z.; Zhang, Q.; Zheng, P.; Sun, J.; Ma, Y. Comprehensive Improvement of Sample Preparation Methodologies Facilitates Dynamic Metabolomics of Aspergillus niger. Biotechnol. J. 2019, 14, e1800315. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Independent Variable | Sample Weight | Solvent Used | Cell Lysis Method |
|---|---|---|---|---|
| 1 | Sample weight | 10 mg, 20 mg, 50 mg, 100 mg | MeOH | Bead beating (5 ms−1, 60 s) |
| 2 | Extraction solvent | 50 mg | MeOH, 1:1 MeOH/H2O, 2:1 CHCl3/MeOH | Bead beating (5 ms−1, 60 s) |
| 3 | Cell lysis method | 50 mg | MeOH | Bead beating (5 ms−1, 60 s), sonication (40 kHz) freeze-thaw cycle (24 h) |
| 4 | Sample-to-solvent ratio | 50 mg | MeOH | Bead beating (5 ms−1, 60 s) |
| Variable | HC n = 20 | CD n = 20 | CoD n = 20 |
|---|---|---|---|
| Gender | |||
| Female (%) | 45 | 40 | 60 |
| Male (%) | 55 | 60 | 40 |
| Age (range) | 6.6 (2.3–13.7) | 12.3 (7.6–14.8) | 9.2 (4.0–14.8) |
| BMI z-score | 0.3 | −0.7 | 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelly, P.E.; Ng, H.J.; Farrell, G.; McKirdy, S.; Russell, R.K.; Hansen, R.; Rattray, Z.; Gerasimidis, K.; Rattray, N.J.W. An Optimised Monophasic Faecal Extraction Method for LC-MS Analysis and Its Application in Gastrointestinal Disease. Metabolites 2022, 12, 1110. https://doi.org/10.3390/metabo12111110
Kelly PE, Ng HJ, Farrell G, McKirdy S, Russell RK, Hansen R, Rattray Z, Gerasimidis K, Rattray NJW. An Optimised Monophasic Faecal Extraction Method for LC-MS Analysis and Its Application in Gastrointestinal Disease. Metabolites. 2022; 12(11):1110. https://doi.org/10.3390/metabo12111110
Chicago/Turabian StyleKelly, Patricia E., H Jene Ng, Gillian Farrell, Shona McKirdy, Richard K. Russell, Richard Hansen, Zahra Rattray, Konstantinos Gerasimidis, and Nicholas J. W. Rattray. 2022. "An Optimised Monophasic Faecal Extraction Method for LC-MS Analysis and Its Application in Gastrointestinal Disease" Metabolites 12, no. 11: 1110. https://doi.org/10.3390/metabo12111110
APA StyleKelly, P. E., Ng, H. J., Farrell, G., McKirdy, S., Russell, R. K., Hansen, R., Rattray, Z., Gerasimidis, K., & Rattray, N. J. W. (2022). An Optimised Monophasic Faecal Extraction Method for LC-MS Analysis and Its Application in Gastrointestinal Disease. Metabolites, 12(11), 1110. https://doi.org/10.3390/metabo12111110