

Acetyl Co-A Carboxylase Inhibition Halts Hyperglycemia Induced Upregulation of De Novo Lipogenesis in Podocytes and Proximal Tubular Cells
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Hyperglycemia Upregulates De Novo Lipogenesis in Healthy Podocytes and Proximal Epithelial Cells In Vitro
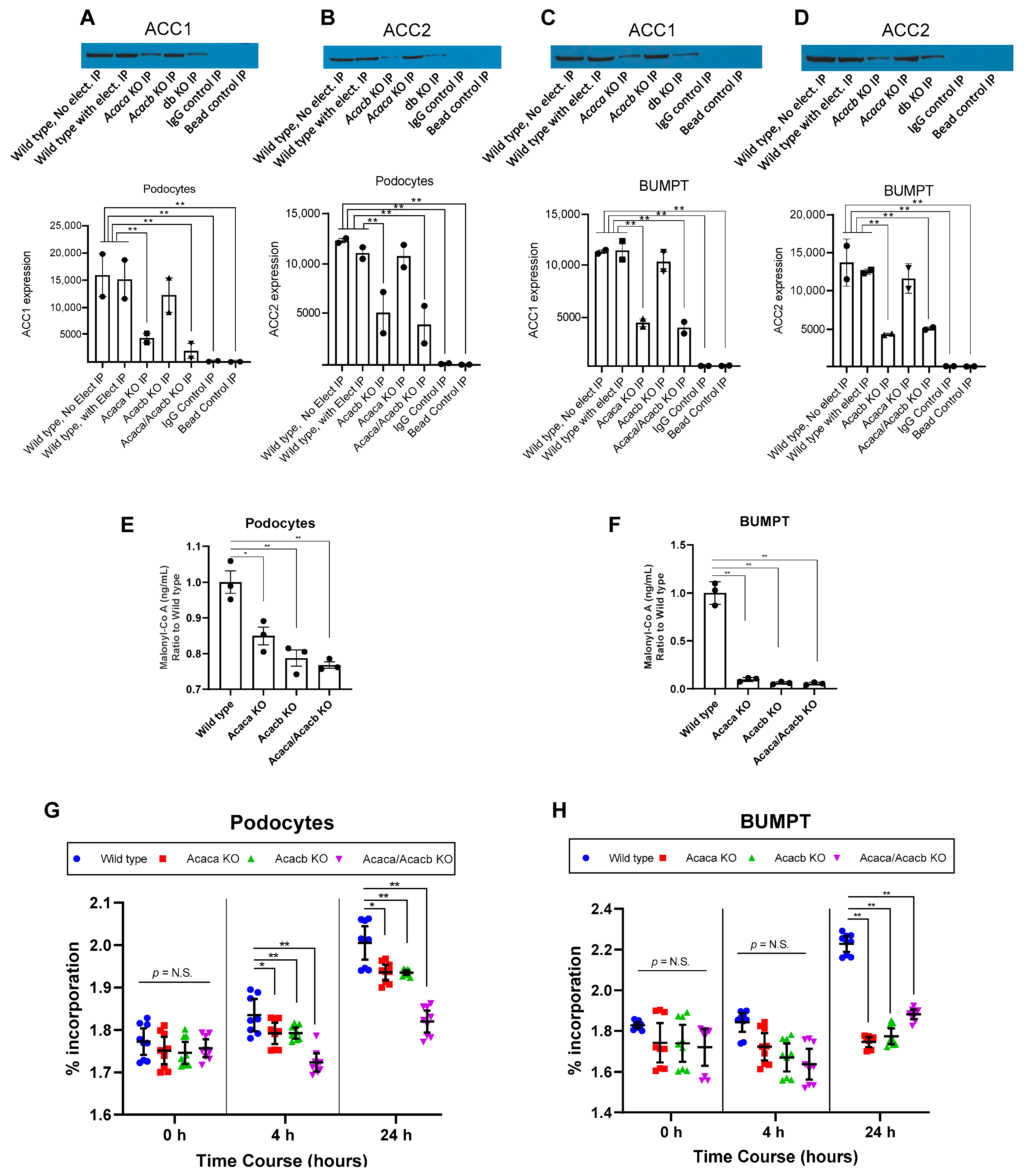
2.2. Acetyl-Co A Carboxylase Encoding Gene Deletion Halts Hyperglycemia Mediated Upregulation of De Novo Lipogenesis in Podocytes and BUMPT Cells In Vitro
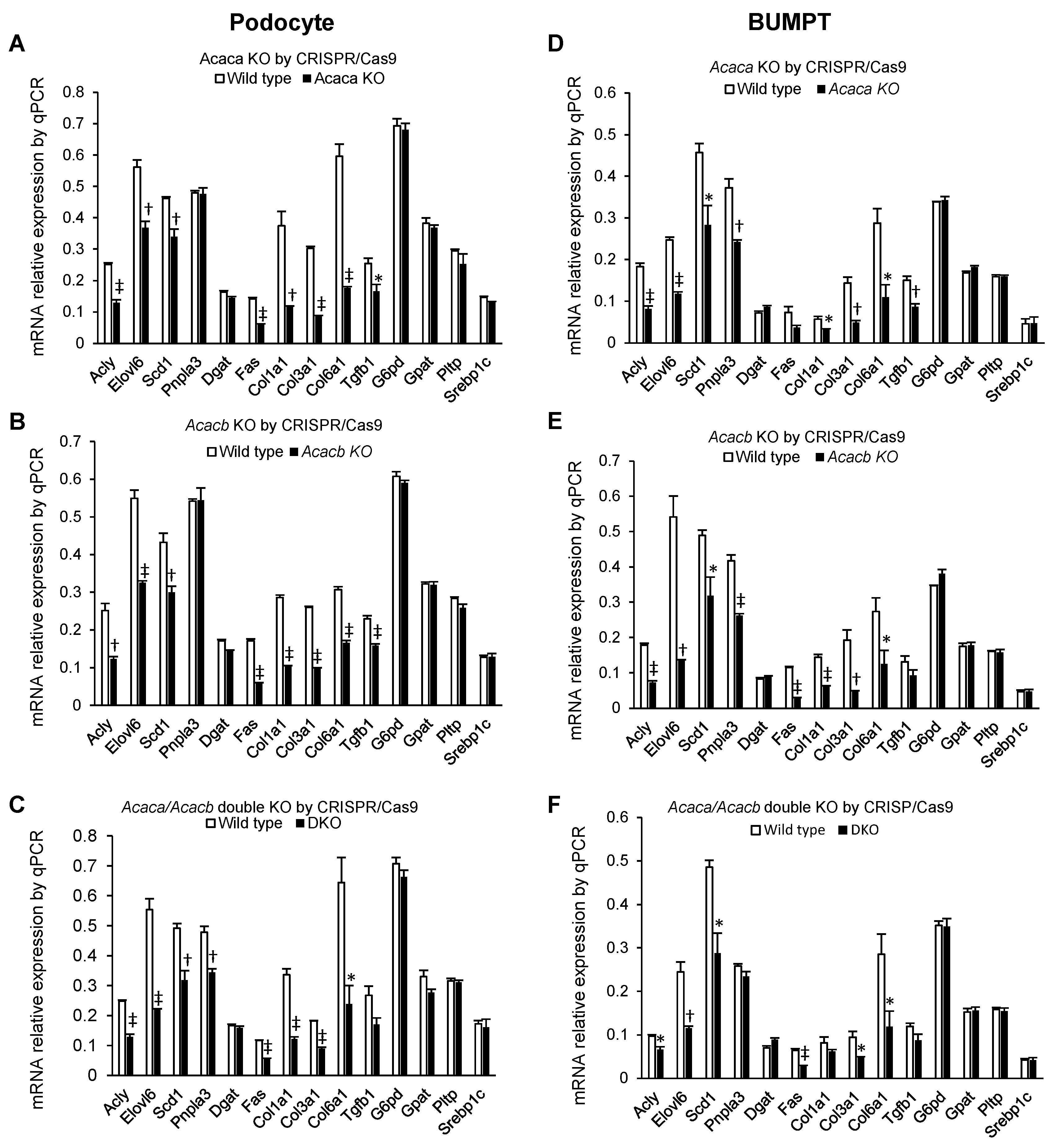
2.3. Alteration of mRNA Markers of Other Genes by Acaca and Acacb KO
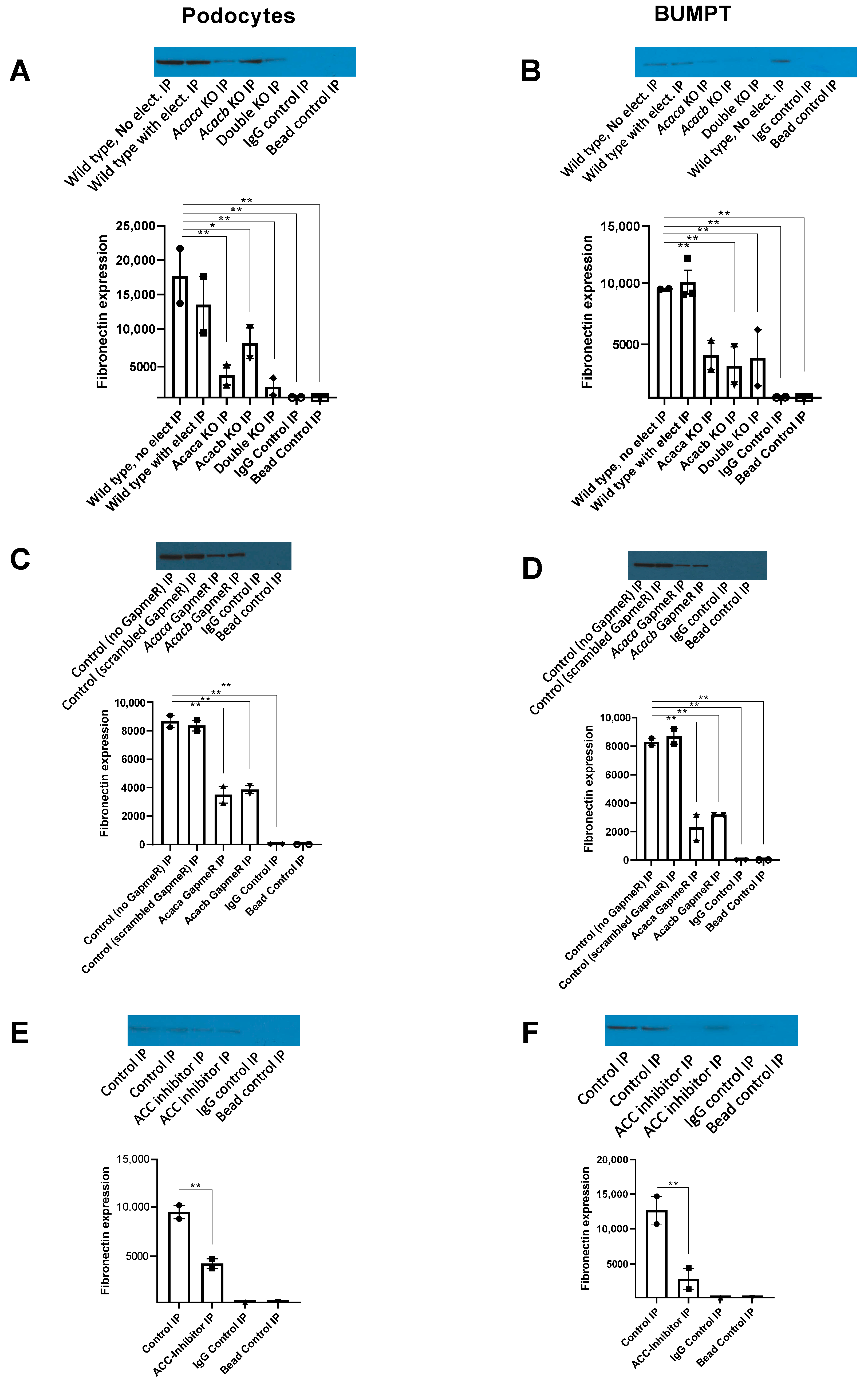
2.4. Down Regulation of Glycemic Mediated De Novo Lipogenesis by GapmeRs in Podocytes and Primary Tubular Epithelial Cells In Vitro
2.5. Alteration of mRNA Markers of Other Genes in GapmeR Mediated Acaca and Acacb Downregulated Cells
2.6. Down Regulation of Hyperglycemia Mediated De Novo Lipogenesis by Pharmacological ACC Inhibitor in Podocytes and Primary Tubular Epithelial Cells In Vitro
2.7. Alteration of mRNA Markers of Other Genes by Pharmacological ACC Inhibitor
2.8. Decreased Expression of Fibronectin with ACC Blockade
3. Discussion
4. Materials and Methods
4.1. Isolation and Culture of Primary Tubular Epithelial Cells
4.2. Mouse Proximal Tubular Cells
4.3. Mouse Podocytes
4.4. Silencing Acaca and Acacb Genes with Different Methods
4.4.1. CRISPR-Cas9 Mediated Knockout of Acaca and Acacb
4.4.2. RNA Interference (Antisense LNA GapmeR) Mediated Acaca and Acacb Silencing
4.5. Relative Expression of mRNA Marker Genes by qPCR
4.6. Pharmacological Inhibition of ACC
4.7. Palmitate Labeling with Labeled Acetate
4.8. Malonyl-CoA Assay
4.9. Immunofluorescence of Cell-Specific Markers
4.10. Immunoprecipitation and Western Blot Analysis
4.11. Lipid Extraction, Liquid Chromatography, and Mass Spectrometry
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- USRDS 2021 Annual Data Report. Available online: https://adr.usrds.org/2021/reference-tables (accessed on 26 January 2022).
- GBD Chronic Kidney Disease Collaboration. Global, Regional, and National Burden of Chronic Kidney Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- United States Renal Data System 2021. Chapter 9: Healthcare Expenditures for Persons with ESRD. Available online: https://adr.usrds.org/2020/end-stage-renal-disease/9-healthcare-expenditures-for-persons-with-esrd (accessed on 26 January 2022).
- HHS FY 2018 Budget in Brief—NIH. Available online: https://www.hhs.gov/about/budget/fy2018/budget-in-brief/nih/index.html#:~:text=The%20Fiscal%20Year%20(FY)%202018,FY%202017%20Continuing%20Resolution%20level (accessed on 26 January 2022).
- USRDS 2018. Incidence, Prevalence, Patient Characteristics, and Treatment Modalities. Available online: https://usrds.org/media/1736/v2_c01_incprev_18_usrds.pdf (accessed on 20 October 2021).
- Collins, A.J. Growth of the ESKD Population: Progress or Peril? J. Am. Soc. Nephrol. 2019, 30, 3–4. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.P.; Morgenstern, H.; Saran, R.; Herman, W.H.; Robinson, B.M. Projecting ESRD Incidence and Prevalence in the United States through 2030. J. Am. Soc. Nephrol. 2019, 30, 127–135. [Google Scholar] [CrossRef]
- Rowley, W.R.; Bezold, C.; Arikan, Y.; Byrne, E.; Krohe, S. Diabetes 2030: Insights from Yesterday, Today, and Future Trends. Popul. Health Manag. 2017, 20, 6–12. [Google Scholar] [CrossRef]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.; Cooper, M.E. Diabetic Kindey Disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Invest. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Afshinnia, F.; Nair, V.; Lin, J.; Rajendiran, T.M.; Soni, T.; Byun, J.; Sharma, K.; Fort, P.E.; Gardner, T.W.; Looker, H.C.; et al. Increased lipogenesis and impaired beta-oxidation predict type 2 diabetic kidney disease progression in American Indians. JCI Insight 2019, 4, e130317. [Google Scholar] [CrossRef]
- Song, Z.; Xiaoli, A.M.; Yang, F. Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients 2018, 10, 1383. [Google Scholar] [CrossRef]
- Thampy, K.G.; Wakil, S.J. Regulation of acetyl-coenzyme A carboxylase. I. Purification and properties of two forms of acetylcoenzyme A carboxylase from rat liver. J. Biol. Chem. 1988, 263, 6447–6453. [Google Scholar] [CrossRef]
- Abu-Elheiga, L.; Brinkley, W.R.; Zhong, L.; Chirala, S.S.; Woldegiorgis, G.; Wakil, S.J. The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. USA 2000, 97, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Thampy, K.G.; Wakil, S.J. Regulation of acetyl-coenzyme A carboxylase. II. Effect of fasting and refeeding on the activity, phosphate content, and aggregation state of the enzyme. J. Biol. Chem. 1988, 263, 6454–6458. [Google Scholar] [CrossRef]
- Hollands, M.A.; Cawthorne, M.A. Important sites of lipogenesis in the mouse other than liver and white adipose tissue. Biochem. J. 1981, 196, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Feng, Y.L.; Chen, H.; Chen, D.Q.; Vaziri, N.D.; Su, W.; Ma, S.X.; Shang, Y.Q.; Mao, J.R.; Yu, X.Y.; Zhang, L.; et al. Activated NF-kappaB/Nrf2 and Wnt/beta-catenin pathways are associated with lipid metabolism in CKD patients with microalbuminuria and macroalbuminuria. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2314–2332. [Google Scholar] [CrossRef]
- Martinez-Garcia, C.; Izquierdo-Lahuerta, A.; Vivas, Y.; Velasco, I.; Yeo, T.K.; Chen, S.; Medina-Gomez, G. Renal Lipotoxicity-Associated Inflammation and Insulin Resistance Affects Actin Cytoskeleton Organization in Podocytes. PLoS ONE 2015, 10, e0142291. [Google Scholar] [CrossRef]
- Lennon, R.; Pons, D.; Sabin, M.A.; Wei, C.; Shield, J.P.; Coward, R.J.; Tavare, J.M.; Mathieson, P.W.; Saleem, M.A.; Welsh, G.I. Saturated fatty acids induce insulin resistance in human podocytes: Implications for diabetic nephropathy. Nephrol. Dial. Transpl. 2009, 24, 3288–3296. [Google Scholar] [CrossRef]
- Jiang, X.S.; Chen, X.M.; Wan, J.M.; Gui, H.B.; Ruan, X.Z.; Du, X.G. Autophagy Protects against Palmitic Acid-Induced Apoptosis in Podocytes in vitro. Sci. Rep. 2017, 7, 42764. [Google Scholar] [CrossRef]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, J.; Lee, H.S. Palmitate induces mitochondrial superoxide generation and activates AMPK in podocytes. J. Cell. Physiol. 2017, 232, 3205–3217. [Google Scholar] [CrossRef] [PubMed]
- Sieber, J.; Lindenmeyer, M.T.; Kampe, K.; Campbell, K.N.; Cohen, C.D.; Hopfer, H.; Mundel, P.; Jehle, A.W. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am. J. Physiol. Ren. Physiol. 2010, 299, F821–F829. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Nam, S.M.; Kim, J.H.; Das, R.; Choi, S.K.; Nguyen, T.T.; Quan, X.; Choi, S.J.; Chung, C.H.; Lee, E.Y.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Tanaka, Y.; Kume, S.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Koya, D.; Haneda, M.; et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim. Biophys. Acta 2014, 1842, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Mu, L.; Yang, Z.; Du, C.; Wu, M.; Song, S.; Yuan, C.; Shi, Y. Carbohydrate response element-binding protein regulates lipid metabolism via mTOR complex1 in diabetic nephropathy. J. Cell. Physiol. 2021, 236, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kume, S.; Maeda, S.; Osawa, N.; Takeda, N.; Chin-Kanasaki, M.; Isshiki, K.; Ugi, S.; Oshima, I.; Uzu, T.; et al. Overexpression of acetyl CoA carboxylase beta exacerbates podocyte injury in the kidney of streptozotocin-induced diabetic mice. Biochem. Biophys. Res. Commun. 2018, 495, 1115–1121. [Google Scholar] [CrossRef]
- Kampe, K.; Sieber, J.; Orellana, J.M.; Mundel, P.; Jehle, A.W. Susceptibility of podocytes to palmitic acid is regulated by fatty acid oxidation and inversely depends on acetyl-CoA carboxylases 1 and 2. Am. J. Physiol. Ren. Physiol. 2014, 306, F401–F409. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, J.; Xin, W.; Chen, L.; Zhao, X.; Lv, Z.; Liu, Y.; Wan, Q. Lipid accumulation is ahead of epithelial-to-mesenchymal transition and therapeutic intervention by acetyl-CoA carboxylase 2 silence in diabetic nephropathy. Metabolism 2014, 63, 716–726. [Google Scholar] [CrossRef]
- Xin, W.; Zhao, X.; Liu, L.; Xu, Y.; Li, Z.; Chen, L.; Wang, X.; Yi, F.; Wan, Q. Acetyl-CoA carboxylase 2 suppression rescues human proximal tubular cells from palmitic acid induced lipotoxicity via autophagy. Biochem. Biophys. Res. Commun. 2015, 463, 364–369. [Google Scholar] [CrossRef]
- Lee, M.; Katerelos, M.; Gleich, K.; Galic, S.; Kemp, B.E.; Mount, P.F.; Power, D.A. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. J. Am. Soc. Nephrol. 2018, 29, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Witters, L.A.; Watts, T.D.; Daniels, D.L.; Evans, J.L. Insulin stimulates the dephosphorylation and activation of acetyl-CoA carboxylase. Proc. Natl. Acad. Sci. USA 1988, 85, 5473–5477. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Shimomura, I.; Bashmakov, Y.; Ikemoto, S.; Horton, J.D.; Brown, M.S.; Goldstein, J.L. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 13656–13661. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 394–406.e6. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Lawitz, E.; Noureddin, M.; DeFronzo, R.; Shulman, G.I. GS-0976 (Firsocostat): An investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 135–141. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Bhandari, B.R.; Ruane, P.J.; Kohli, A.; Harting, E.; Ding, D.; Chuang, J.C.; Huss, R.S.; Chung, C.; Myers, R.P.; et al. Fenofibrate Mitigates Hypertriglyceridemia in Nonalcoholic Steatohepatitis Patients Treated with Cilofexor/Firsocostat. Clin. Gastroenterol. Hepatol. 2022; in press. [Google Scholar]
- Loomba, R.; Kayali, Z.; Noureddin, M.; Ruane, P.; Lawitz, E.J.; Bennett, M.; Wang, L.; Harting, E.; Tarrant, J.M.; McColgan, B.J.; et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1463.e6–1473.e6. [Google Scholar] [CrossRef]
- Neokosmidis, G.; Cholongitas, E.; Tziomalos, K. Acetyl-CoA carboxylase inhibitors in non-alcoholic steatohepatitis: Is there a benefit? World J. Gastroenterol. 2021, 27, 6522–6526. [Google Scholar] [CrossRef]
- Scerbo, D.; Son, N.H.; Sirwi, A.; Zeng, L.; Sas, K.M.; Cifarelli, V.; Schoiswohl, G.; Huggins, L.A.; Gumaste, N.; Hu, Y.; et al. Kidney triglyceride accumulation in the fasted mouse is dependent upon serum free fatty acids. J. Lipid Res. 2017, 58, 1132–1142. [Google Scholar] [CrossRef]
- Bienholz, A.; Al-Taweel, A.; Roeser, N.F.; Kribben, A.; Feldkamp, T.; Weinberg, J.M. Substrate modulation of fatty acid effects on energization and respiration of kidney proximal tubules during hypoxia/reoxygenation. PLoS ONE 2014, 9, e94584. [Google Scholar] [CrossRef]
- Geng, H.; Lan, R.; Wang, G.; Siddiqi, A.R.; Naski, M.C.; Brooks, A.I.; Barnes, J.L.; Saikumar, P.; Weinberg, J.M.; Venkatachalam, M.A. Inhibition of autoregulated TGFbeta signaling simultaneously enhances proliferation and differentiation of kidney epithelium and promotes repair following renal ischemia. Am. J. Pathol. 2009, 174, 1291–1308. [Google Scholar] [CrossRef] [PubMed]
- Mundel, P.; Reiser, J.; Zuniga Mejia Borja, A.; Pavenstadt, H.; Davidson, G.R.; Kriz, W.; Zeller, R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp. Cell Res. 1997, 236, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayampilly, P.; Roeser, N.; Rajendiran, T.M.; Pennathur, S.; Afshinnia, F. Acetyl Co-A Carboxylase Inhibition Halts Hyperglycemia Induced Upregulation of De Novo Lipogenesis in Podocytes and Proximal Tubular Cells. Metabolites 2022, 12, 940. https://doi.org/10.3390/metabo12100940
Kayampilly P, Roeser N, Rajendiran TM, Pennathur S, Afshinnia F. Acetyl Co-A Carboxylase Inhibition Halts Hyperglycemia Induced Upregulation of De Novo Lipogenesis in Podocytes and Proximal Tubular Cells. Metabolites. 2022; 12(10):940. https://doi.org/10.3390/metabo12100940
Chicago/Turabian StyleKayampilly, Pradeep, Nancy Roeser, Thekkelnaycke M Rajendiran, Subramaniam Pennathur, and Farsad Afshinnia. 2022. "Acetyl Co-A Carboxylase Inhibition Halts Hyperglycemia Induced Upregulation of De Novo Lipogenesis in Podocytes and Proximal Tubular Cells" Metabolites 12, no. 10: 940. https://doi.org/10.3390/metabo12100940
APA StyleKayampilly, P., Roeser, N., Rajendiran, T. M., Pennathur, S., & Afshinnia, F. (2022). Acetyl Co-A Carboxylase Inhibition Halts Hyperglycemia Induced Upregulation of De Novo Lipogenesis in Podocytes and Proximal Tubular Cells. Metabolites, 12(10), 940. https://doi.org/10.3390/metabo12100940