
Ubiquitous Aberration in Cholesterol Metabolism across Pancreatic Ductal Adenocarcinoma

 ,
,  ,
,  , , and
, , and 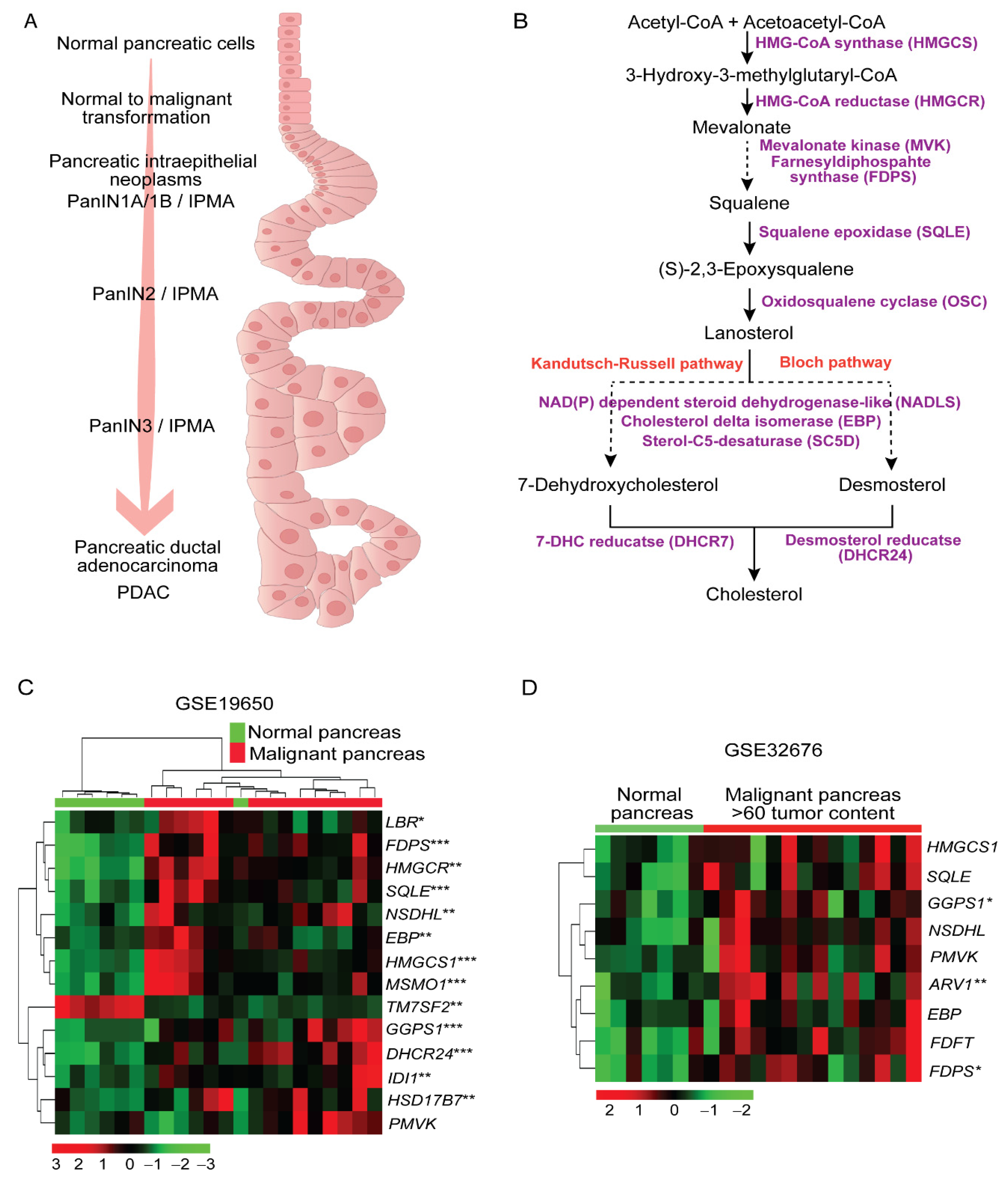{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Higher Expression of Cholesterol Synthesis Genes in Human Pancreatic Ductal Adenocarcinoma
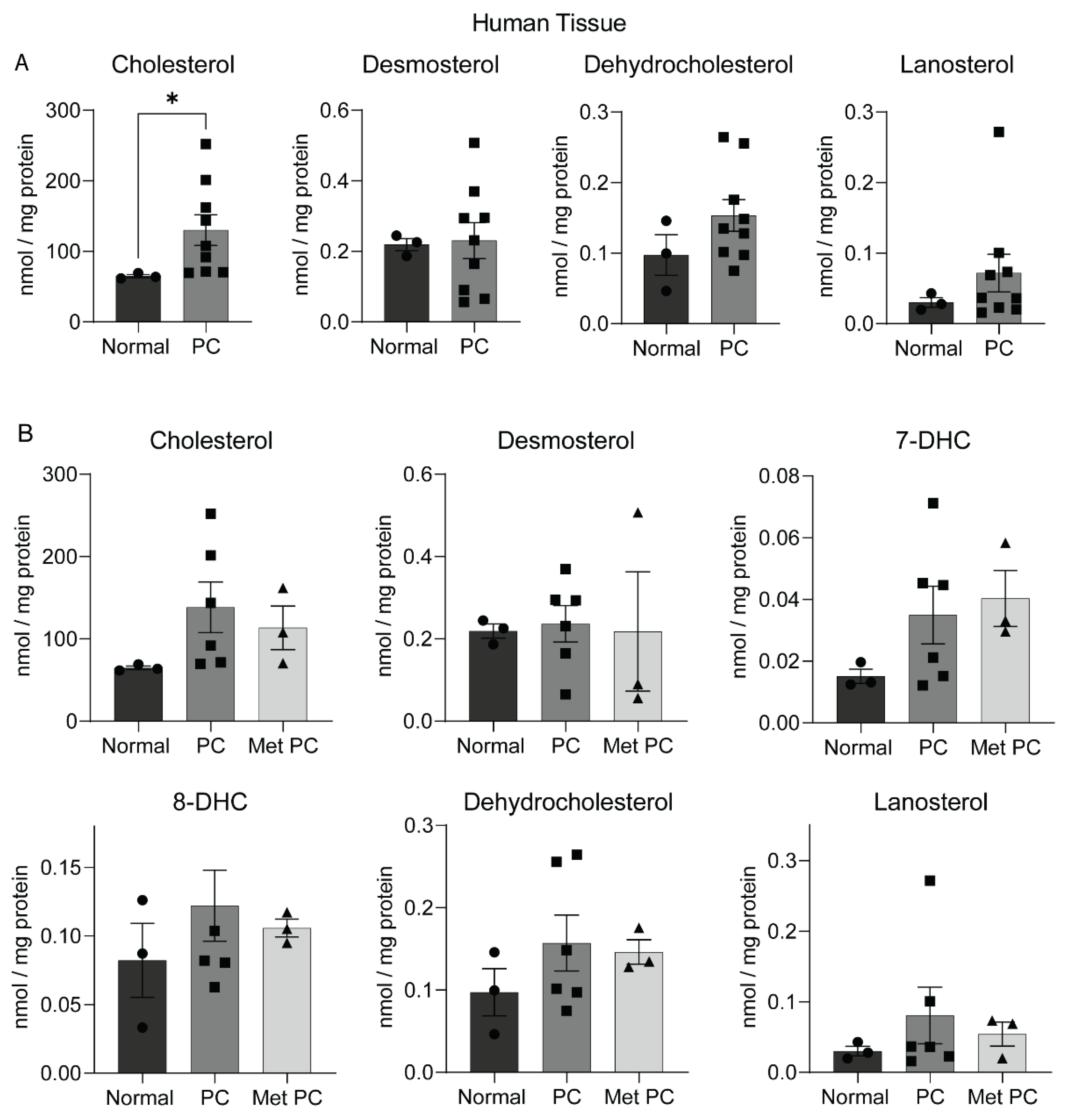
2.2. Sterol Alterations in Human Pancreatic Tumors
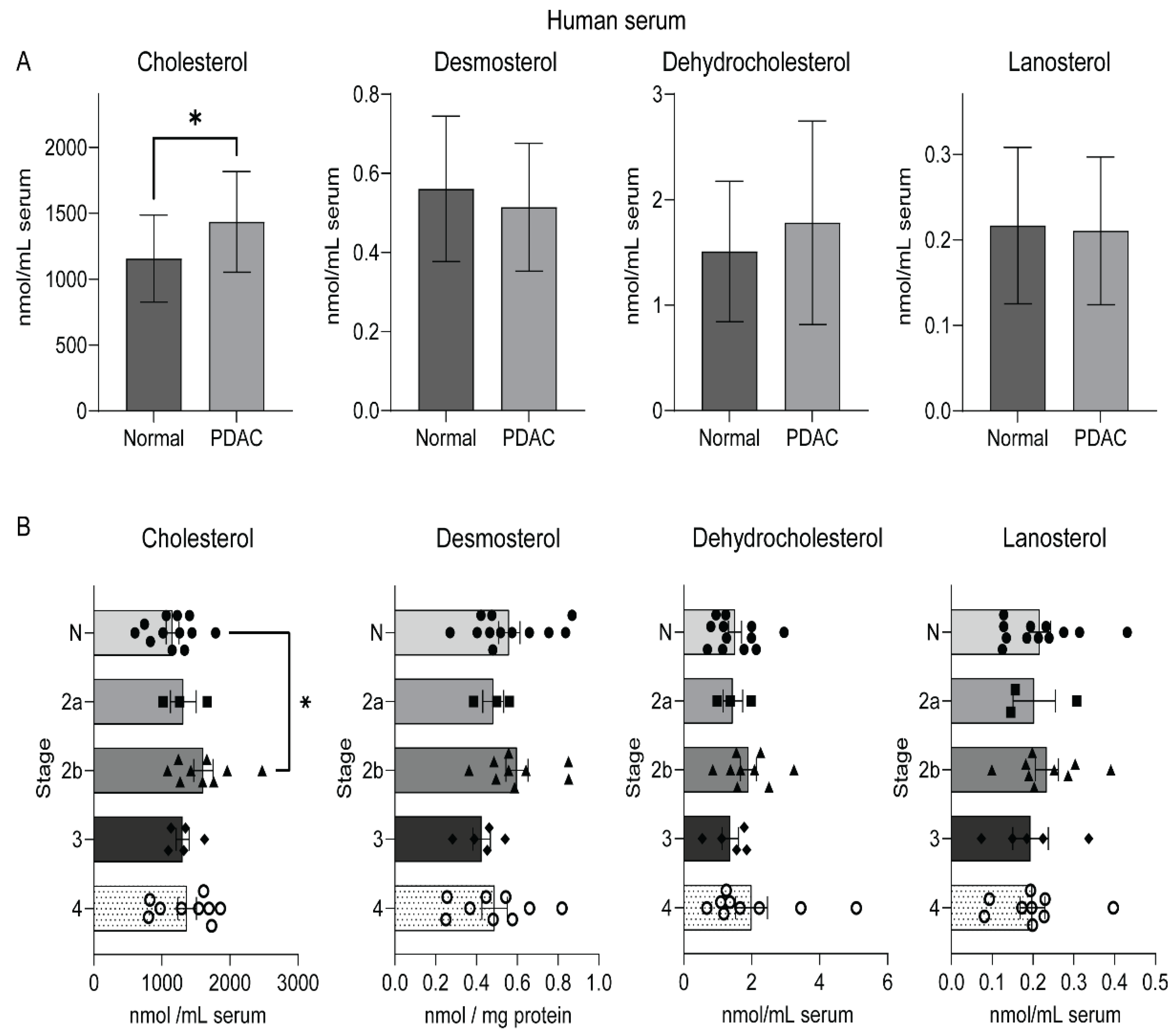
2.3. An Elevated Level of Serum Cholesterol in Human Pancreatic Cancer
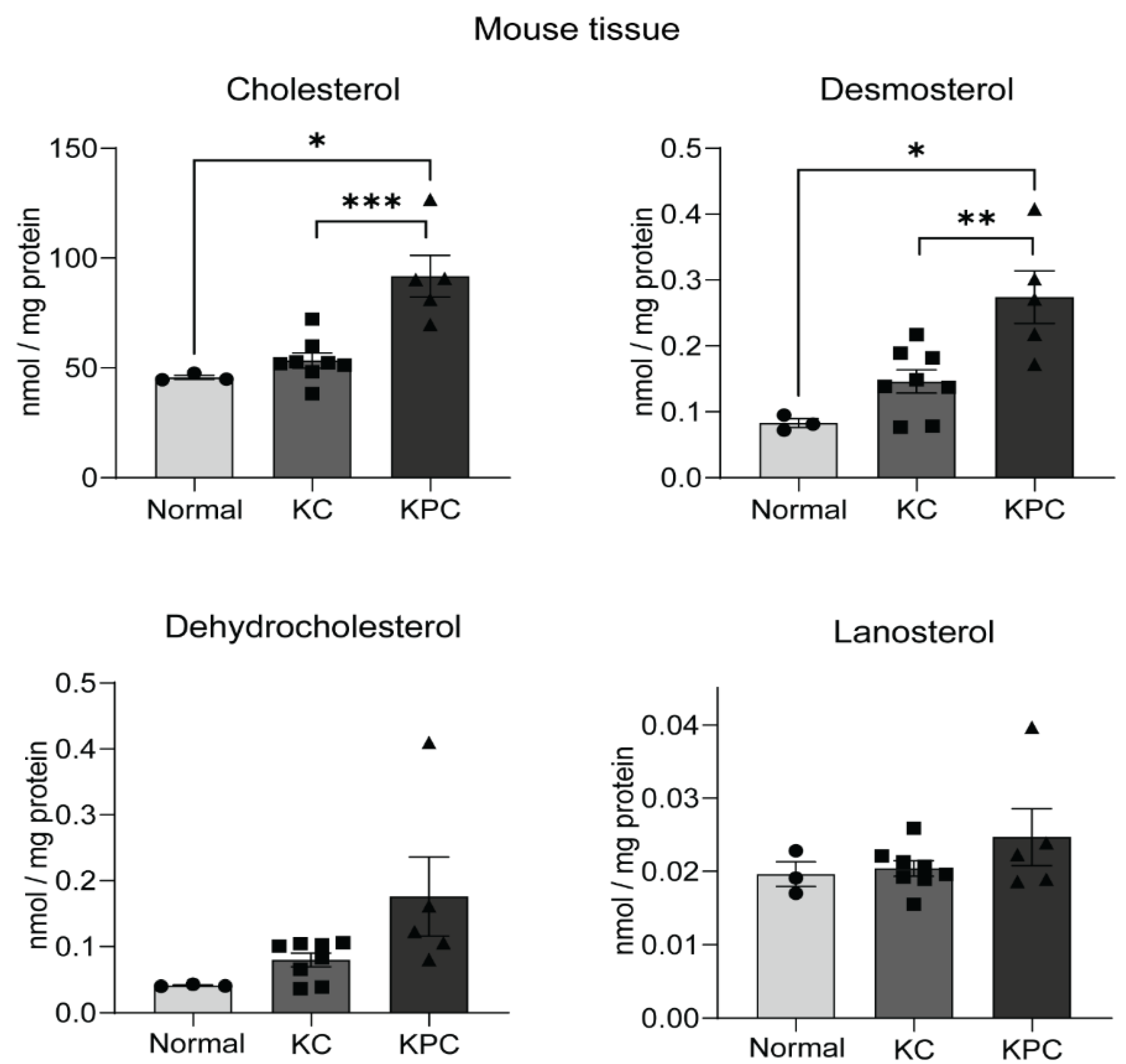
2.4. Sterol Upregulation in Different Mouse Models of PDAC
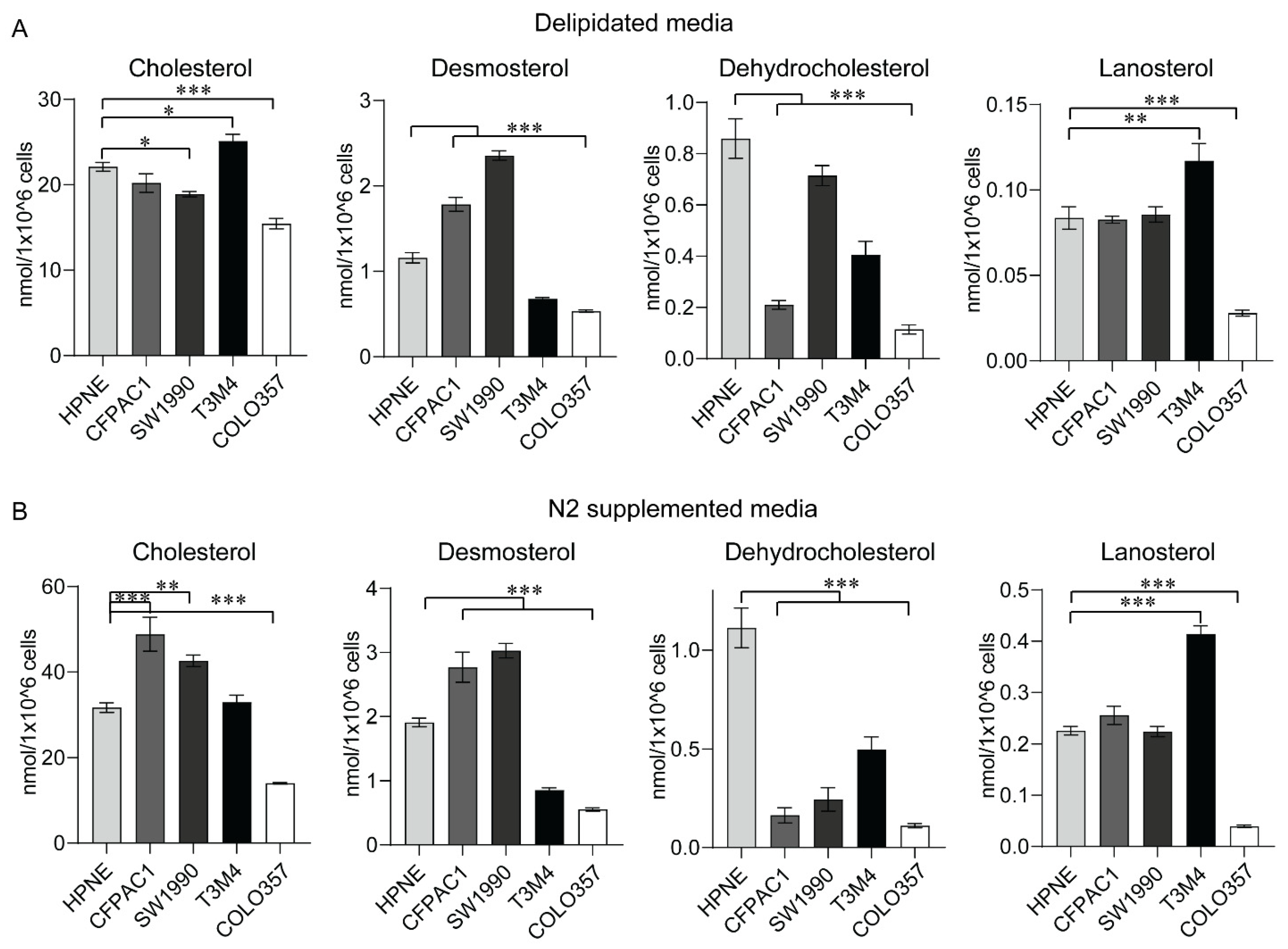
2.5. Sterol Levels from In Vitro Models of PC
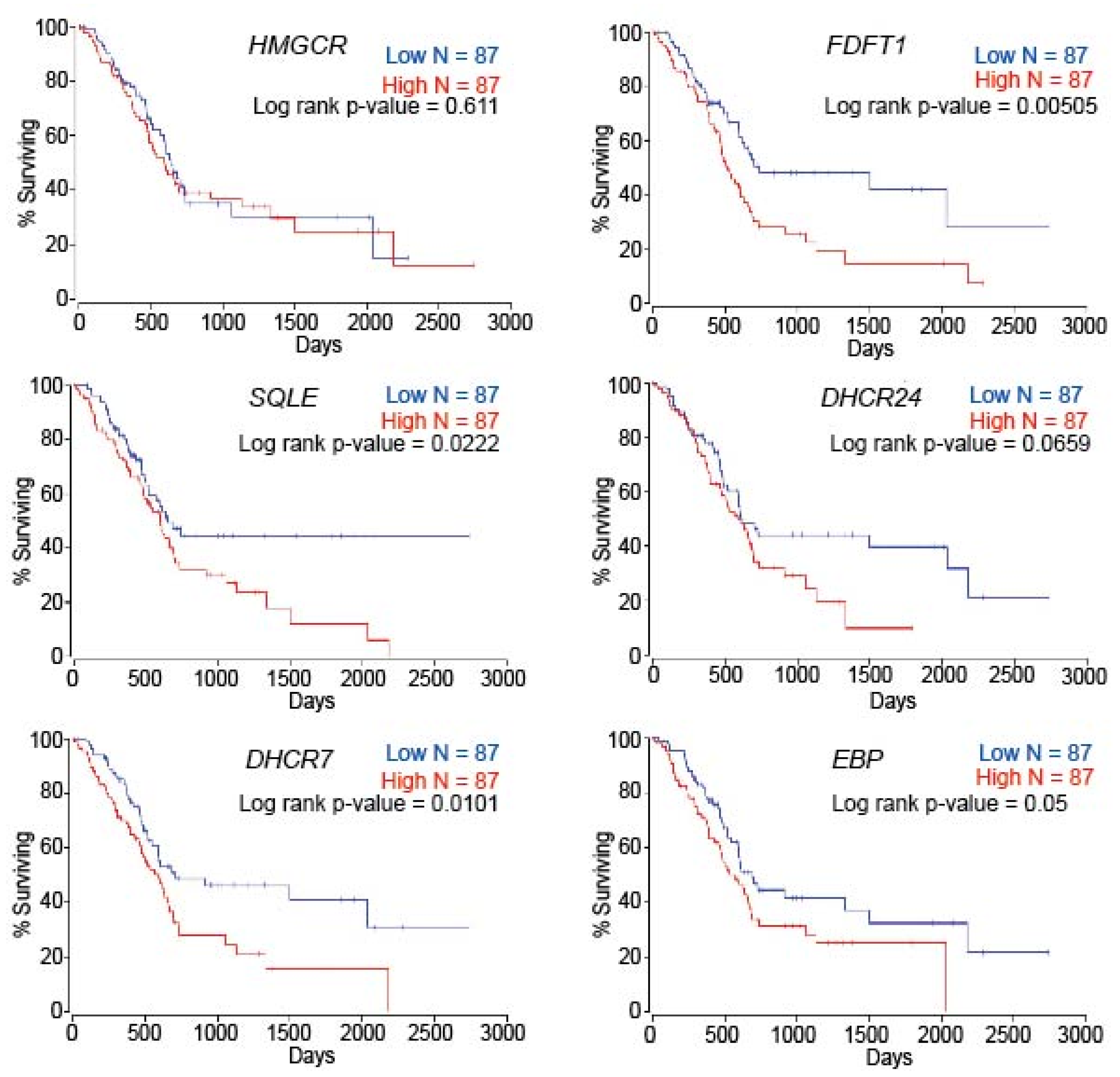
2.6. Patient Survival Is Dependent on Cholesterol Metabolism-Associated Genes in PC
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Mice and Ethics Statement
4.3. Plasma and Pancreatic Tissue Preparation
4.4. Cell Culture and Preparation
4.5. Metabolite Extraction from Cell Lines
4.6. LC–MS/MS (SRM) Analyses
4.7. Gene Expression and Survival Analyses
4.8. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, W.G.; Wu, M.; Bowen, R.; Zheng, M.; Fitch, W.L.; Pai, R.K.; Wodziak, D.; Visser, B.C.; Poultsides, G.A.; Norton, J.A.; et al. Metabolomic-derived novel cyst fluid biomarkers for pancreatic cysts: Glucose and kynurenine. Gastrointest. Endosc. 2013, 78, 295–302.e2. [Google Scholar] [CrossRef]
- Li, H.Y.; Appelbaum, F.R.; Willman, C.L.; Zager, R.A.; Banker, D.E. Cholesterol-modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood 2003, 101, 3628–3634. [Google Scholar] [CrossRef]
- Steck, T.L.; Lange, Y. Cell cholesterol homeostasis: Mediation by active cholesterol. Trends Cell Biol. 2010, 20, 680–687. [Google Scholar] [CrossRef]
- Silvente-Poirot, S.; Poirot, M. Cholesterol and Cancer, in the Balance. Science 2014, 343, 1445–1446. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. How Cells Handle Cholesterol. Science 2000, 290, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.; Rentero, C.; Conway, J.R.; Murray, R.Z.; Timpson, P.; Enrich, C.; Grewal, T. The cross-talk of LDL-cholesterol with cell motility: Insights from the Niemann Pick Type C1 mutation and altered integrin trafficking. Cell Adhes. Migr. 2015, 9, 384–391. [Google Scholar] [CrossRef][Green Version]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Blackford, A.L.; Molin, M.D.; Wolfgang, C.L.; Goggins, M. Time to progression of pancreatic ductal adenocarcinoma from low-to-high tumour stages. Gut 2015, 64, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.-S.; Hwang, C.-I.; Somerville, T.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20. [Google Scholar] [CrossRef]
- Rachagani, S.; Senapati, S.; Chakraborty, S.; Ponnusamy, M.P.; Kumar, S.; Smith, L.M.; Jain, M.; Batra, S.K. Activated KrasG12D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin. Br. J. Cancer 2011, 104, 1038–1048. [Google Scholar] [CrossRef][Green Version]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Gabitova, L.; Restifo, D.; Gorin, A.; Manocha, K.; Handorf, E.; Yang, D.-H.; Cai, K.Q.; Klein-Szanto, A.J.; Cunningham, D.; Kratz, L.E.; et al. Endogenous Sterol Metabolites Regulate Growth of EGFR/KRAS-Dependent Tumors via LXR. Cell Rep. 2015, 12, 1927–1938. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gu, D.; Lee, S.S.-Y.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef]
- Karasinska, J.M.; Topham, J.T.; Kalloger, S.E.; Jang, G.H.; Denroche, R.E.; Culibrk, L.; Williamson, L.M.; Wong, H.-L.; Lee, M.K.; O’Kane, G.M.; et al. Altered Gene Expression along the Glycolysis–Cholesterol Synthesis Axis Is Associated with Outcome in Pancreatic Cancer. Clin. Cancer Res. 2019, 26, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Souchek, J.J.; Baine, M.J.; Lin, C.; Rachagani, S.; Gupta, S.; Kaur, S.; Lester, K.; Zheng, D.; Chen, S.; Smith, L.; et al. Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br. J. Cancer 2014, 111, 1139–1149. [Google Scholar] [CrossRef]
- Jamnagerwalla, J.; Howard, L.E.; Allott, E.H.; Vidal, A.C.; Moreira, D.M.; Castro-Santamaria, R.; Andriole, G.L.; Freeman, M.R.; Freedland, S.J. Serum cholesterol and risk of high-grade prostate cancer: Results from the REDUCE study. Prostate Cancer Prostatic Dis. 2018, 21, 252–259. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Dembek, A.; Höring, M.; Schymik, H.S.; Dahlem, C.; Sultan, A.; Wirth, N.; Al-Fityan, S.; Diesel, B.; Gasparoni, G.; et al. Dysregulation of cholesterol homeostasis in human lung cancer tissue and tumour-associated macrophages. EBioMedicine 2021, 72, 103578. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Mitsche, M.A.; McDonald, J.G.; Hobbs, H.H.; Cohen, J.C. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. eLife 2015, 4, e07999. [Google Scholar] [CrossRef]
- Qin, W.-H.; Yang, Z.-S.; Li, M.; Chen, Y.; Zhao, X.-F.; Qin, Y.-Y.; Song, J.-Q.; Wang, B.-B.; Yuan, B.; Cui, X.-L.; et al. High Serum Levels of Cholesterol Increase Antitumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice. Gastroenterology 2020, 158, 1713–1727. [Google Scholar] [CrossRef]
- Rachagani, S.; Macha, M.; Menning, M.S.; Dey, P.; Pai, P.; Smith, L.M.; Mo, Y.-Y.; Batra, S.K. Changes in microRNA (miRNA) expression during pancreatic cancer development and progression in a genetically engineered KrasG12D;Pdx1-Cre mouse (KC) model. Oncotarget 2015, 6, 40295–40309. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xu, L.; Lamberson, C.; Haas, D.; Korade, Z.; Porter, N.A. A highly sensitive method for analysis of 7-dehydrocholesterol for the study of Smith-Lemli-Opitz syndrome. J. Lipid Res. 2014, 55, 329–337. [Google Scholar] [CrossRef]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.; Korade, Z.; Mirnics, K. Cholesterol Biosynthesis and Uptake in Developing Neurons. ACS Chem. Neurosci. 2019, 10, 3671–3681. [Google Scholar] [CrossRef] [PubMed]
- Genaro-Mattos, T.C.; Tallman, K.A.; Allen, L.; Anderson, A.; Mirnics, K.; Korade, Z.; Porter, N.A. Dichlorophenyl piperazines, including a recently-approved atypical antipsychotic, are potent inhibitors of DHCR7, the last enzyme in cholesterol biosynthesis. Toxicol. Appl. Pharmacol. 2018, 349, 21–28. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, V.; Dugnani, E.; Liberati, D.; Marra, P.; Citro, A.; Canu, T.; Policardi, M.; Valla, L.; Esposito, A.; Piemonti, L. Glucose metabolism during tumorigenesis in the genetic mouse model of pancreatic cancer. Acta Diabetol. 2019, 56, 1013–1022. [Google Scholar] [CrossRef]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef]
- Fiorenza, A.M.; Branchi, A.; Cardenà, A.; Molgora, M.; Rovellini, A.; Sommariva, D. Serum cholesterol levels in patients with cancer. Int. J. Clin. Lab. Res. 1996, 26, 37–42. [Google Scholar] [CrossRef]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.-X.; Hruban, R.H.; Adsay, V.; Tuveson, D.A.; et al. KrasG12D and Smad4/Dpc4 Haploinsufficiency Cooperate to Induce Mucinous Cystic Neoplasms and Invasive Adenocarcinoma of the Pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef]
- Yang, J.; Wang, L.; Jia, R. Role of de novo cholesterol synthesis enzymes in cancer. J. Cancer 2020, 11, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Lillemoe, K.D.; Yeo, C.J.; Warfield, P.B.; Hruban, R.H. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am. J. Surg. Pathol. 1998, 22, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Klöppel, G.; Lingenthal, G.; Von Bülow, M.; Kern, H. Histological and fine structural features of pancreatic ductal adenocarcinomas in relation to growth and prognosis: Studies in xenografted tumours and clinico-histopathological correlation in a series of 75 cases. Histopathology 1985, 9, 841–856. [Google Scholar] [CrossRef]
- Kugel, S.; Hingorani, S.R. Cholesterol Biosynthesis Influences Subtype Specificity and Plasticity in Pancreas Cancer. Cancer Cell 2020, 38, 443–445. [Google Scholar] [CrossRef]
- Kandutsch, A.; Russell, A.E. Preputial gland tumor sterols. 3. A metabolic pathway from lanosterol to cholesterol. J. Biol. Chem. 1960, 235, 2256–2261. [Google Scholar] [CrossRef]
- Haney, S.L.; Varney, M.L.; Chhonker, Y.S.; Shin, S.; Mehla, K.; Crawford, A.J.; Smith, H.J.; Smith, L.M.; Murry, D.J.; Hollingsworth, M.A.; et al. Inhibition of geranylgeranyl diphosphate synthase is a novel therapeutic strategy for pancreatic ductal adenocarcinoma. Oncogene 2019, 38, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Seshacharyulu, P.; Rachagani, S.; Muniyan, S.; Siddiqui, J.A.; Cruz, E.; Sharma, S.; Krishnan, R.; Killips, B.J.; Sheinin, Y.; Lele, S.M.; et al. FDPS cooperates with PTEN loss to promote prostate cancer progression through modulation of small GTPases/AKT axis. Oncogene 2019, 38, 5265–5280. [Google Scholar] [CrossRef]
- Agabiti, S.S.; Li, J.; Wiemer, A.J. Geranylgeranyl diphosphate synthase inhibition induces apoptosis that is dependent upon GGPP depletion, ERK phosphorylation and caspase activation. Cell Death Dis. 2017, 8, e2678. [Google Scholar] [CrossRef]
- Brown, D.N.; Caffa, I.; Cirmena, G.; Piras, D.; Garuti, A.; Gallo, M.; Alberti, S.; Nencioni, A.; Ballestrero, A.; Zoppoli, G. Squalene epoxidase is a bona fide oncogene by amplification with clinical relevance in breast cancer. Sci. Rep. 2016, 6, 19435. [Google Scholar] [CrossRef]
- Cirmena, G.; Franceschelli, P.; Isnaldi, E.; Ferrando, L.; De Mariano, M.; Ballestrero, A.; Zoppoli, G. Squalene epoxidase as a promising metabolic target in cancer treatment. Cancer Lett. 2018, 425, 13–20. [Google Scholar] [CrossRef]
- Walsh, C.A.; Akrap, N.; Garre, E.; Magnusson, Y.; Harrison, H.; Andersson, D.; Jonasson, E.; Rafnsdottir, S.; Choudhry, H.; Buffa, F.; et al. The mevalonate precursor enzyme HMGCS1 is a novel marker and key mediator of cancer stem cell enrichment in luminal and basal models of breast cancer. PLoS ONE 2020, 15, e0236187. [Google Scholar] [CrossRef]
- Wang, I.-H.; Huang, T.-T.; Chen, J.-L.; Chu, L.-W.; Ping, Y.-H.; Hsu, K.-W.; Huang, K.-H.; Fang, W.-L.; Lee, H.-C.; Chen, C.-F.; et al. Mevalonate Pathway Enzyme HMGCS1 Contributes to Gastric Cancer Progression. Cancers 2020, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Chushi, L.; Wei, W.; Kangkang, X.; Yongzeng, F.; Ning, X.; Xiaolei, C. HMGCR is up-regulated in gastric cancer and promotes the growth and migration of the cancer cells. Gene 2016, 587, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Bjarnadottir, O.; Feldt, M.; Inasu, M.; Bendahl, P.-O.; Elebro, K.; Kimbung, S.; Borgquist, S. Statin use, HMGCR expression, and breast cancer survival—The Malmö Diet and Cancer Study. Sci. Rep. 2020, 10, 558. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Stopsack, K.H.; Allott, E.H.; Gerke, T.; Giovannucci, E.L.; Mucci, L.A.; Kantoff, P.W. Intratumoral Sterol-27-Hydroxylase (CYP27A1) Expression in Relation to Cholesterol Synthesis and Vitamin D Signaling and Its Association with Lethal Prostate Cancer. Cancer Epidemiol. Prev. Biomark. 2019, 28, 1052–1058. [Google Scholar] [CrossRef]
- Sivanand, S.; Heiden, M.G.V. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell 2020, 37, 147–156. [Google Scholar] [CrossRef]
- Mormile, R. Total Serum Cholesterol and Pancreatic Cancer Risk: What Is the Link? Pathol. Oncol. Res. 2019, 26, 1361. [Google Scholar] [CrossRef]
- Chen, W.C.-Y.; Boursi, B.; Mamtani, R.; Yang, Y.-X. Total Serum Cholesterol and Pancreatic Cancer: A Nested Case–Control Study. Cancer Epidemiol. Prev. Biomark. 2019, 28, 363–369. [Google Scholar] [CrossRef]
- Guillaumond, F.; Iovanna, J.L.; Vasseur, S. Pancreatic tumor cell metabolism: Focus on glycolysis and its connected metabolic pathways. Arch. Biochem. Biophys. 2014, 545, 69–73. [Google Scholar] [CrossRef]
- Wang, W.-J. Association of cholesterol with risk of pancreatic cancer: A meta-analysis. World J. Gastroenterol. 2015, 21, 3711–3719. [Google Scholar] [CrossRef] [PubMed]
- Feltrin, S.; Ravera, F.; Traversone, N.; Ferrando, L.; Bedognetti, D.; Ballestrero, A.; Zoppoli, G. Sterol synthesis pathway inhibition as a target for cancer treatment. Cancer Lett. 2020, 493, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Terblanche, M.; Adhikari, N.K. Statin research in critical illness: Hampered by poor trial design? Crit. Care 2009, 13, 1015. [Google Scholar] [CrossRef] [PubMed][Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunda, V.; Genaro-Mattos, T.C.; Kaushal, J.B.; Chirravuri-Venkata, R.; Natarajan, G.; Mallya, K.; Grandgenett, P.M.; Mirnics, K.; Batra, S.K.; Korade, Z.; et al. Ubiquitous Aberration in Cholesterol Metabolism across Pancreatic Ductal Adenocarcinoma. Metabolites 2022, 12, 47. https://doi.org/10.3390/metabo12010047
Gunda V, Genaro-Mattos TC, Kaushal JB, Chirravuri-Venkata R, Natarajan G, Mallya K, Grandgenett PM, Mirnics K, Batra SK, Korade Z, et al. Ubiquitous Aberration in Cholesterol Metabolism across Pancreatic Ductal Adenocarcinoma. Metabolites. 2022; 12(1):47. https://doi.org/10.3390/metabo12010047
Chicago/Turabian StyleGunda, Venugopal, Thiago C. Genaro-Mattos, Jyoti B. Kaushal, Ramakanth Chirravuri-Venkata, Gopalakrishnan Natarajan, Kavita Mallya, Paul M. Grandgenett, Karoly Mirnics, Surinder K. Batra, Zeljka Korade, and et al. 2022. "Ubiquitous Aberration in Cholesterol Metabolism across Pancreatic Ductal Adenocarcinoma" Metabolites 12, no. 1: 47. https://doi.org/10.3390/metabo12010047
APA StyleGunda, V., Genaro-Mattos, T. C., Kaushal, J. B., Chirravuri-Venkata, R., Natarajan, G., Mallya, K., Grandgenett, P. M., Mirnics, K., Batra, S. K., Korade, Z., & Rachagani, S. (2022). Ubiquitous Aberration in Cholesterol Metabolism across Pancreatic Ductal Adenocarcinoma. Metabolites, 12(1), 47. https://doi.org/10.3390/metabo12010047