
Metabolomics Defines Complex Patterns of Dyslipidaemia in Juvenile-SLE Patients Associated with Inflammation and Potential Cardiovascular Disease Risk

 , , and
, , and 
Abstract

1. Introduction
2. Results
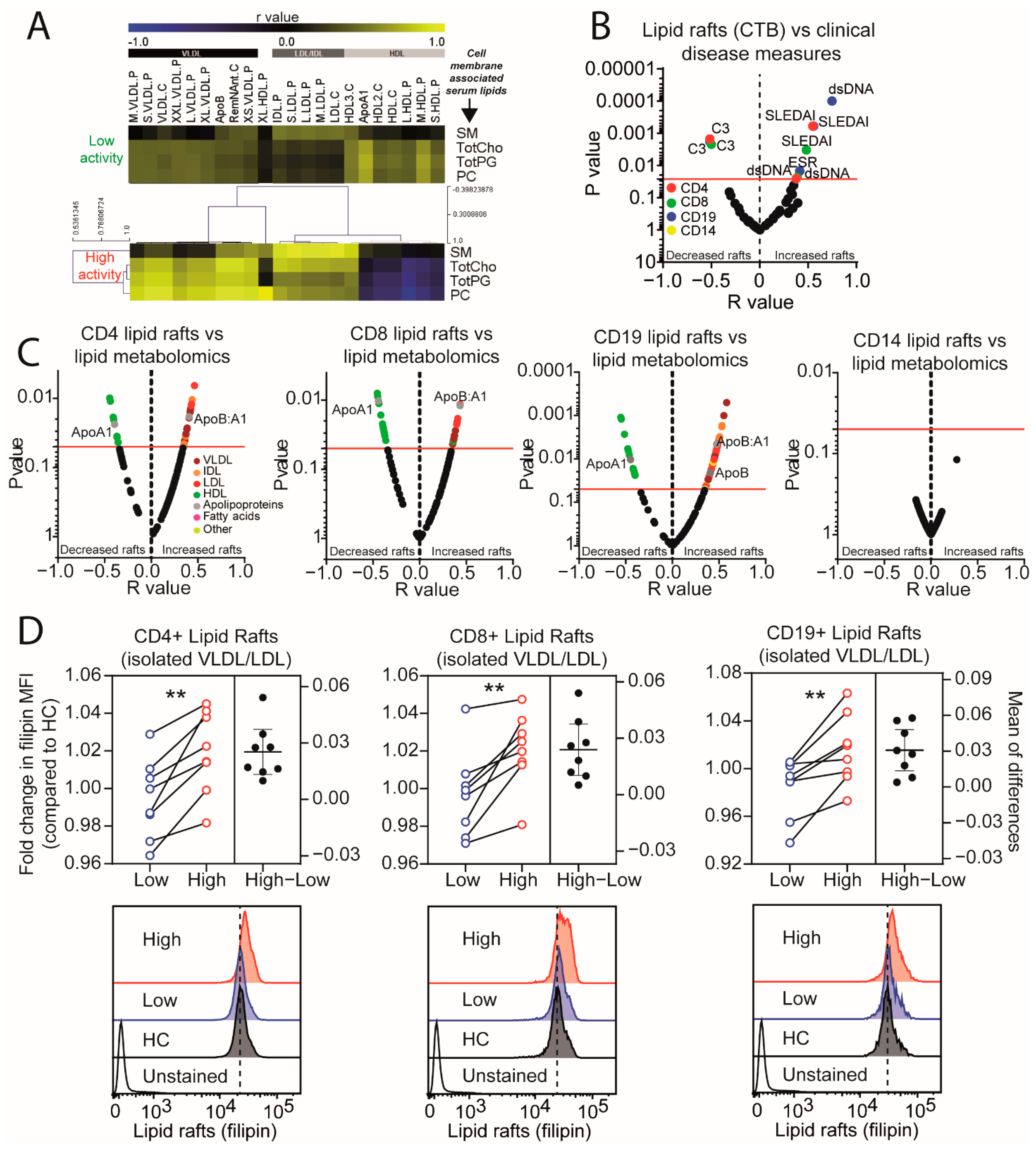
2.1. Dyslipidaemia in JSLE Patients Is Associated with Reduced Atheroprotective Small HDL Lipoproteins
2.2. Atherogenic Lipoproteins Are Associated with Increased Disease Activity in JSLE
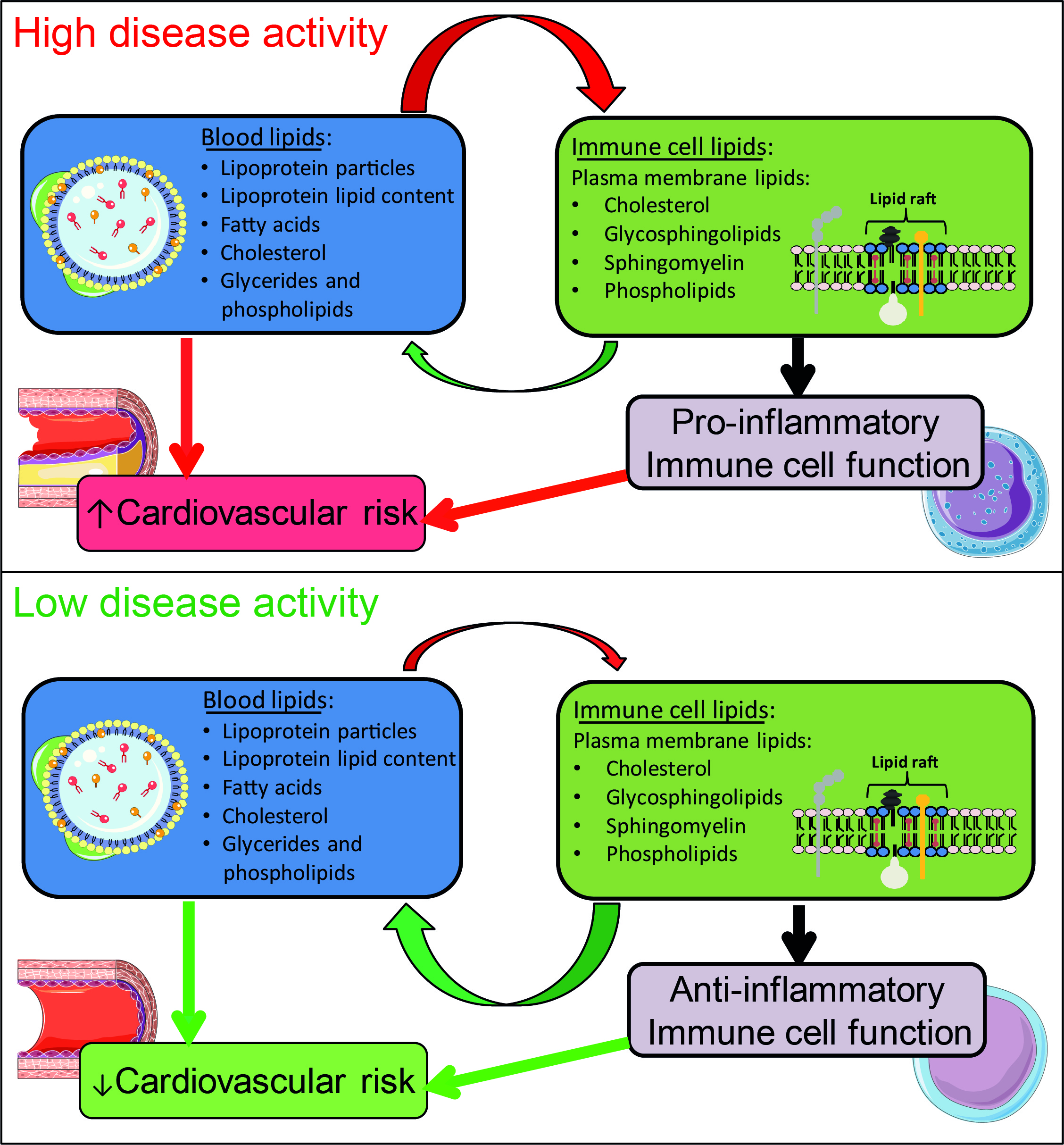
2.3. Lipid Raft Signalling Platforms Correlate with Lipoprotein Expression in Active JSLE Patients
3. Discussion
4. Materials and Methods
4.1. Patients and Control Samples
4.2. Metabolomics
4.3. Metabolomics Data Analysis
4.4. Flow Cytometry
4.5. VLDL Isolation and Cell Culture
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kone-Paut, I.; Piram, M.; Guillaume, S.; Tran, T.A. Lupus in adolescence. Lupus 2007, 16, 606–612. [Google Scholar] [CrossRef]
- Ambrose, N.; Morgan, T.A.; Galloway, J.; Ionnoau, Y.; Beresford, M.W.; Isenberg, D.A. Differences in disease phenotype and severity in SLE across age groups. Lupus 2016, 25, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Urowitz, M.B.; Bookman, A.A.; Koehler, B.E.; Gordon, D.A.; Smythe, H.A.; Ogryzlo, M.A. The bimodal mortality pattern of systemic lupus erythematosus. Am. J. Med. 1976, 60, 221–225. [Google Scholar] [CrossRef]
- Michel, V.; Bakovic, M. Lipid rafts in health and disease. Biol. Cell 2007, 99, 129–140. [Google Scholar] [CrossRef]
- Quinlan, C.; Kari, J.; Pilkington, C.; Deanfield, J.; Shroff, R.; Marks, S.D.; Tullus, K. The vascular phenotype of children with systemic lupus erythematosus. Pediatr. Nephrol. 2015, 30, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Barsalou, J.; Bradley, T.J.; Silverman, E.D. Cardiovascular risk in pediatric-onset rheumatological diseases. Arthritis Res. Ther. 2013, 15, 212. [Google Scholar] [CrossRef] [PubMed]
- Schanberg, L.E.; Sandborg, C.; Barnhart, H.X.; Ardoin, S.P.; Yow, E.; Evans, G.W.; Mieszkalski, K.L.; Ilowite, N.T.; Eberhard, A.; Levy, D.M.; et al. Premature atherosclerosis in pediatric systemic lupus erythematosus: Risk factors for increased carotid intima-media thickness in the atherosclerosis prevention in pediatric lupus erythematosus cohort. Arthritis Rheum. 2009, 60, 1496–1507. [Google Scholar] [CrossRef]
- Stary, H.C. Evolution and progression of atherosclerotic lesions in coronary-arteries of children and young-adults. Arteriosclerosis 1989, 9, I19–I32. [Google Scholar]
- Lemaire-Ewing, S.; Lagrost, L.; Neel, D. Lipid rafts: A signalling platform linking lipoprotein metabolism to atherogenesis. Atherosclerosis 2012, 221, 303–310. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Grossman, J.; Skaggs, B.; FitzGerald, J.; Sahakian, L.; Ragavendra, N.; Charles-Schoeman, C.; Watson, K.; Wong, W.K.; Volkmann, E.; et al. Dysfunctional proinflammatory high-density lipoproteins confer increased risk of atherosclerosis in women with systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, P.N.; Beyene, J.; Benseler, S.M.; Sarkissian, T.; Silverman, E.D. Predictors of lipid abnormalities in children with new-onset systemic lupus erythematosus. J. Rheumatol. 2007, 34, 2112–2119. [Google Scholar] [PubMed]
- Waddington, K.E.; Robinson, G.A.; Rubio-Cuesta, B.; Chrifi-Alaoui, E.; Andreone, S.; Poon, K.-S.; Ivanova, I.; Martin-Gutierrez, L.; Owen, D.M.; Jury, E.C.; et al. LXR directly regulates glycosphingolipid synthesis and affects human CD4+ T cell function. Proc. Natl. Acad. Sci. USA 2021, 118, e2017394118. [Google Scholar] [CrossRef]
- Jury, E.C.; Flores-Borja, F.; Kabouridis, P.S. Lipid rafts in T cell signalling and disease. Semin. Cell Dev. Biol. 2007, 18, 608–615. [Google Scholar] [CrossRef]
- Miguel, L.; Owen, D.M.; Lim, C.; Liebig, C.; Evans, J.; Magee, A.I.; Jury, E.C. Primary human CD4+ T cells have diverse levels of membrane lipid order that correlate with their function. J. Immunol. 2011, 186, 3505–3516. [Google Scholar] [CrossRef]
- Jury, E.C.; Kabouridis, P.S.; Flores-Borja, F.; Mageed, R.A.; Isenberg, D.A. Altered lipid raft–associated signaling and ganglioside expression in T lymphocytes from patients with systemic lupus erythematosus. J. Clin. Investig. 2004, 113, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.; Deepak, S.; Miguel, L.; Hall, C.J.; Isenberg, D.A.; Magee, A.I.; Butters, T.; Jury, E.C. Normalizing glycosphingolipids restores function in CD4+ T cells from lupus patients. J. Clin. Investig. 2014, 124, 712–724. [Google Scholar] [CrossRef]
- Jury, E.C.; Isenberg, D.A.; Mauri, C.; Ehrenstein, M.R. Atorvastatin restores Lck expression and lipid raft-associated signaling in T cells from patients with systemic lupus erythematosus. J. Immunol. 2006, 177, 7416–7422. [Google Scholar] [CrossRef] [PubMed]
- Pohl, J.; Ring, A.; Korkmaz, U.; Ehehalt, R.; Stremmel, W. FAT/CD36-mediated long-chain fatty acid uptake in adipocytes requires plasma membrane rafts. Mol. Biol. Cell 2005, 16, 24–31. [Google Scholar] [CrossRef]
- Fernandes das Neves, M.; Jury, E.C.; Delgado Alves, J. High density lipoprotein influences CD4 T cell proliferation in Systemic Lupus Erythematosus and increases TGF-β1 expression: A potential role in the protection from atherosclerosis and autoimmunity. Lupus 2021, 30, 2019–2021. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.S.; Farewell, V.T.; Isenberg, D.A.; Griffiths, B.; Teh, L.S.; Bruce, I.N.; Ahmad, Y.; Rahman, A.; Prabu, A.; Akil, M.; et al. The use of Systemic Lupus Erythematosus Disease Activity Index-2000 to define active disease and minimal clinically meaningful change based on data from a large cohort of systemic lupus erythematosus patients. Rheumatology 2011, 50, 982–988. [Google Scholar] [CrossRef]
- Mikdashi, J.; Nived, O. Measuring disease activity in adults with systemic lupus erythematosus: The challenges of administrative burden and responsiveness to patient concerns in clinical research. Arthritis Res. Ther. 2015, 17, 183. [Google Scholar] [CrossRef] [PubMed]
- VLDL Cholesterol. Available online: https://labtestsonline.org/tests/vldl-cholesterol (accessed on 16 December 2021).
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Barshop, N.J.; Sirlin, C.B.; Schwimmer, J.B.; Lavine, J.E. Review article: Epidemiology, pathogenesis and potential treatments of paediatric non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 28, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Frishberg, A.; van den Munckhof, I.; Ter Horst, R.; Schraa, K.; Joosten, L.A.B.; Rutten, J.H.W.; Iancu, A.C.; Dregoesc, I.M.; Tigu, B.A.; Netea, M.G.; et al. An integrative model of cardiometabolic traits identifies two types of metabolic syndrome. Elife 2021, 10, e61710. [Google Scholar] [CrossRef]
- Storey, S.M.; Gallegos, A.M.; Atshaves, B.P.; McIntosh, A.L.; Martin, G.G.; Parr, R.D.; Landrock, K.K.; Mer, A.B.; Ball, J.M.; Schroeder, F. Selective cholesterol dynamics between lipoproteins and caveolae/lipid rafts. Biochemistry 2007, 46, 13891–13906. [Google Scholar] [CrossRef][Green Version]
- Schmidt, N.M.; Wing, P.A.C.; Diniz, M.O.; Pallett, L.J.; Swadling, L.; Harris, J.M.; Burton, A.R.; Jeffery-Smith, A.; Zakeri, N.; Amin, O.E.; et al. Targeting human Acyl-CoA:cholesterol acyltransferase as a dual viral and T cell metabolic checkpoint. Nat. Commun. 2021, 12, 2814. [Google Scholar] [CrossRef] [PubMed]
- Soep, J.B.; Mietus-Snyder, M.; Malloy, M.J.; Witztum, J.L.; von Scheven, E. Assessment of atherosclerotic risk factors and endothelial function in children and young adults with pediatric-onset systemic lupus erythematosus. Arthritis Rheum. 2004, 51, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Chung, H.T.; Chang, C.J.; Yeh, K.W.; Chen, L.C.; Huang, J.L. Lymphopenia is a risk factor in the progression of carotid intima-media thickness in juvenile-onset systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 3766–3775. [Google Scholar] [CrossRef] [PubMed]
- Lilleby, V.; Haugen, M.; Mørkrid, L.; Frey Frøslie, K.; Holven, K.B.; Førre, O. Body composition, lipid and lipoprotein levels in childhood-onset systemic lupus erythematosus. Scand. J. Rheumatol. 2007, 36, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Simental-Mendía, L.E.; Simental-Mendía, M.; Sánchez-García, A.; Linden-Torres, E. Effect of hydroxychloroquine on lipid levels: A systematic review and meta-analysis. Curr. Pharm. Des. 2021, 27, 4133–4139. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yu, M.; Morin, E.E.; Kang, J.; Kaplan, M.J.; Schwendeman, A. High-Density Lipoprotein in Lupus: Disease Biomarkers and Potential Therapeutic Strategy. Arthritis Rheumatol. 2020, 72, 20–30. [Google Scholar] [CrossRef]
- Batuca, J.R.; Ames, P.R.; Isenberg, D.A.; Alves, J.D. Antibodies toward high-density lipoprotein components inhibit paraoxonase activity in patients with systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 2007, 1108, 137–146. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. Effect of inflammation on HDL structure and function. Curr. Opin. Lipidol. 2016, 27, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Fernandes das Neves, M.; Batuca, J.R.; Delgado Alves, J. The role of high-density lipoprotein in the regulation of the immune response: Implications for atherosclerosis and autoimmunity. Immunology 2021, 164, 231–241. [Google Scholar] [CrossRef]
- Hewing, B.; Landmesser, U. LDL, HDL, VLDL, and CVD Prevention: Lessons from Genetics? Curr. Cardiol. Rep. 2015, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Cicco, S.; Prete, M.; Solimando, A.G.; Susca, N.; Crudele, L.; Buonavoglia, A.; Colonna, P.; Dammacco, F.; Vacca, A.; et al. Early echocardiographic detection of left ventricular diastolic dysfunction in patients with systemic lupus erythematosus asymptomatic for cardiovascular disease. Clin. Exp. Med. 2020, 20, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Juonala, M.; Viikari, J.S.; Kähönen, M.; Solakivi, T.; Helenius, H.; Jula, A.; Marniemi, J.; Taittonen, L.; Laitinen, T.; Nikkari, T.; et al. Childhood levels of serum apolipoproteins B and A-I predict carotid intima-media thickness and brachial endothelial function in adulthood: The cardiovascular risk in young Finns study. J. Am. Coll. Cardiol. 2008, 52, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Ojanen, X.; Cheng, R.; Törmäkangas, T.; Rappaport, N.; Wilmanski, T.; Wu, N.; Fung, E.; Nedelec, R.; Sebert, S.; Vlachopoulos, D.; et al. Towards early risk biomarkers: Serum metabolic signature in childhood predicts cardio-metabolic risk in adulthood. EBioMedicine 2021, 72, 103611. [Google Scholar] [CrossRef]
- Robinson, G.A.; Waddington, K.E.; Coelewij, L.; Peng, J.; Naja, M.; Wincup, C.; Radziszewska, A.; Peckham, H.; Isenberg, D.A.; Ioannou, Y.; et al. Increased apolipoprotein-B:A1 ratio predicts cardiometabolic risk in patients with juvenile onset SLE. EBioMedicine 2021, 65, 103243. [Google Scholar] [CrossRef]
- Waddington, K.E.; Jury, E.C.; Pineda-Torra, I. Liver X receptors in immune cell function in humans. Biochem. Soc. Trans. 2015, 43, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.A.; Waddington, K.E.; Pineda-Torra, I.; Jury, E.C. Transcriptional Regulation of T-Cell Lipid Metabolism: Implications for Plasma Membrane Lipid Rafts and T-Cell Function. Front. Immunol. 2017, 8, 1636. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Yuan, S.G.; Peng, D.Q.; Zhao, S.P. HDL and ApoA-I inhibit antigen presentation-mediated T cell activation by disrupting lipid rafts in antigen presenting cells. Atherosclerosis 2012, 225, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Beamonte, R.; Lou-Bonafonte, J.M.; Martinez-Gracia, M.V.; Osada, J. Sphingomyelin in High-Density Lipoproteins: Structural Role and Biological Function. Int. J. Mol. Sci. 2013, 14, 7716–7741. [Google Scholar] [CrossRef]
- Waddington, K.E.; Papadaki, A.; Coelewij, L.; Adriani, M.; Nytrova, P.; Havrdova, E.K.; Fogdell-Hahn, A.; Farrell, R.; Donnes, P.; Pineda-Torra, I.; et al. Using Serum Metabolomics to Predict Development of Anti-drug Antibodies in Multiple Sclerosis Patients Treated with IFN beta. Front. Immunol. 2020, 11, 1527. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651. [Google Scholar] [CrossRef] [PubMed]
- Kloudova, A.; Guengerich, F.P.; Soucek, P. The Role of Oxysterols in Human Cancer. Trends Endocrinol. Metab. 2017, 28, 485–496. [Google Scholar] [CrossRef]
- Tulk, B.M.; Kapadia, S.; Edwards, J.C. CLIC1 inserts from the aqueous phase into phospholipid membranes, where it functions as an anion channel. Am. J. Physiol. Cell Physiol. 2002, 282, C1103–C1112. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Fernandez, R.; Coll, R.C.; Kearney, J.; Breit, S.; O’Neill, L.A.J. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1 beta transcription and activate the NLRP3 inflammasome. J. Biol. Chem. 2017, 292, 12077–12087. [Google Scholar] [CrossRef]
- Abud-Mendoza, C. Statins in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, e60. [Google Scholar] [CrossRef]
- Yu, H.-H.; Chen, P.-C.; Yang, Y.-H.; Wang, L.-C.; Lee, J.-H.; Lin, Y.-T.; Chiang, B.-L. Statin reduces mortality and morbidity in systemic lupus erythematosus patients with hyperlipidemia: A nationwide population-based cohort study. Atherosclerosis 2015, 243, 11–18. [Google Scholar] [CrossRef]
- Ruiz-Limon, P.; Barbarroja, N.; Perez-Sanchez, C.; Aguirre, M.A.; Bertolaccini, M.L.; Khamashta, M.A.; Rodriguez-Ariza, A.; Almadén, Y.; Segui, P.; Khraiwesh, H.; et al. Atherosclerosis and cardiovascular disease in systemic lupus erythematosus: Effects of in vivo statin treatment. Ann. Rheum. Dis. 2015, 74, 1450–1458. [Google Scholar] [CrossRef]
- Erkan, D.; Willis, R.; Murthy, V.L.; Basra, G.; Vega, J.; Ruiz-Limón, P.; Carrera, A.L.; Papalardo, E.; Martínez-Martínez, L.A.; González, E.B.; et al. A prospective open-label pilot study of fluvastatin on proinflammatory and prothrombotic biomarkers in antiphospholipid antibody positive patients. Ann. Rheum. Dis. 2014, 73, 1176–1180. [Google Scholar] [CrossRef]
- Ardoin, S.P.; Schanberg, L.E.; Sandborg, C.I.; Barnhart, H.X.; Evans, G.W.; Yow, E.; Mieszkalski, K.L.; Ilowite, N.T.; Eberhard, A.; Imundo, L.F.; et al. Secondary analysis of APPLE study suggests atorvastatin may reduce atherosclerosis progression in pubertal lupus patients with higher C reactive protein. Ann. Rheum. Dis. 2014, 73, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.A.; Kiani, A.N.; Post, W.; Christopher-Stine, L.; Magder, L.S. Lupus Atherosclerosis Prevention Study (LAPS). Ann. Rheum. Dis. 2011, 70, 760–765. [Google Scholar] [CrossRef]
- Schanberg, L.E.; Sandborg, C.; Barnhart, H.X.; Ardoin, S.P.; Yow, E.; Evans, G.W.; Mieszkalski, K.L.; Ilowite, N.T.; Eberhard, A.; Imundo, L.F.; et al. Use of atorvastatin in systemic lupus erythematosus in children and adolescents. Arthritis Rheum. 2012, 64, 285–296. [Google Scholar] [CrossRef]
- van Vollenhoven, R.F.; Voskuyl, A.; Morand, E.; Aranow, C. Remission in SLE: Closing in on the target. Ann. Rheum. Dis. 2015, 74, 2103–2106. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, D.A.; Merrill, J.T. Why, why, why de-lupus (does so badly in clinical trials). Expert Rev. Clin. Immunol. 2016, 12, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Waddington, K.; Adriani, M.; Torra, I.P.; Ioannou, J.; Jury, E. Stratification of patients with juvenile-onset systemic lupus erythematosus using immune and metabolic phenotyping. Rheumatology 2017, 56, 39. [Google Scholar] [CrossRef][Green Version]
- Gibson, C.M.; Kastelein, J.J.P.; Phillips, A.T.; Aylward, P.E.; Yee, M.K.; Tendera, M.; Nicholls, S.J.; Pocock, S.; Goodman, S.G.; Alexander, J.H.; et al. Rationale and design of ApoA-I Event Reducing in Ischemic Syndromes II (AEGIS-II): A phase 3, multicenter, double-blind, randomized, placebo-controlled, parallel-group study to investigate the efficacy and safety of CSL112 in subjects after acute myocardial infarction. Am. Heart J. 2021, 231, 121–127. [Google Scholar] [PubMed]
- da Silva, S.G.L.; Terreri, M.T.; Abad, T.T.O.; Machado, D.; Fonseca, F.L.A.; Hix, S.; Suano-Souza, F.I.; Sarni, R.O.S.; Len, C.A. The effect of nutritional intervention on the lipid profile and dietary intake of adolescents with juvenile systemic lupus erythematosus: A randomized, controlled trial. Lupus 2018, 27, 820–827. [Google Scholar] [CrossRef]
- Clark, W.F.; Parbtani, A.; Huff, M.W.; Reid, B.; Holub, B.J.; Falardeau, P. Omega-3 fatty-acid dietary supplementation in systemic lupus-erythematosus. Kidney Int. 1989, 3, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Ilowite, N.T.; Copperman, N.; Leicht, T.; Kwong, T.; Jacobson, M.S. Effects of dietary modification and fish-oil supplementation on dyslipoproteinemia in pediatric systemic lupus-erythematosus. J. Rheumatol. 1995, 22, 1347–1351. [Google Scholar] [PubMed]
- Fan, Y.Y.; Ly, L.H.; Barhoumi, R.; McMurray, D.N.; Chapkin, R.S. Dietary docosahexaenoic acid suppresses T cell protein kinase C theta lipid raft recruitment and IL-2 production. J. Immunol. 2004, 173, 6151–6160. [Google Scholar] [CrossRef]
- Coelewij, L.; Waddington, K.E.; Robinson, G.A.; Chocano, E.; McDonnell, T.; Farinha, F.; Peng, J.; Donnes, P.; Smith, E.; Croca, S.; et al. Serum Metabolomic Signatures Can Predict Subclinical Atherosclerosis in Patients with Systemic Lupus Erythematosus. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1446–1458. [Google Scholar] [CrossRef]
- Robinson, G.A.; Peng, J.; Peckham, H.; Radziszewska, A.; Butler, G.; Pineda-Torra, I.; Jury, E.C.; Ciurtin, C. Sex hormones drive changes in lipoprotein metabolism. iScience 2021, 24, 103257. [Google Scholar] [CrossRef] [PubMed]
- Novikova, D.S.; Popkova, T.V.; Lukina, G.V.; Luchikhina, E.L.; Karateev, D.E.; Volkov, A.V.; Novikov, A.A.; Aleksandrova, E.N.; Nasonov, E.L. The Effects of Rituximab on Lipids, Arterial Stiffness, and Carotid Intima-Media Thickness in Rheumatoid Arthritis. J. Korean Med. Sci. 2016, 31, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Andy Liaw, M.W. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2014. Available online: http://www.R.-project.org/ (accessed on 18 November 2021).
- Wang, M.K. forestplotNMR: Create Custom Forestplots for NMR Metabolomics Data, R package version 0.2.1; Available online: https://rdrr.io/github/mariakalimeri/forestplot/ (accessed on 5 July 2021).
- Kuhn, M. The Caret Package. Available online: http://topepo.github.io/caret/index.html (accessed on 20 November 2021).
- Waddington, K.E.; Pineda-Torra, I.; Jury, E.C. Analyzing T-Cell Plasma Membrane Lipids by Flow Cytometry. Methods Mol. Biol. 2019, 1951, 209–216. [Google Scholar] [PubMed]
- Gu, Z.G.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC | JSLE | Clinically Inactive JSLE (+/− Serological Activity) | Clinically Active JSLE | p-Value | |
|---|---|---|---|---|---|
| Total number | 32 | 65 | 43 | 22 | − |
| Female:Male | 17:15 | 53:12 | 36:7 | 17:5 | 0.0086 |
| Age, mean (range) | 19 (16–25) | 19.3 (13–25) | 19.3 (13–24) | 19.2 (14–25) | 0.8436 * |
| BMI, median (IQR) | 23.1 (20–24.5) | 22.4 (20.3–26.7) | 22.4 (20.3–26.6) | 22.5 (20.4–27.1) | 0.5416 * |
| Race, number (%) | |||||
| White | 14 (44) | 15 (23) | 11 (26) | 4 (18) | 0.1141 |
| Asian | 10 (31) | 27 (42) | 18 (42) | 9 (41) | 0.7667 |
| Black | 1 (3) | 17 (26) | 11 (26) | 6 (27) | 0.0447 |
| Other/unknown | 5 (16) | 6 (9) | 4 (9) | 2 (9) | 0.7726 |
| Disease characteristics, mean (range) | |||||
| Age of diagnosis | − | 12.1 (0–18) | 12.2 (2–17) | 11.9 (0–18) | 0.9676 * |
| Disease duration | − | 7.0 (0–21) | 6.8 (2–21) | 7.3 (0–17) | 0.9119 * |
| SLEDAI | − | 2.5 (0–10) | 0.8 (0–2) | 5.8 (4–10) | <0.0001 * |
| Organ involvement (historical), n (%) | |||||
| Neurological | − | 12 (18) | 9 (21) | 3 (14) | 0.7732 |
| Serositis | − | 8 (12) | 4 (9) | 4 (18) | 0.5877 |
| Cutaneous | − | 56 (86) | 39 (91) | 17 (77) | 0.3331 |
| Haematological | − | 28 (43) | 17 (40) | 11 (50) | 0.7225 |
| Musculoskeletal | − | 53 (82) | 36 (84) | 17 (77) | 0.8179 |
| Renal | − | 20 (31) | 10 (23) | 10 (45) | 0.1857 |
| Serology, median (IQR) | |||||
| dsDNA (IU/mL) (NR = <50) | − | 23 (3–141.5) | 8 (2–40) | 326 (88.5–587) | 0.1116 * |
| Positive ENA (number, %) | − | 40 (62) | 26 (60) | 14 (64) | 0.9696 * |
| CRP (mg/L) (NR < 10) | − | 1 (0.6–2.7) | 1.1 (0.6–2.35) | 0.7 (0.6–3.55) | 0.7148 * |
| C3 (g/L) (NR = 0.9–1.8) | − | 0.98 (0.74–1.18) | 1.1 (0.92–1.2) | 0.76 (0.58–0.97) | 0.0009 * |
| LC (109/L) (NR = 1.3–3.5) | − | 1.24 (1.2–1.7) | 1.37 (1.03–2.02) | 1.19 (0.93–1.44) | 0.0634 * |
| Albumin (g/L) (NR = 34–48) | − | 42.3 (40.0–44.5) | 42.7 (40.6–45.4) | 42.2 (33.9–44.5) | 0.0194 |
| Clinical lipids, median (IQR) | |||||
| Cholesterol (NR < 5mmol/L) | − | 4.0 (3.4–4.4) | 3.9 (3.4–4.5) | 4.0 (3.45–4.3) | 0.6686 * |
| Cholesterol, number (% outside NR) | − | 5.0 (8) | 3 (7) | 2 (9) | 0.9552 |
| Triglycerides (NR < 3mmol/L) | − | 0.8 (0.53–1.15) | 0.7 (0.5–1.0) | 0.9 (0.65–1.7) | 0.6034 * |
| Triglycerides, number (% outside NR) | − | 4 (6) | 2 (5) | 2 (9) | 0.7801 |
| HDL-C (NR > 1mmol/L) | − | 1.5 (1.2–1.7) | 1.5 (1.2–1.7) | 1.4 (1.15–1.6) | 0.4333 * |
| HDL-C, number (% outside NR) | − | 5 (8) | 3 (7) | 2 (9) | 0.9552 |
| LDL-C (NR < 3mmol/L) | − | 2.1 (1.6–2.4) | 2 (1.5–2.4) | 2.2 (1.65–2.45) | 0.5229 * |
| LDL-C, number (% outside NR) | − | 6 (9) | 4 (9) | 2 (9) | 0.9996 |
| Current treatment, n (%) | |||||
| Hydroxychloroquine | − | 60 (92) | 39 (91) | 21 (95) | 0.7930 |
| Mycophenolate mofetil | − | 25 (38) | 15 (35) | 10 (45) | 0.7092 |
| Prednisolone | − | 31 (48) | 19 (44) | 12 (55) | 0.7312 |
| Vitamin D | − | 12 (18) | 7 (16) | 5 (23) | 0.8179 |
| Methotrexate | − | 6 (9) | 5 (12) | 1 (5) | 0.6468 |
| Azathioprine | − | 15 (23) | 6 (14) | 9 (41) | 0.0509 |
| Rituximab in the last year | − | 0 (0) | 0 (0) | 0 (0) | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, G.A.; Peng, J.; Pineda-Torra, I.; Ciurtin, C.; Jury, E.C. Metabolomics Defines Complex Patterns of Dyslipidaemia in Juvenile-SLE Patients Associated with Inflammation and Potential Cardiovascular Disease Risk. Metabolites 2022, 12, 3. https://doi.org/10.3390/metabo12010003
Robinson GA, Peng J, Pineda-Torra I, Ciurtin C, Jury EC. Metabolomics Defines Complex Patterns of Dyslipidaemia in Juvenile-SLE Patients Associated with Inflammation and Potential Cardiovascular Disease Risk. Metabolites. 2022; 12(1):3. https://doi.org/10.3390/metabo12010003
Chicago/Turabian StyleRobinson, George A., Junjie Peng, Ines Pineda-Torra, Coziana Ciurtin, and Elizabeth C. Jury. 2022. "Metabolomics Defines Complex Patterns of Dyslipidaemia in Juvenile-SLE Patients Associated with Inflammation and Potential Cardiovascular Disease Risk" Metabolites 12, no. 1: 3. https://doi.org/10.3390/metabo12010003
APA StyleRobinson, G. A., Peng, J., Pineda-Torra, I., Ciurtin, C., & Jury, E. C. (2022). Metabolomics Defines Complex Patterns of Dyslipidaemia in Juvenile-SLE Patients Associated with Inflammation and Potential Cardiovascular Disease Risk. Metabolites, 12(1), 3. https://doi.org/10.3390/metabo12010003