
Novel Ubiquitin Specific Protease-13 Inhibitors Alleviate Neurodegenerative Pathology

 , , and
, , and
Abstract
:1. Introduction
2. Results
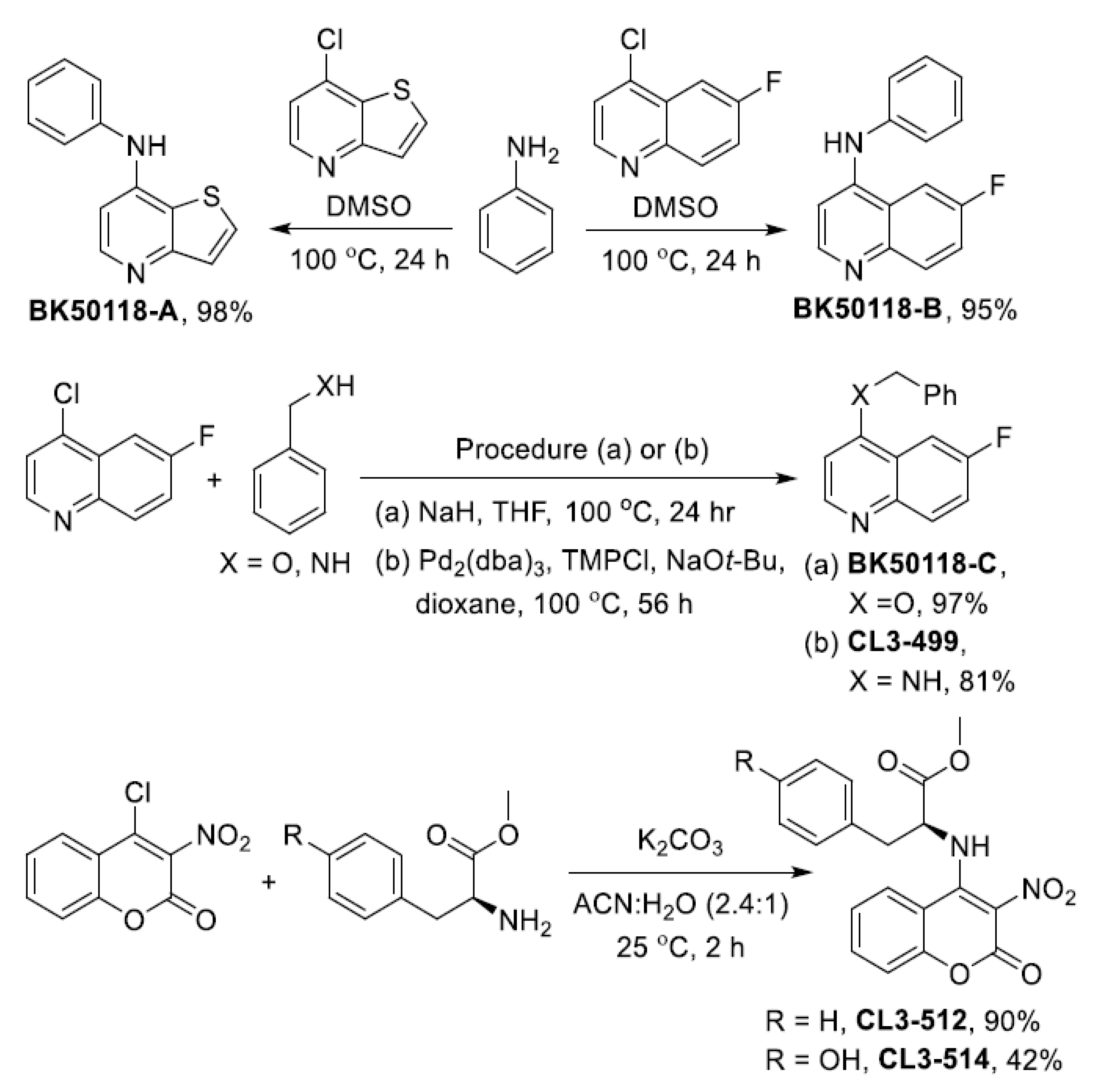
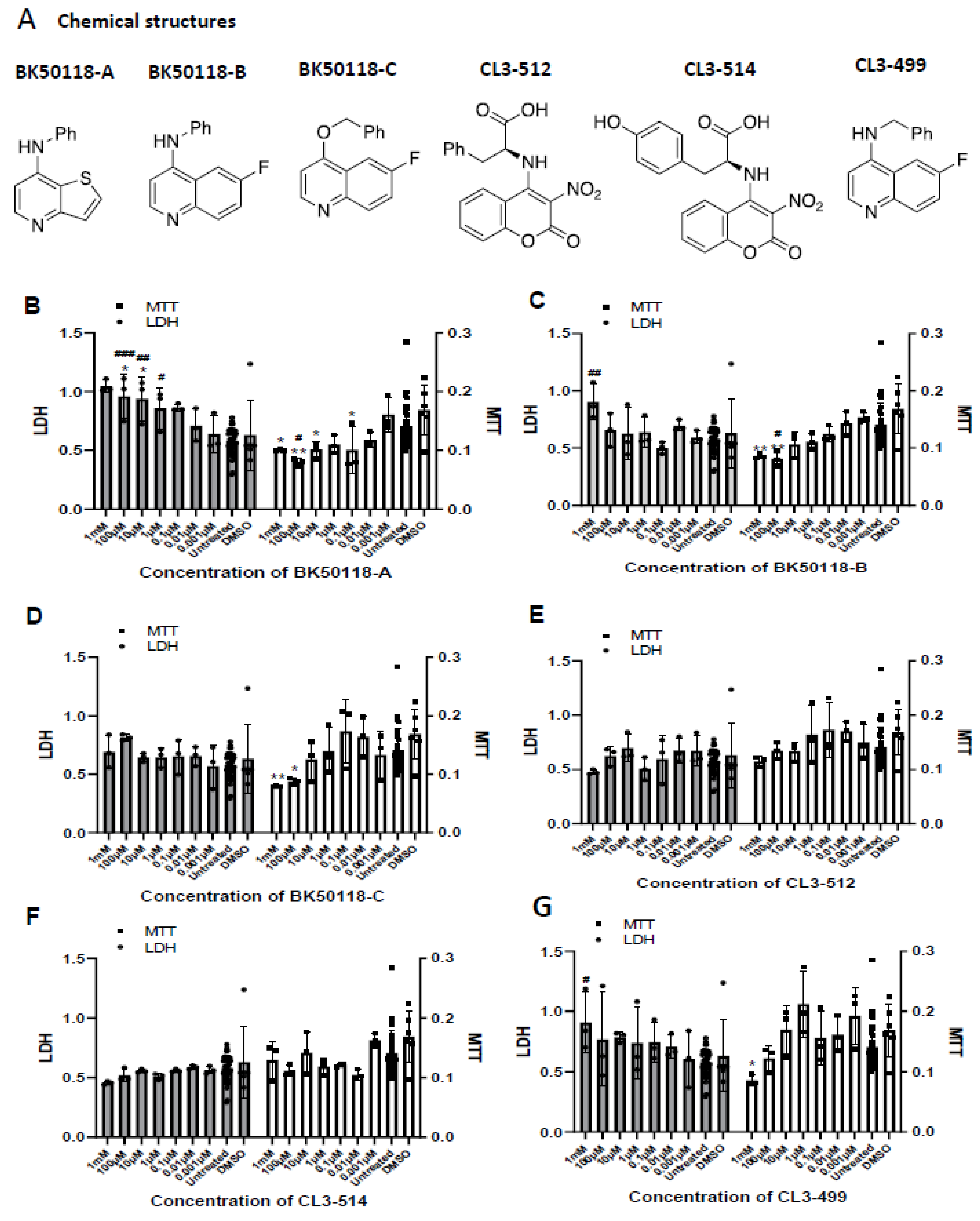
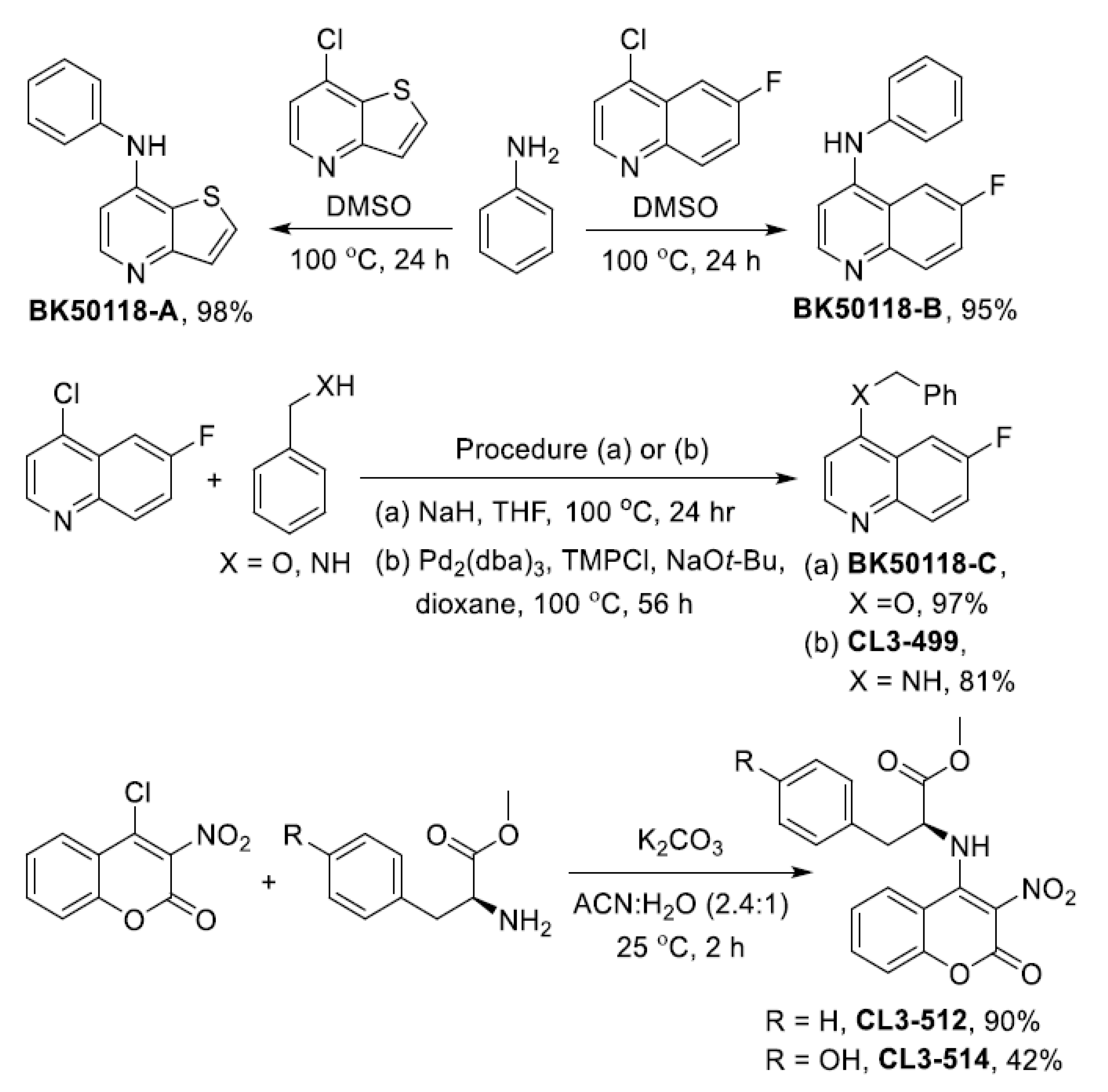
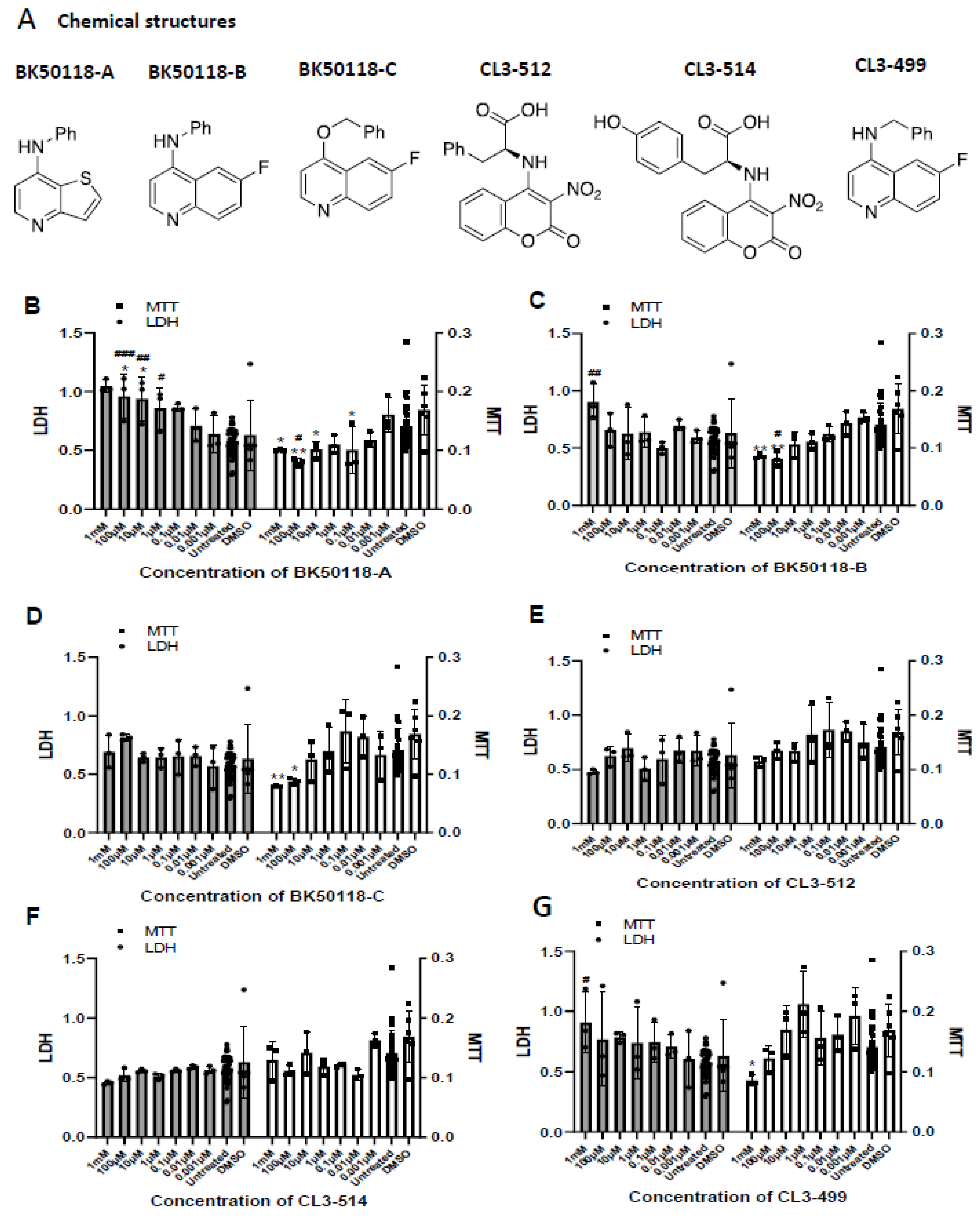
2.1. Chemical Synthesis and Cell Viability Assays
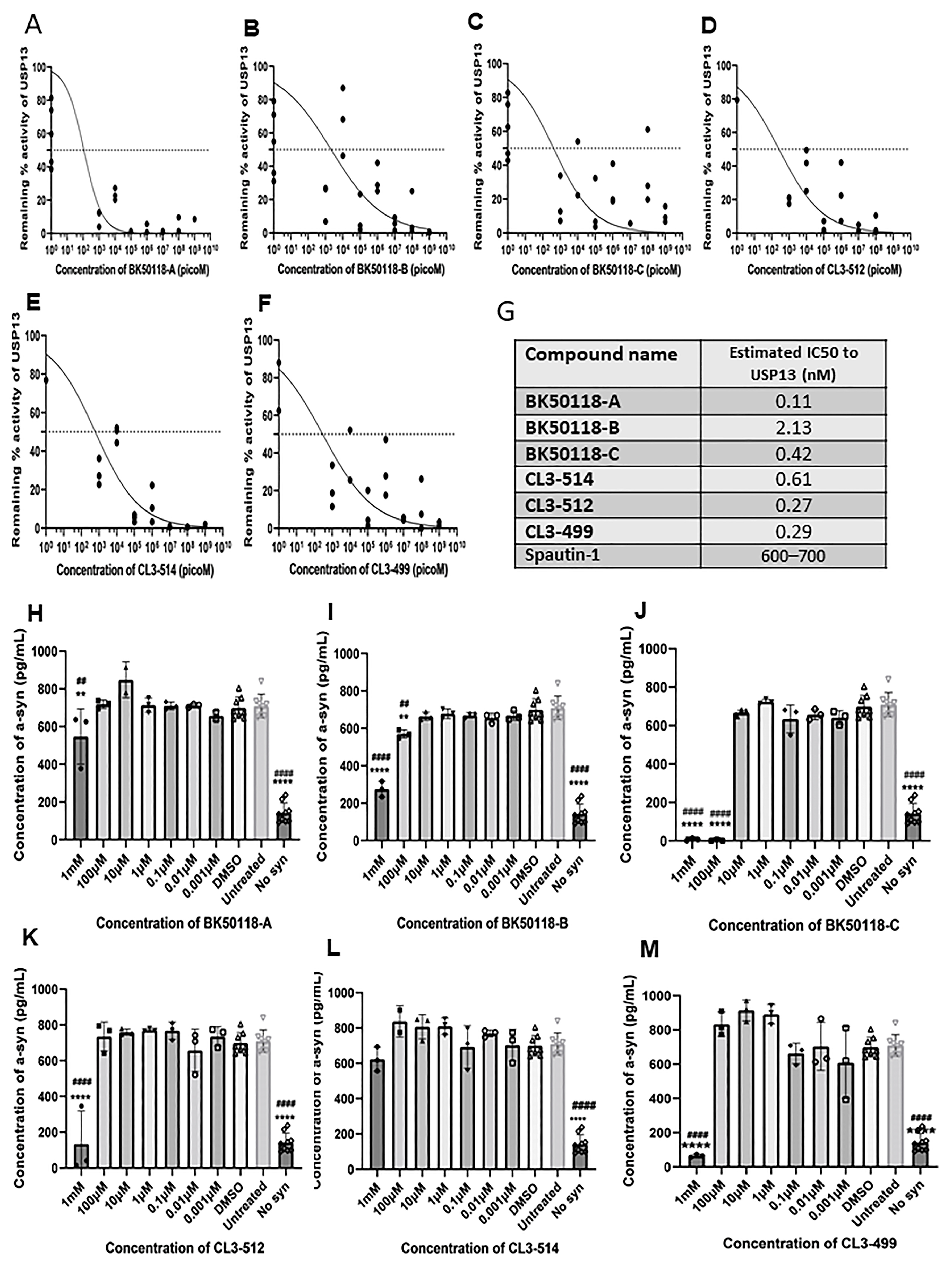
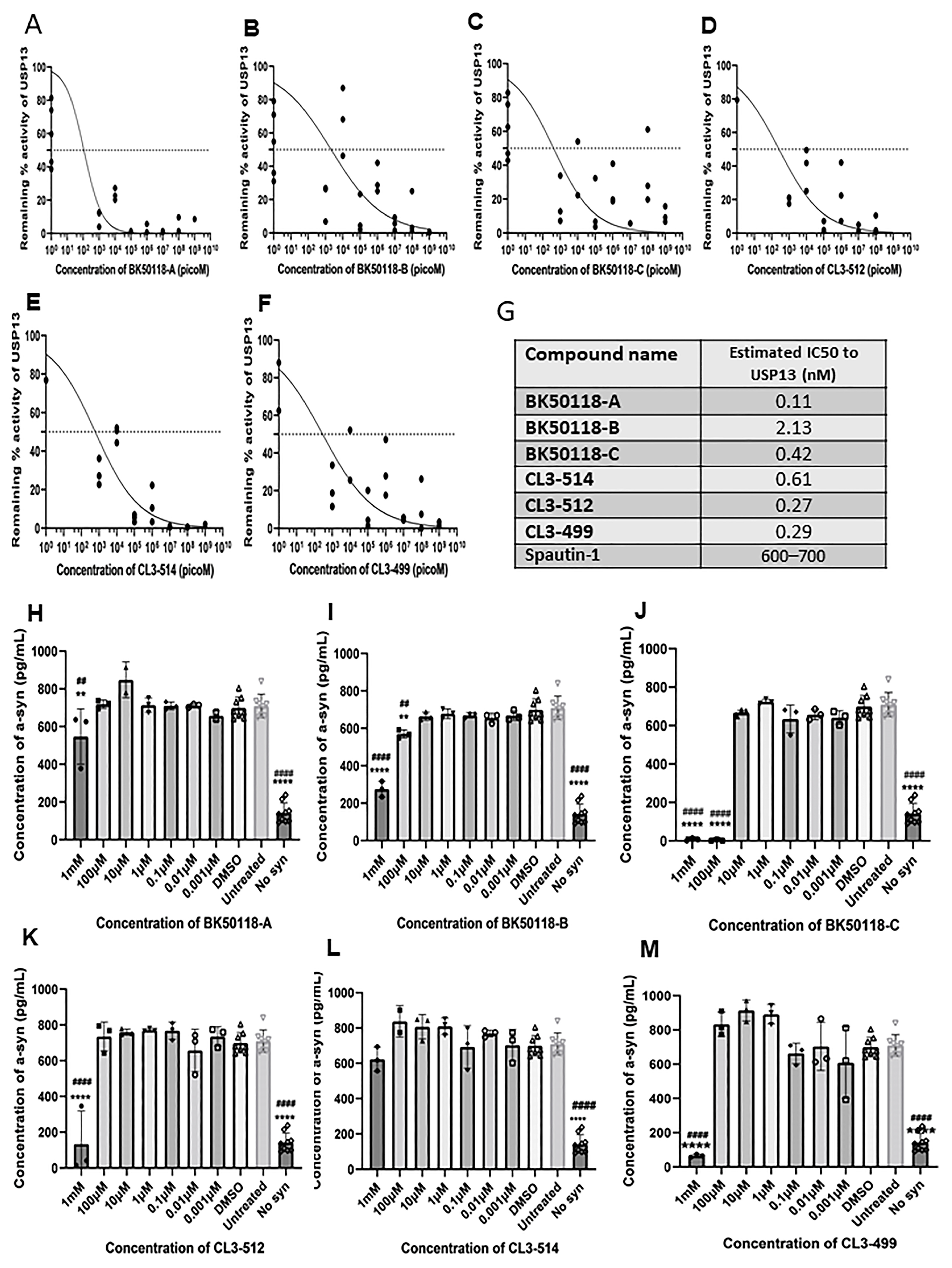
2.2. Novel Small Molecules Are Potent USP13 Inhibitors
2.3. Absorption, Distribution, Metabolism and Excretion (ADME) Studies
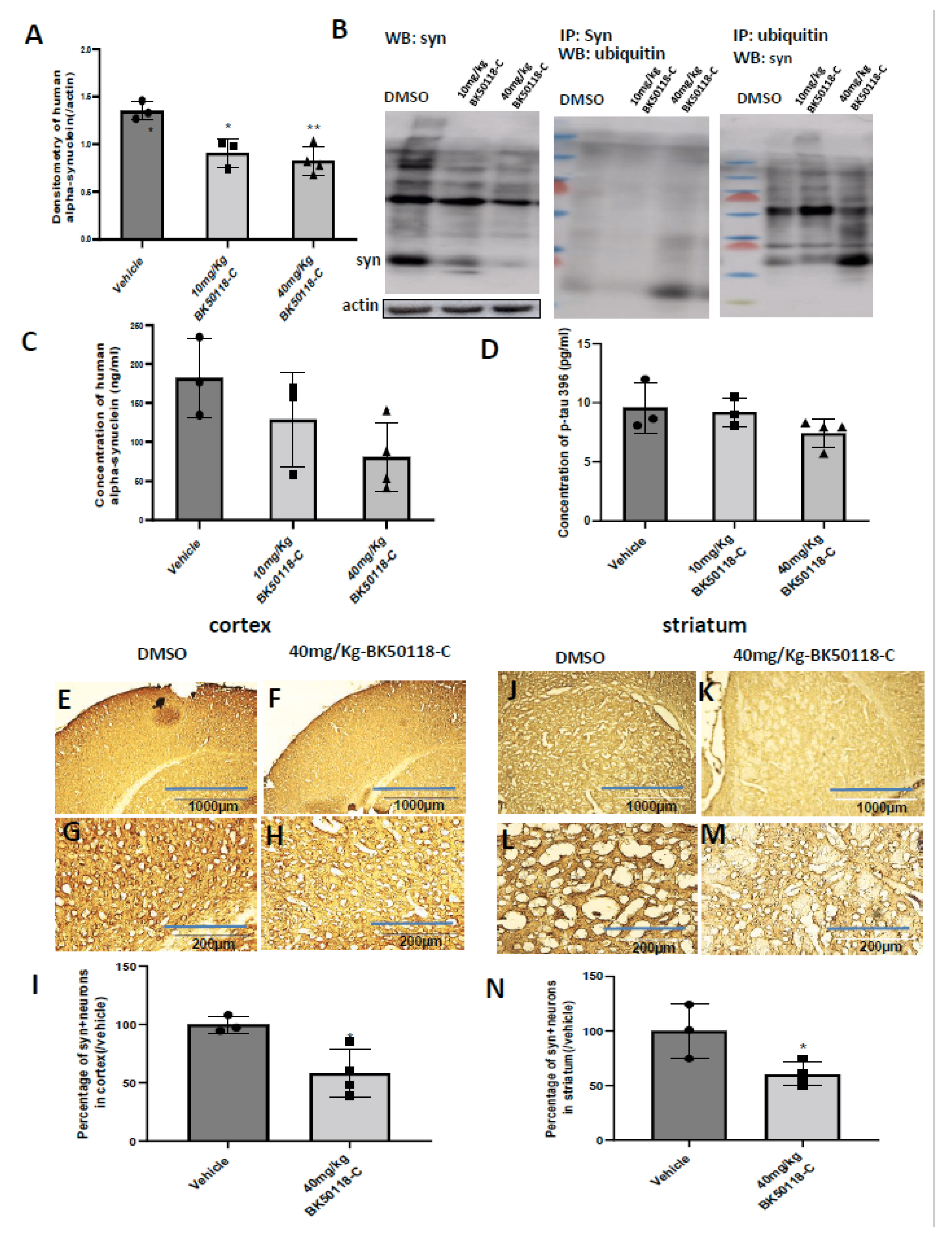
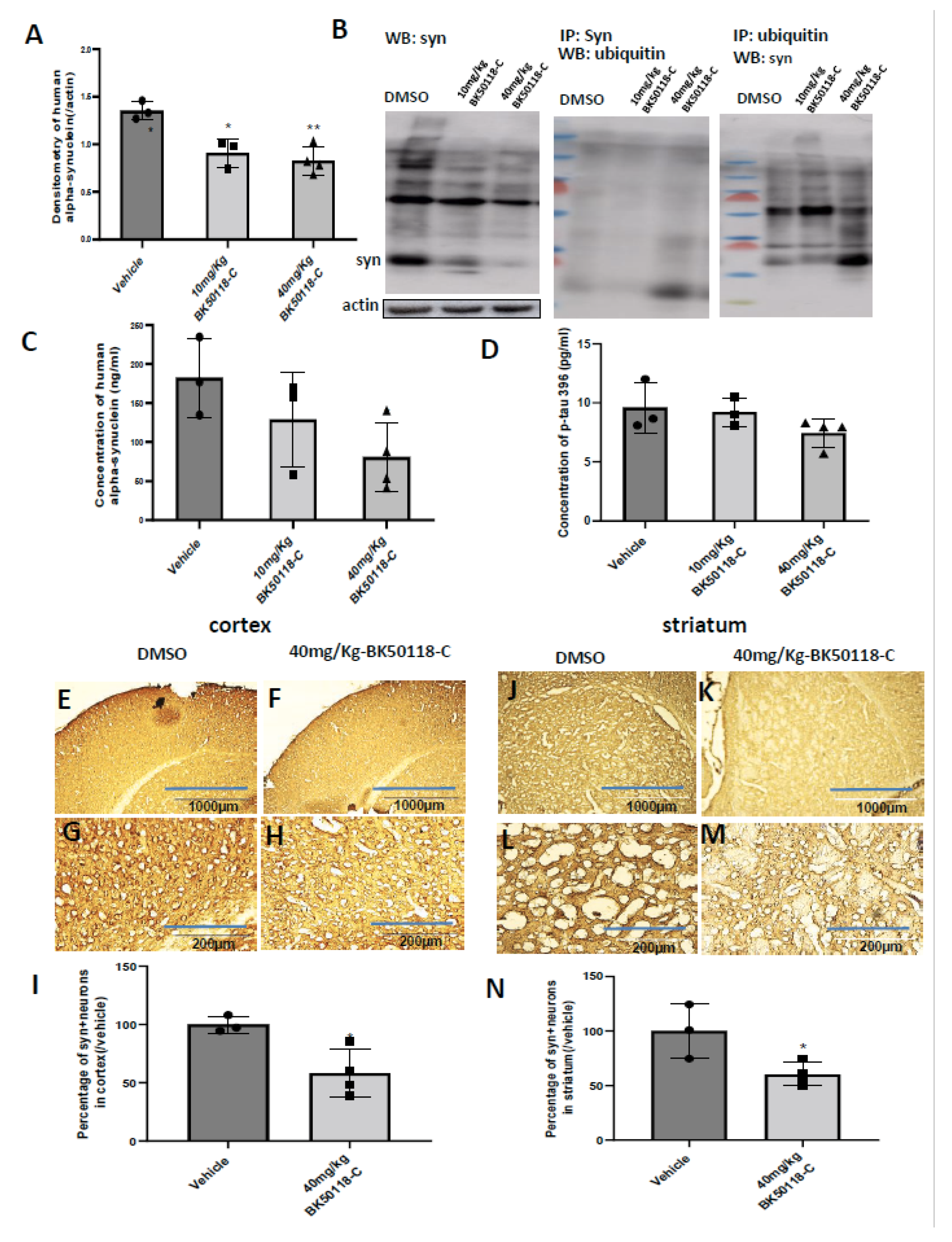
2.4. BK50118-C Reduces Alpha-Synuclein, Increases Alpha-Synuclein Ubiquitination and Improves Neuronal Survival in Mice
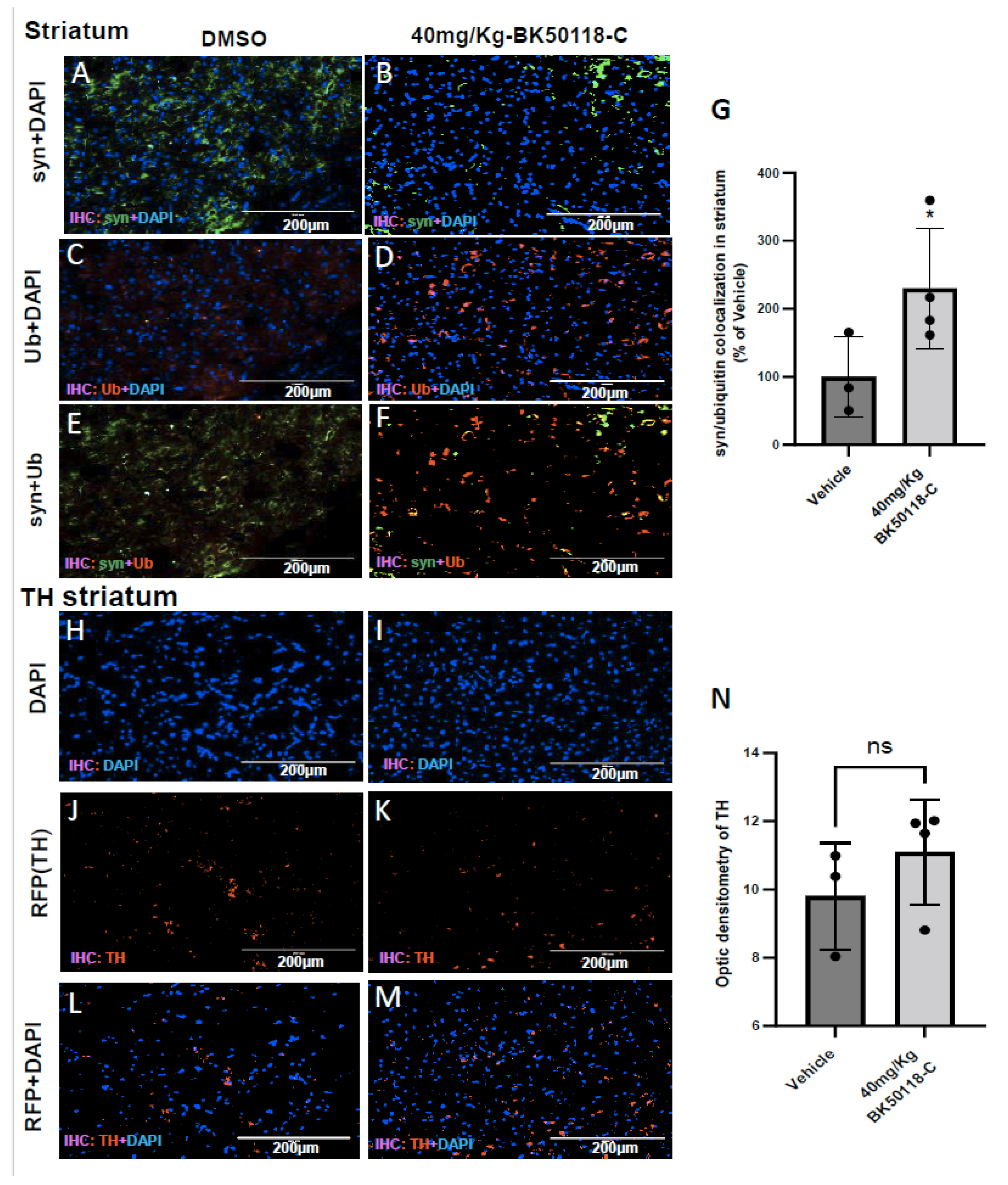
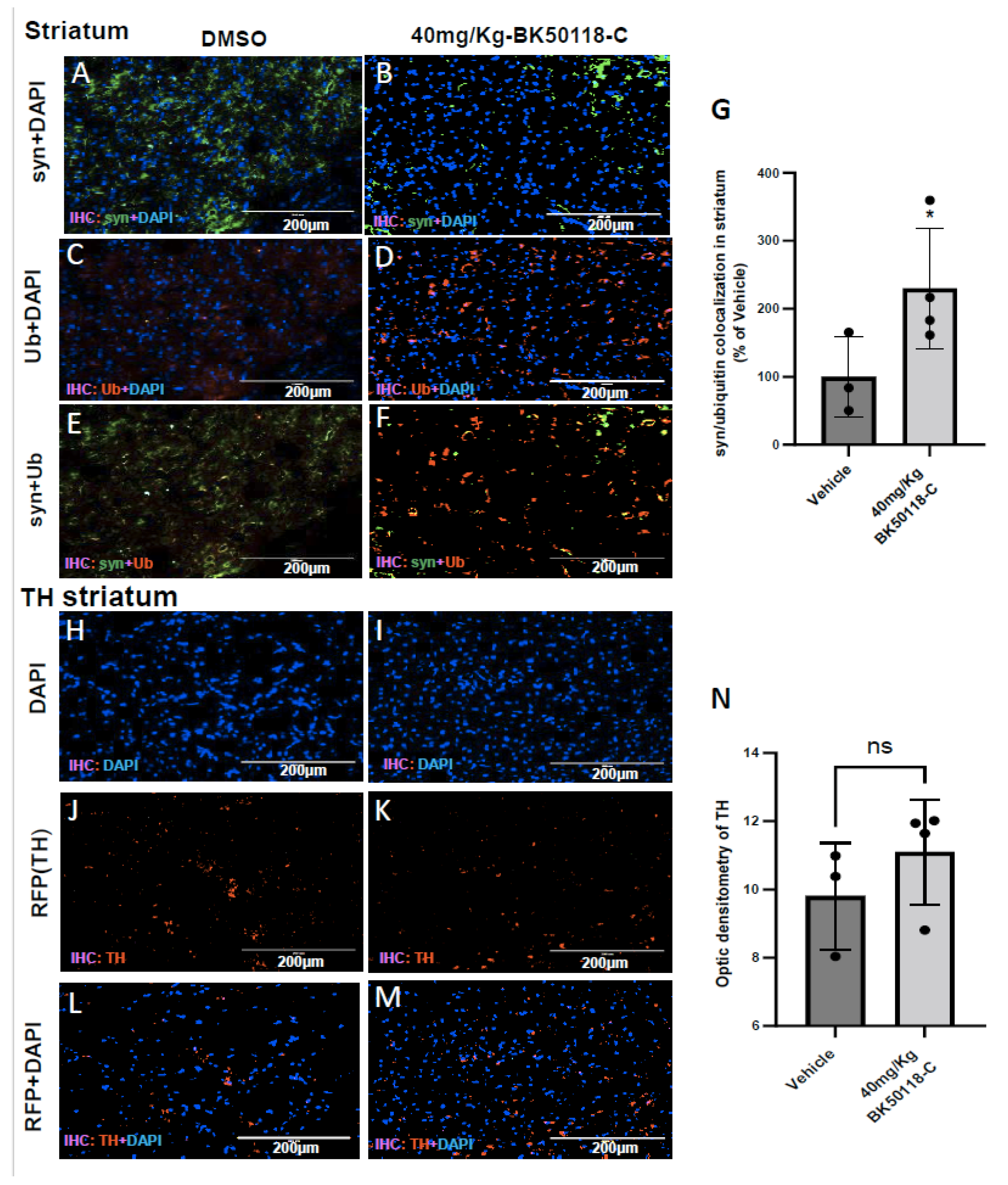
2.5. BK50118-C Increases Alpha-Synuclein Ubiquitination and Has Minimal Effects on Tyrosine Hydroxylase Levels in Striatum of TgA53T Mice
3. Discussion
4. Materials and Methods
4.1. Transgenic Mice, Stereotaxic Surgery and Treatment
4.2. Chemical Synthesis of USP13 Inhibitors
4.3. Cell LINES, Transfection and Treatment
4.4. Western Blot Analysis
4.5. Enzyme-Linked Immunosorbent ASSAY (ELISA)
4.6. Immunoprecipitation (IP)
4.7. Immunohistology
4.8. Nissl and Silver Staining
4.9. Pharmacokinetics Studies
4.10. Statistical and Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mevissen, T.E.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Jacomin, A.C.; Taillebourg, E.; Fauvarque, M.O. Deubiquitinating Enzymes Related to Autophagy: New Therapeutic Opportunities? Cells 2018, 7, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-H.; Zhou, C.-J.; Zhou, Z.-R.; Song, A.-X.; Hu, H.-Y. Domain Analysis Reveals That a Deubiquitinating Enzyme USP13 Performs Non-Activating Catalysis for Lys63-Linked Polyubiquitin. PLoS ONE 2011, 6, e29362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzny, D.M.; Scherer, S.E.; Kaul, R.; Wang, J.; Yu, J.; Sudbrak, R.; Buhay, C.J.; Chen, R.; Cree, A.; Ding, Y.; et al. The DNA sequence, annotation and analysis of human chromosome 3. Nature 2006, 440, 1194–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Park. Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandelwal, P.J.; Moussa, C.E.-H. The relationship between parkin and protein aggregation in neurodegenerative diseases. Front. Psychiatry 2010, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Herrmann, F.R.; Bussiere, T.; Bouras, C.; Kovari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- Ethell, D.W. An Amyloid-Notch Hypothesis for Alzheimer’s Disease. Neuroscientist 2010, 16, 614–617. [Google Scholar] [CrossRef]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A.; Boland, B.; Kumar, A.; Lee, S.; et al. Autophagy Induction and Autophagosome Clearance in Neurons: Relationship to Autophagic Pathology in Alzheimer’s Disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [Green Version]
- Kegel, K.B.; Kim, M.; Sapp, E.; McIntyre, C.; Castano, J.G.; Aronin, N.; DiFiglia, M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 2000, 20, 7268–7278. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Moussa, C.E.-H. Parkin Is Dispensable for Mitochondrial Function, but Its Ubiquitin Ligase Activity Is Critical for Macroautophagy and Neurotransmitters: Therapeutic Potential beyond Parkinson’s Disease. Neurodegener. Dis. 2015, 15, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Franjie, A.; Moussa, C.E. Tyrosine kinase inhibition increases functional parkin- B eclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med. 2013, 5, 1247–1262. [Google Scholar] [CrossRef]
- Liu, X.; Hebron, M.L.; Mulki, S.; Wang, C.; Lekah, E.; Ferrante, D.; Shi, W.; Kurd-Misto, B.; Moussa, C. Ubiquitin Specific Protease 13 Regulates Tau Accumulation and Clearance in Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 72, 425–441. [Google Scholar] [CrossRef]
- Liu, X.; Hebron, M.; Shi, W.; Lonskaya, I.; Moussa, C.E.-H. Ubiquitin specific protease-13 independently regulates parkin ubiquitination and alpha-synuclein clearance in alpha-synucleinopathies. Hum. Mol. Genet. 2018, 28, 548–560. [Google Scholar] [CrossRef]
- Xie, X.; Matsumoto, S.; Endo, A.; Fukushima, T.; Kawahara, H.; Saeki, Y.; Komada, M. Deubiquitinases USP5 and USP13 are recruited to and regulate heat-induced stress granules by deubiquitinating activities. J. Cell Sci. 2018, 131, jcs.210856. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 Controls the Levels of p53 by Regulating the Deubiquitination Activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J.; Feng, X.; Huang, X.-Y.; Huang, Z.; Huang, Y.; Li, C.; Li, G.; Nong, S.; Wu, R.; Huang, Y.; et al. Spautin-1 Ameliorates Acute Pancreatitis via Inhibiting Impaired Autophagy and Alleviating Calcium Overload. Mol. Med. 2016, 22, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Neuronal α-Synucleinopathy with Severe Movement Disorder in Mice Expressing A53T Human α-Synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Wills, J.; Credle, J.; Haggerty, T.; Lee, J.-H.; Oaks, A.W.; Sidhu, A. Tauopathic Changes in the Striatum of A53T α-Synuclein Mutant Mouse Model of Parkinson’s Disease. PLoS ONE 2011, 6, e17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Moussa, C. Regulatory Role of Ubiquitin Specific Protease-13 (USP13) in Misfolded Protein Clearance in Neurodegenerative Diseases. Neuroscience 2021, 460, 161–166. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Sharpe, K.; Weerasinghe, P.; Algarzae, N.K.; Shekoyan, A.R.; Moussa, C.E.-H. Parkin Ubiquitinates Tar-DNA Binding Protein-43 (TDP-43) and Promotes Its Cytosolic Accumulation via Interaction with Histone Deacetylase 6 (HDAC6). J. Biol. Chem. 2013, 288, 4103–4115. [Google Scholar] [CrossRef] [Green Version]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E.-H. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 2013, 92, 373–386. [Google Scholar] [CrossRef]
- Xie, W.; Jin, S.; Wu, Y.; Xian, H.; Tian, S.; Liu, D.-A.; Guo, Z.; Cui, J. Auto-ubiquitination of NEDD4-1 Recruits USP13 to Facilitate Autophagy through Deubiquitinating VPS34. Cell Rep. 2020, 30, 2807–2819.e4. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Jin, S.; Cui, J. The NEDD4-USP13 axis facilitates autophagy via deubiquitinating PIK3C3. Autophagy 2020, 16, 1150–1151. [Google Scholar] [CrossRef]
- Chen, M.; Gutierrez, G.; Ronai, Z.A. Ubiquitin-recognition protein Ufd1 couples the endoplasmic reticulum (ER) stress response to cell cycle control. Proc. Natl. Acad. Sci. USA 2011, 108, 9119–9124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, S.; Fang, J.; Wang, S.; Deng, B.; Zhu, L. MicroRNA-135b regulates the stability of PTEN and promotes glycolysis by targeting USP13 in human colorectal cancers. Oncol. Rep. 2014, 33, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Shin, S.K.; Jiang, Y.; Choi, W.H.; Hong, C.; Kim, D.-E.; Lee, M.J. Facilitated Tau Degradation by USP14 Aptamers via Enhanced Proteasome Activity. Sci. Rep. 2015, 5, 10757. [Google Scholar] [CrossRef]
- Wang, P.; Joberty, G.; Buist, A.; Vanoosthuyse, A.; Stancu, I.-C.; Vasconcelos, B.; Pierrot, N.; Faelth-Savitski, M.; Kienlen-Campard, P.; Octave, J.-N.; et al. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 2017, 133, 731–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexopoulou, Z.; Lang, J.; Perrett, R.M.; Elschami, M.; Hurry, M.; Kim, H.T.; Mazaraki, D.; Szabo, A.; Kessler, B.; Goldberg, A.L.; et al. Deubiquitinase Usp8 regulates α-synuclein clearance and modifies its toxicity in Lewy body disease. Proc. Natl. Acad. Sci. USA 2016, 113, E4688–E4697. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.V.; Oppermann, F.S.; Rauen, M.J.; Fog, K.; Schmidt, T.; Schmidt, J.; Harmuth, T.; Hartmann-Petersen, R.; Thirstrup, K. Mass spectrometry analyses of normal and polyglutamine expanded ataxin-3 reveal novel interaction partners involved in mitochondrial function. Neurochem. Int. 2018, 112, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Todi, S.V.; Scaglione, K.; Blount, J.R.; Basrur, V.; Conlon, K.P.; Pastore, A.; Elenitoba-Johnson, K.; Paulson, H.L. Activity and Cellular Functions of the Deubiquitinating Enzyme and Polyglutamine Disease Protein Ataxin-3 Are Regulated by Ubiquitination at Lysine 117. J. Biol. Chem. 2010, 285, 39303–39313. [Google Scholar] [CrossRef] [Green Version]
- He, W.-T.; Zheng, X.-M.; Zhang, Y.-H.; Gao, Y.; Song, A.-X.; van der Goot, G.; Hu, H.-Y. Cytoplasmic Ubiquitin-Specific Protease 19 (USP19) Modulates Aggregation of Polyglutamine-Expanded Ataxin-3 and Huntingtin through the HSP90 Chaperone. PLoS ONE 2016, 11, e0147515. [Google Scholar] [CrossRef]
- Niu, K.; Fang, H.; Chen, Z.; Zhu, Y.; Tan, Q.; Wei, D.; Li, Y.; Balajee, A.S.; Zhao, Y. USP33 deubiquitinates PRKN/parkin and antagonizes its role in mitophagy. Autophagy 2019, 16, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Salazar, C.; Hincapié, L.M.R.; Ruiz, L.M. The Interplay among PINK1/PARKIN/Dj-1 Network during Mitochondrial Quality Control in Cancer Biology: Protein Interaction Analysis. Cells 2018, 7, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C.; Xia, K.; Jiang, W.; Ronai, Z.; Zhuang, X.; et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Investig. 2009, 119, 650–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BK50118-C | Dosage (mg/kg, I.P) | 10.00 |
| Total drug injected (nmol) | 1185.77 | |
| Brain | Cmax (nM) | 81.49 ± 69.97 |
| Cmax (ng/mL) | 20.62 ± 17.70 | |
| Tmax (h) | 1.00 | |
| AUC (nM. h) | 164.3 ± 39.49 | |
| T1/2 (elimination) (h) | 2.32 | |
| Serum | Dosage (mg/Kg) | 10.00 |
| Cmax (nM) | 354.63 ± 272 | |
| Cmax (ng/mL) | 89.72 ± 68.81 | |
| Tmax (h) | 1.00 | |
| AUC (nM. h) | 599.4 ± 142.9 | |
| T1/2 (elimination) (h) | 1.84 | |
| Ratio of Serum/Brain (%) | 28.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Balaraman, K.; Lynch, C.C.; Hebron, M.; Wolf, C.; Moussa, C. Novel Ubiquitin Specific Protease-13 Inhibitors Alleviate Neurodegenerative Pathology. Metabolites 2021, 11, 622. https://doi.org/10.3390/metabo11090622
Liu X, Balaraman K, Lynch CC, Hebron M, Wolf C, Moussa C. Novel Ubiquitin Specific Protease-13 Inhibitors Alleviate Neurodegenerative Pathology. Metabolites. 2021; 11(9):622. https://doi.org/10.3390/metabo11090622
Chicago/Turabian StyleLiu, Xiaoguang, Kaluvu Balaraman, Ciarán C. Lynch, Michaeline Hebron, Christian Wolf, and Charbel Moussa. 2021. "Novel Ubiquitin Specific Protease-13 Inhibitors Alleviate Neurodegenerative Pathology" Metabolites 11, no. 9: 622. https://doi.org/10.3390/metabo11090622
APA StyleLiu, X., Balaraman, K., Lynch, C. C., Hebron, M., Wolf, C., & Moussa, C. (2021). Novel Ubiquitin Specific Protease-13 Inhibitors Alleviate Neurodegenerative Pathology. Metabolites, 11(9), 622. https://doi.org/10.3390/metabo11090622