
Substantively Lowered Levels of Pantothenic Acid (Vitamin B5) in Several Regions of the Human Brain in Parkinson’s Disease Dementia

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Cohort Characterisation
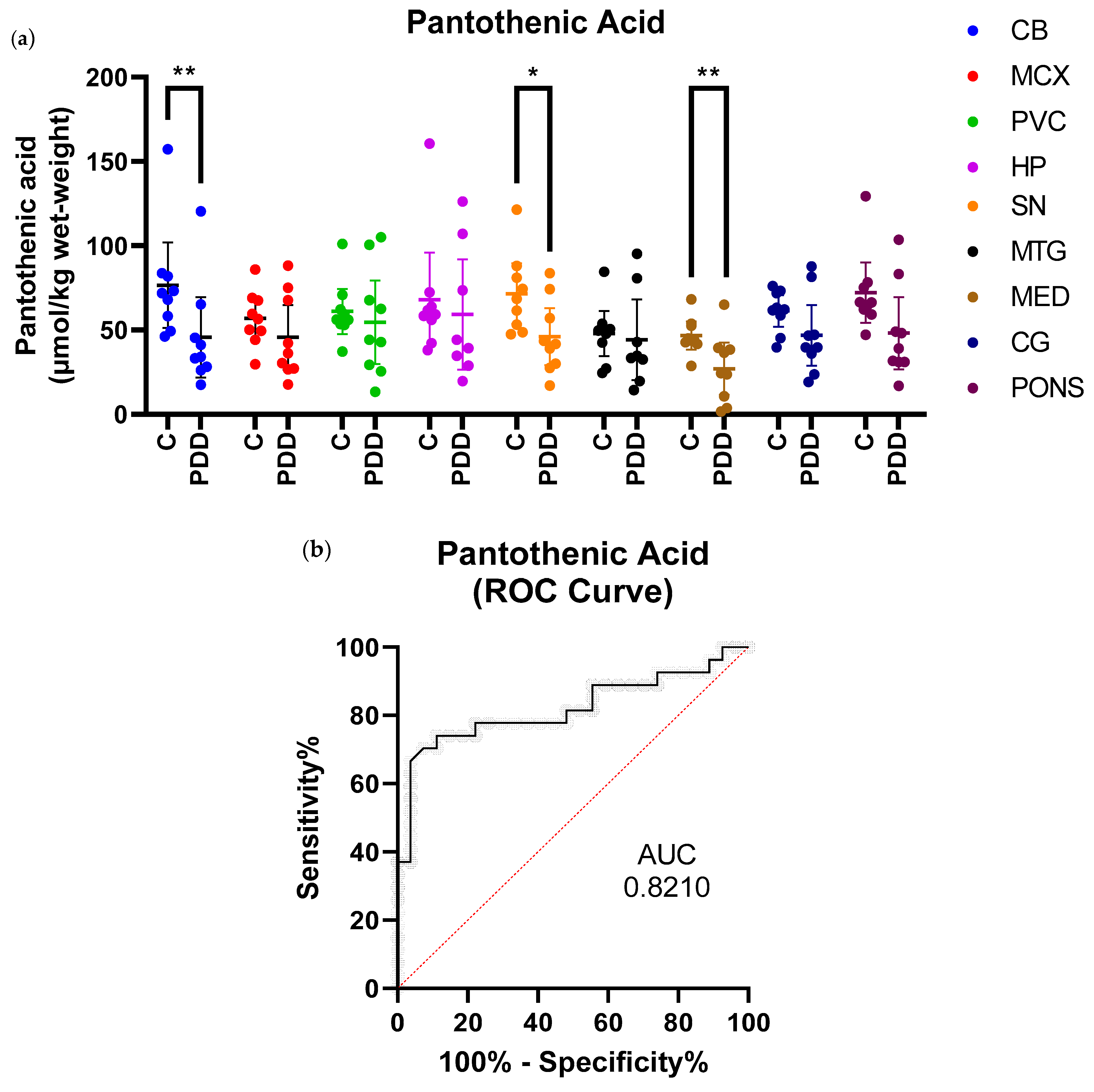
2.2. Pantothenic Acid Analysis
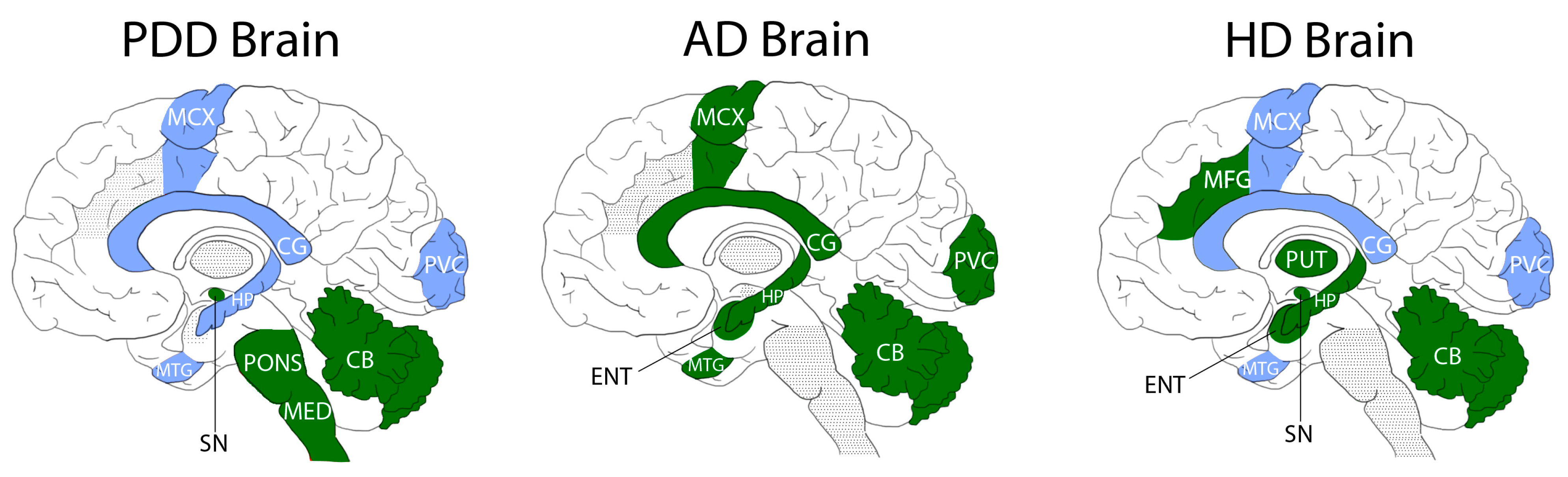
2.3. Comparisons with ADD and HD
3. Discussion
4. Materials and Methods
4.1. Tissue for Pantothenic Acid Quantification
4.2. Diagnosis and Severity of PDD Cases
4.3. Pantothenic Acid Quantification
4.4. UHPLC-MS/MS Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Rietdijk, C.D.; Perez-Pardo, P.; Garssen, J.; van Wezel, R.J.; Kraneveld, A.D. Exploring Braak’s Hypothesis of Parkinson’s Disease. Front. Neurol. 2017, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. To stage, or not to stage. Curr. Opin. Neurobiol. 2020, 61, 10–22. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Mason, S.L.; Evans, J.R.; Foltynie, T.; Brayne, C.; Robbins, T.W.; Barker, R.A. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1258–1264. [Google Scholar] [CrossRef] [Green Version]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 2008, 23, 837–844. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Cummings, J. The Role of Neuropsychiatric Symptoms in Research Diagnostic Criteria for Neurodegenerative Diseases. Am. J. Geriatr. Psychiatry 2020, 29, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Fifel, K.; Videnovic, A. Circadian and Sleep Dysfunctions in Neurodegenerative Disorders—An Update. Front. Neurosci. 2020, 14, 627330. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.; Sung, J.; An, R.; Hernandez, M.E.; Sosnoff, J.J. Gait variability in people with neurological disorders: A systematic review and meta-analysis. Hum. Mov. Sci. 2016, 47, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Ding, L.J.; Li, F.F.; Han, Y.; Mu, L. Neurodegeneration and cognition in Parkinson’s disease: A review. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2275–2281. [Google Scholar]
- Mestre, T.A.; Bachoud-Levi, A.C.; Marinus, J.; Stout, J.C.; Paulsen, J.S.; Como, P.; Duff, K.; Sampaio, C.; Goetz, C.G.; Cubo, E.; et al. Rating scales for cognition in Huntington’s disease: Critique and recommendations. Mov. Disord. 2018, 33, 187–195. [Google Scholar] [CrossRef]
- Gueli, M.C.; Taibi, G. Alzheimer’s disease: Amino acid levels and brain metabolic status. Neurol. Sci. 2013, 34, 1575–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Begley, P.; Church, S.J.; Patassini, S.; Hollywood, K.A.; Jullig, M.; Curtis, M.A.; Waldvogel, H.J.; Faull, R.L.; Unwin, R.D.; et al. Graded perturbations of metabolism in multiple regions of human brain in Alzheimer’s disease: Snapshot of a pervasive metabolic disorder. Biochim. Biophys. Acta 2016, 1862, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Patassini, S.; Begley, P.; Reid, S.J.; Xu, J.; Church, S.J.; Curtis, M.; Dragunow, M.; Waldvogel, H.J.; Unwin, R.D.; Snell, R.G.; et al. Identification of elevated urea as a severe, ubiquitous metabolic defect in the brain of patients with Huntington’s disease. Biochem. Biophys. Res. Commun. 2015, 468, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Handley, R.R.; Reid, S.J.; Brauning, R.; Maclean, P.; Mears, E.R.; Fourie, I.; Patassini, S.; Cooper, G.J.S.; Rudiger, S.R.; McLaughlan, C.J.; et al. Brain urea increase is an early Huntington’s disease pathogenic event observed in a prodromal transgenic sheep model and HD cases. Proc. Natl. Acad. Sci. USA 2017, 114, E11293–E11302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Begley, P.; Church, S.J.; Patassini, S.; McHarg, S.; Kureishy, N.; Hollywood, K.A.; Waldvogel, H.J.; Liu, H.; Zhang, S.; et al. Elevation of brain glucose and polyol-pathway intermediates with accompanying brain-copper deficiency in patients with Alzheimer’s disease: Metabolic basis for dementia. Sci. Rep. 2016, 6, 27524. [Google Scholar] [CrossRef]
- Patassini, S.; Begley, P.; Xu, J.; Church, S.J.; Reid, S.J.; Kim, E.H.; Curtis, M.A.; Dragunow, M.; Waldvogel, H.J.; Snell, R.G.; et al. Metabolite mapping reveals severe widespread perturbation of multiple metabolic processes in Huntington’s disease human brain. Biochim. Biophys. Acta 2016, 1862, 1650–1662. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Ansoleaga, B.; Jove, M.; Schluter, A.; Garcia-Esparcia, P.; Moreno, J.; Pujol, A.; Pamplona, R.; Portero-Otin, M.; Ferrer, I. Deregulation of purine metabolism in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.R.; Clark, C.; Ammann, W.; Stoessl, A.J.; Shtybel, W.; Hayden, M.R. Cortical glucose metabolism in Huntington’s disease. Neurology 1992, 42, 223–229. [Google Scholar] [CrossRef]
- Xu, J.; Patassini, S.; Begley, P.; Church, S.; Waldvogel, H.J.; Faull, R.L.M.; Unwin, R.D.; Cooper, G.J.S. Cerebral deficiency of vitamin B5 (d-pantothenic acid; pantothenate) as a potentially-reversible cause of neurodegeneration and dementia in sporadic Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2020, 527, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Patassini, S.; Begley, P.; Xu, J.; Church, S.J.; Kureishy, N.; Reid, S.J.; Waldvogel, H.J.; Faull, R.L.M.; Snell, R.G.; Unwin, R.D.; et al. Cerebral Vitamin B5 (D-Pantothenic Acid) Deficiency as a Potential Cause of Metabolic Perturbation and Neurodegeneration in Huntington’s Disease. Metabolites 2019, 9, 113. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, S.J. Defective pantothenate metabolism and neurodegeneration. Biochem. Soc. Trans. 2014, 42, 1063–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholefield, M.; Church, S.J.; Xu, J.; Robinson, A.C.; Gardiner, N.J.; Roncaroli, F.; Hooper, N.M.; Unwin, R.D.; Cooper, G.J.S. Effects of Alterations of Post-Mortem Delay and Other Tissue-Collection Variables on Metabolite Levels in Human and Rat Brain. Metabolites 2020, 10, 438. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ahn, S.Y.; Lee, H.A.; Won, K.S.; Chang, H.W.; Oh, J.S.; Kim, H.W. Dietary intake of pantothenic acid is associated with cerebral amyloid burden in patients with cognitive impairment. Food Nutr. Res. 2018, 62, 1415. [Google Scholar] [CrossRef] [Green Version]
- Baldini, F.; Hertel, J.; Sandt, E.; Thinnes, C.C.; Neuberger-Castillo, L.; Pavelka, L.; Betsou, F.; Kruger, R.; Thiele, I.; Consortium, N.-P. Parkinson’s disease-associated alterations of the gut microbiome predict disease-relevant changes in metabolic functions. BMC Biol. 2020, 18, 62. [Google Scholar] [CrossRef]
- Vascellari, S.; Palmas, V.; Melis, M.; Pisanu, S.; Cusano, R.; Uva, P.; Perra, D.; Madau, V.; Sarchioto, M.; Oppo, V.; et al. Gut Microbiota and Metabolome Alterations Associated with Parkinson’s Disease. mSystems 2020, 5, e00561-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.X.; Dai, S.X.; Guo, Y.C.; Han, F.F.; Zheng, J.J.; Li, G.H.; Huang, J.F. Meta-Analysis of Parkinson’s Disease and Alzheimer’s Disease Revealed Commonly Impaired Pathways and Dysregulation of NRF2-Dependent Genes. J. Alzheimer’s Dis. 2017, 56, 1525–1539. [Google Scholar] [CrossRef]
- Gibson, G.E.; Kingsbury, A.E.; Xu, H.; Lindsay, J.G.; Daniel, S.; Foster, O.J.; Lees, A.J.; Blass, J.P. Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson’s disease. Neurochem. Int. 2003, 43, 129–135. [Google Scholar] [CrossRef]
- Willkommen, D.; Lucio, M.; Moritz, F.; Forcisi, S.; Kanawati, B.; Smirnov, K.S.; Schroeter, M.; Sigaroudi, A.; Schmitt-Kopplin, P.; Michalke, B. Metabolomic investigations in cerebrospinal fluid of Parkinson’s disease. PLoS ONE 2018, 13, e0208752. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. Glucose, glycolysis, and neurodegenerative diseases. J. Cell Physiol. 2020, 235, 7653–7662. [Google Scholar] [CrossRef] [PubMed]
- Foltynie, T. Glycolysis as a therapeutic target for Parkinson’s disease. Lancet Neurol. 2019, 18, 1072–1074. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Koeppe, R.A.; Minoshima, S.; Giordani, B.; Albin, R.L.; Frey, K.A.; Kuhl, D.E. Cerebral glucose metabolic features of Parkinson disease and incident dementia: Longitudinal study. J. Nucl. Med. 2011, 52, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, F.; Ballarini, T.; Neumann, J.; Schroeter, M.L. FDG-PET hypometabolism is more sensitive than MRI atrophy in Parkinson’s disease: A whole-brain multimodal imaging meta-analysis. Neuroimage Clin. 2019, 21, 101594. [Google Scholar] [CrossRef]
- Gonzalez-Redondo, R.; Garcia-Garcia, D.; Clavero, P.; Gasca-Salas, C.; Garcia-Eulate, R.; Zubieta, J.L.; Arbizu, J.; Obeso, J.A.; Rodriguez-Oroz, M.C. Grey matter hypometabolism and atrophy in Parkinson’s disease with cognitive impairment: A two-step process. Brain 2014, 137, 2356–2367. [Google Scholar] [CrossRef] [Green Version]
- Bohnen, N.I.; Minoshima, S.; Giordani, B.; Frey, K.A.; Kuhl, D.E. Motor correlates of occipital glucose hypometabolism in Parkinson’s disease without dementia. Neurology 1999, 52, 541–546. [Google Scholar] [CrossRef]
- Selnes, P.; Stav, A.L.; Johansen, K.K.; Bjornerud, A.; Coello, C.; Auning, E.; Kalheim, L.; Almdahl, I.S.; Hessen, E.; Zetterberg, H.; et al. Impaired synaptic function is linked to cognition in Parkinson’s disease. Ann. Clin. Transl. Neurol. 2017, 4, 700–713. [Google Scholar] [CrossRef] [Green Version]
- Ismail, N.; Kureishy, N.; Church, S.J.; Scholefield, M.; Unwin, R.D.; Xu, J.; Patassini, S.; Cooper, G.J.S. Vitamin B5 (d-pantothenic acid) localizes in myelinated structures of the rat brain: Potential role for cerebral vitamin B5 stores in local myelin homeostasis. Biochem. Biophys. Res. Commun. 2020, 522, 220–225. [Google Scholar] [CrossRef]
- Fitzgerald, E.; Murphy, S.; Martinson, H.A. Alpha-Synuclein Pathology and the Role of the Microbiota in Parkinson’s Disease. Front. Neurosci. 2019, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamguney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Scheperjans, F.; Derkinderen, P.; Borghammer, P. The Gut and Parkinson’s Disease: Hype or Hope? J. Parkinson’s Dis. 2018, 8, S31–S39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Hustad, E.; Aasly, J.O. Clinical and Imaging Markers of Prodromal Parkinson’s Disease. Front. Neurol. 2020, 11, 395. [Google Scholar] [CrossRef]
- Gellersen, H.M.; Guo, C.C.; O’Callaghan, C.; Tan, R.H.; Sami, S.; Hornberger, M. Cerebellar atrophy in neurodegeneration-a meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hallett, M. The cerebellum in Parkinson’s disease. Brain 2013, 136, 696–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Gender (% Male) | Age at Death (Years) | PMD (Hours) | |
|---|---|---|---|
| Controls (n = 9) | 44 | 70 (61–79) | 19.8 (12.5–26.0) |
| SN Controls (n = 9) † | 44 | 70 (62–79) | 20.6 (10.8–26.0) |
| Cases (n = 9) † | 66 | 73 (61–81) | 14.6 (4.3–21.9) * |
| Region | Controls (n = 9) (µmol/kg Wet-Weight) | PDD Cases (n = 9) (µmol/kg Wet-Weight) | Fold-Change | p-Value |
|---|---|---|---|---|
| CB | 76.7 (51.3–102.0) | 45.7 (21.8–69.7) | 0.6 | 0.008 |
| MCX | 56.9 (44.5–69.4) | 45.7 (26.6–64.9) | 0.8 | 0.3 |
| PVC | 61.1 (47.7–74.5) | 54.6 (29.8–79.4) | 0.9 | 0.5 |
| HP | 68.1 (40.2–96.0) | 59.3 (26.6–91.9) | 0.9 | 0.4 |
| SN | 71.6 (53.6–89.6) | 46.0 (29.2–62.9) | 0.6 | 0.02 |
| MTG | 47.9 (34.6–61.2) | 44.2 (20.2–68.3) | 0.9 | 0.4 |
| MED | 46.8 (38.5–55.0) | 27.1 (11.5–42.6) | 0.6 | 0.008 |
| CG | 61.3 (51.9–70.8) | 46.9 (28.9–64.9) | 0.8 | 0.1 |
| PONS | 72.2 (54.3–90.1) | 48.2 (26.8–69.7) | 0.7 | 0.0503 |
| Region | Fold-Change PDD | Fold-Change AD (Xu et al., 2016) | Fold-Change HD (Patassini et al., 2015) |
|---|---|---|---|
| CB | 0.6 | 0.5 | 0.6 |
| MCX | 0.8 | 0.3 | 0.6 |
| PVC | 0.9 | 0.4 | 0.5 |
| HP | 0.9 | 0.5 | 0.5 |
| SN | 0.6 | - | 0.6 |
| MTG | 0.9 | 0.5 | 0.6 |
| MED | 0.6 | - | - |
| CG | 0.8 | 0.5 | 0.5 |
| PONS | 0.7 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scholefield, M.; Church, S.J.; Xu, J.; Patassini, S.; Hooper, N.M.; Unwin, R.D.; Cooper, G.J.S. Substantively Lowered Levels of Pantothenic Acid (Vitamin B5) in Several Regions of the Human Brain in Parkinson’s Disease Dementia. Metabolites 2021, 11, 569. https://doi.org/10.3390/metabo11090569
Scholefield M, Church SJ, Xu J, Patassini S, Hooper NM, Unwin RD, Cooper GJS. Substantively Lowered Levels of Pantothenic Acid (Vitamin B5) in Several Regions of the Human Brain in Parkinson’s Disease Dementia. Metabolites. 2021; 11(9):569. https://doi.org/10.3390/metabo11090569
Chicago/Turabian StyleScholefield, Melissa, Stephanie J. Church, Jingshu Xu, Stefano Patassini, Nigel M. Hooper, Richard D. Unwin, and Garth J. S. Cooper. 2021. "Substantively Lowered Levels of Pantothenic Acid (Vitamin B5) in Several Regions of the Human Brain in Parkinson’s Disease Dementia" Metabolites 11, no. 9: 569. https://doi.org/10.3390/metabo11090569
APA StyleScholefield, M., Church, S. J., Xu, J., Patassini, S., Hooper, N. M., Unwin, R. D., & Cooper, G. J. S. (2021). Substantively Lowered Levels of Pantothenic Acid (Vitamin B5) in Several Regions of the Human Brain in Parkinson’s Disease Dementia. Metabolites, 11(9), 569. https://doi.org/10.3390/metabo11090569