
Metabolomics-Based Screening of Inborn Errors of Metabolism: Enhancing Clinical Application with a Robust Computational Pipeline

 , ,
, ,
Abstract
:1. Introduction
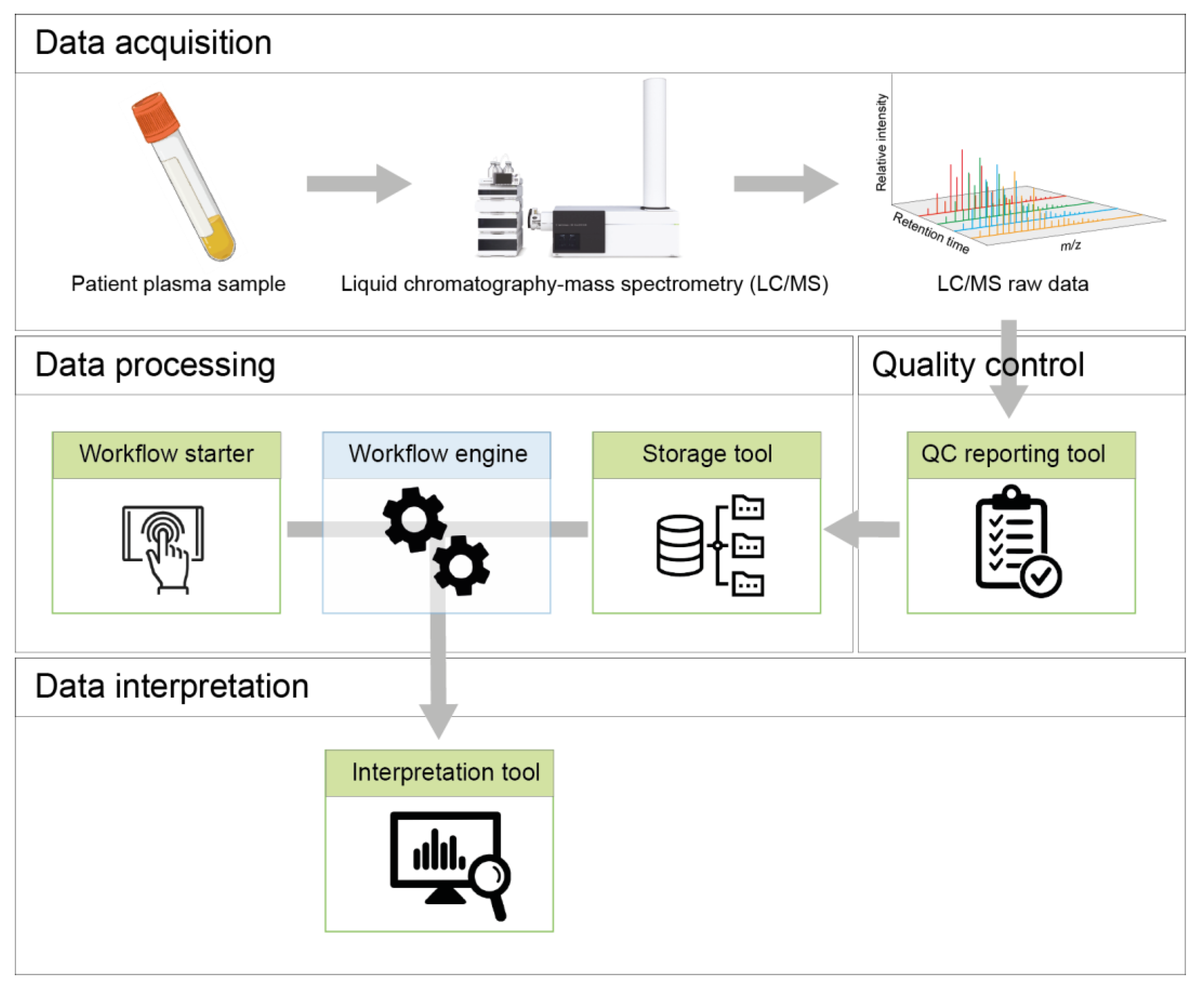
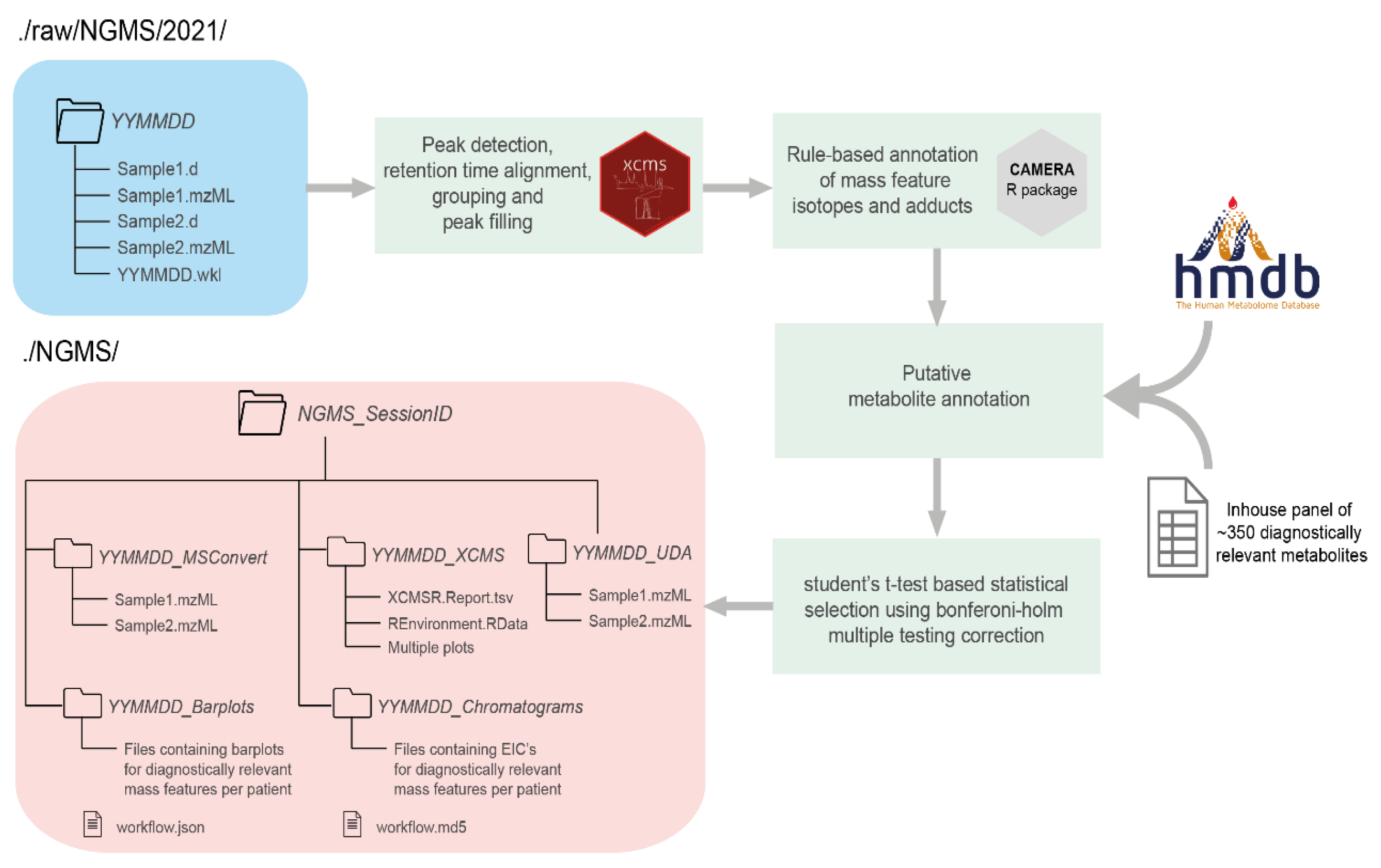
2. Pipeline Design and Architecture
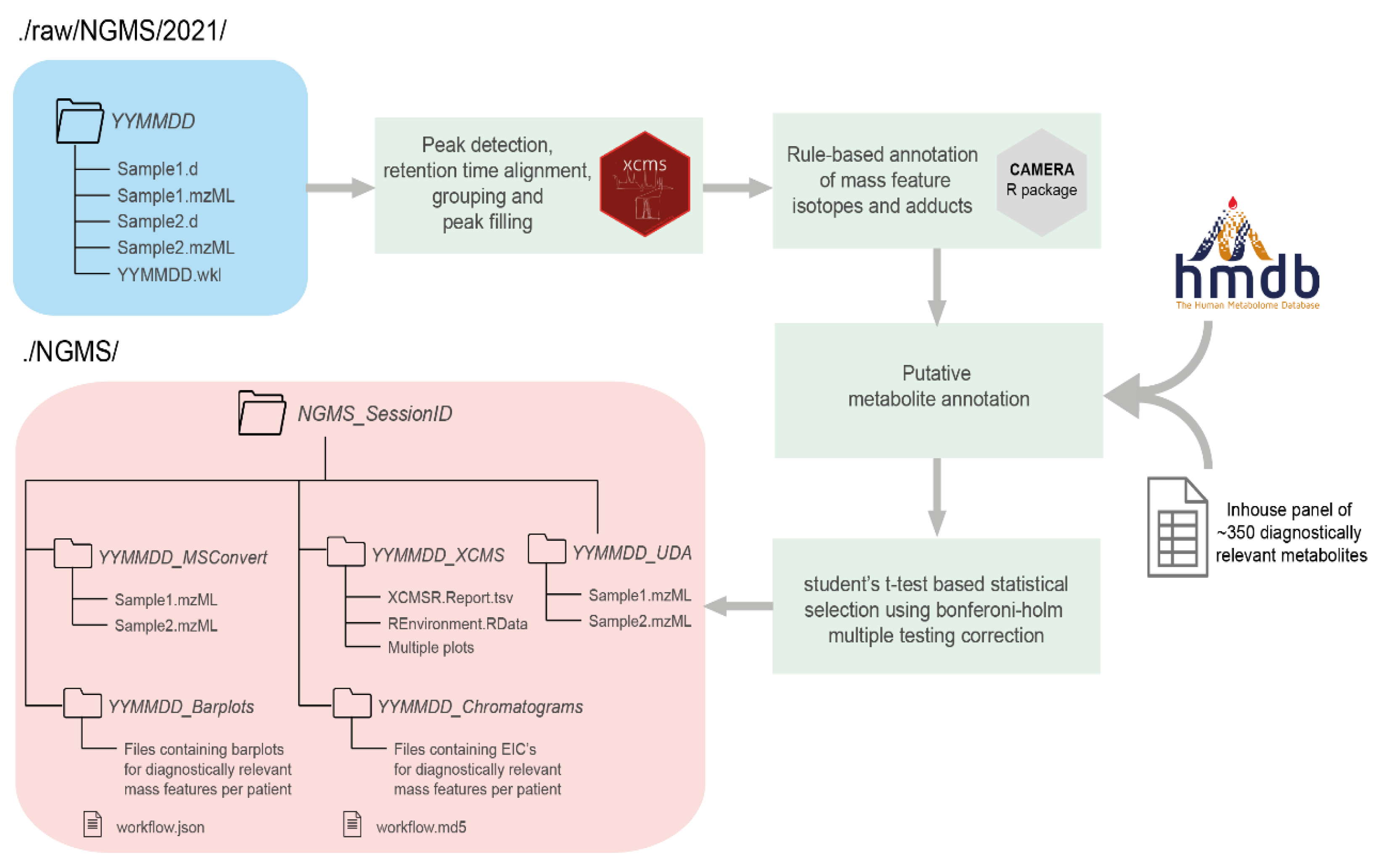
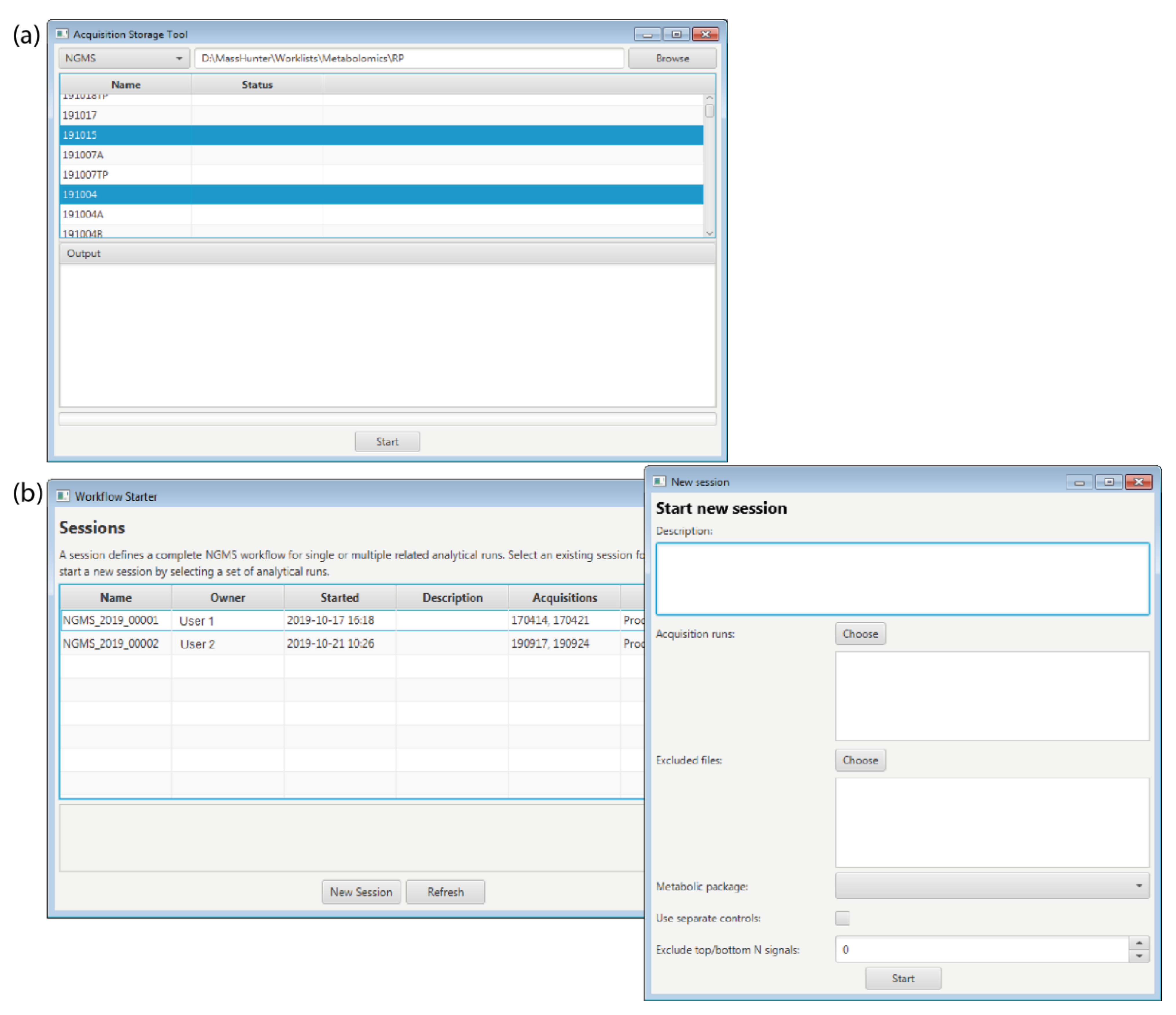
2.1. Storage Tool
2.2. Workflow Starter
2.3. Workflow Engine
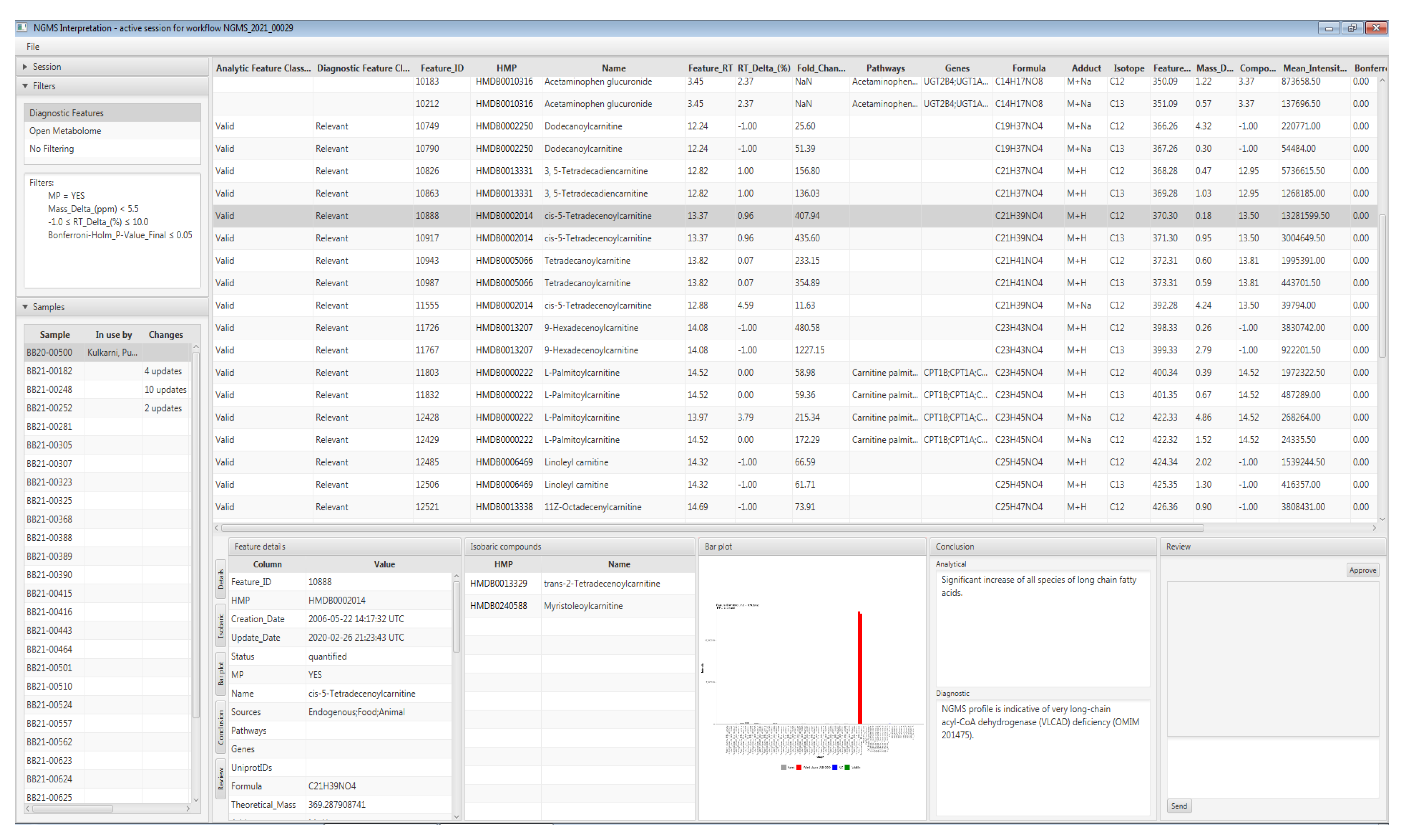
3. Data Interpretation
- A list of measured patient samples available to the current session;
- A comprehensive per sample feature table;
- ○
- Concatenating positive and negative ion mode results;
- ○
- Including mass spectrometry data, statistical metrics, and links to third party metabolite and pathway databases;
- ○
- With unmodifiable result columns, to safeguard result integrity;
- ○
- Users can add per-feature annotations describing diagnostic relevance based on assigned user roles (i.e., data analyst, clinical laboratory specialist) and the changes are logged in an audit trail;
- Bar plot-based visualization for comparing the detected sample features against quality control, validation, or other patient samples;
- A collaborative review and approval process of each sample for patient diagnosis based on configurable user roles; and
- Real-time updates to each sample’s status as users collaboratively browse, annotate and review features.
4. Validation and Quality Control
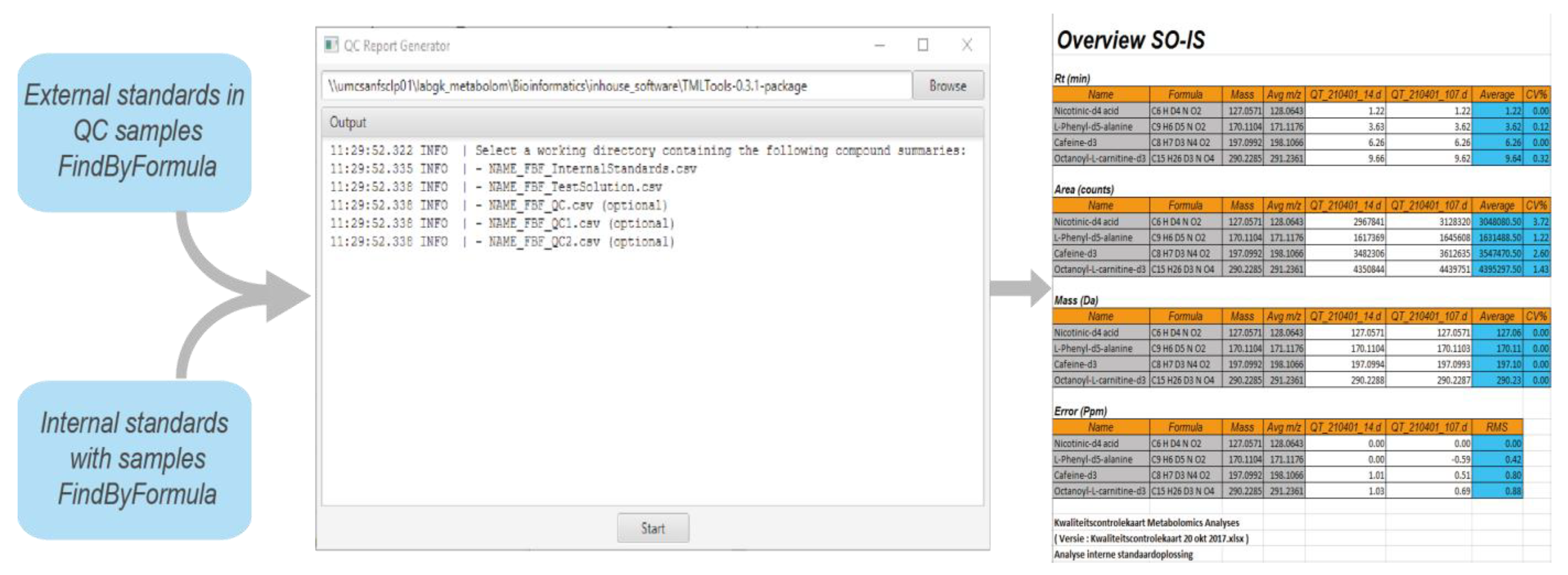
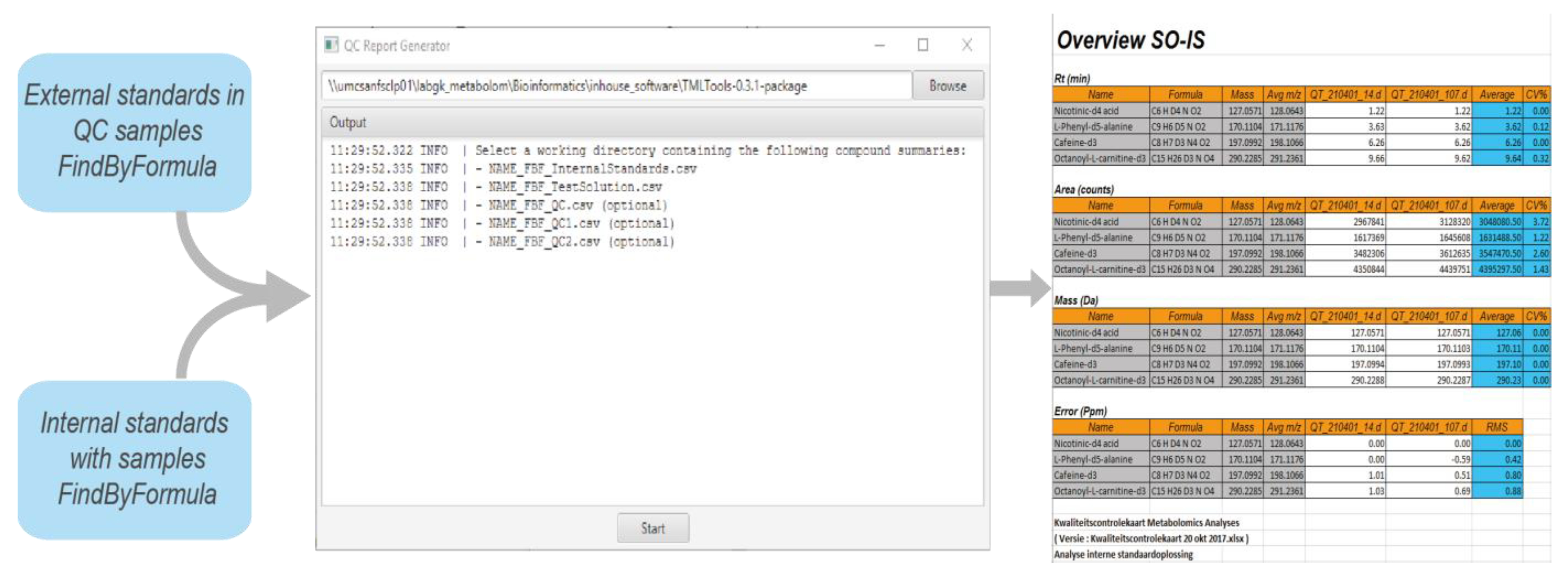
4.1. Quality Control of Data
- Repeatability of retention time;
- Repeatability of response;
- Mass accuracy.
4.2. Validation of Data Processing
4.3. Validation of Pipeline Releases
5. Case Study: Very Long-Chain acyl-CoA Dehydrogenase Deficiency in an Adult Patient
6. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almontashiri, N.A.M.; Zha, L.; Young, K.; Law, T.; Kellogg, M.D.; Bodamer, O.A.; Peake, R.W.A. Clinical Validation of Targeted and Untargeted Metabolomics Testing for Genetic Disorders: A 3 Year Comparative Study. Sci. Rep. 2020, 10, 9382. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.T.; Showalter, M.R.; Fiehn, O. Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites 2019, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.D.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted Metabolomic Analysis for the Clinical Screening of Inborn Errors of Metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonte, R.; Bongaerts, M.; Demirdas, S.; Langendonk, J.G.; Huidekoper, H.H.; Williams, M.; Onkenhout, W.; Jacobs, E.H.; Blom, H.J.; Ruijter, G.J.G. Untargeted Metabolomics-Based Screening Method for Inborn Errors of Metabolism Using Semi-Automatic Sample Preparation with an UHPLC-Orbitrap-MS Platform. Metabolites 2019, 9, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebani, A.; Abily-Donval, L.; Afonso, C.; Marret, S.; Bekri, S. Clinical Metabolomics: The New Metabolic Window for Inborn Errors of Metabolism Investigations in the Post-Genomic Era. Int. J. Mol. Sci. 2016, 17, 1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations. Int. J. Mol. Sci. 2016, 17, 1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.D.; Wittmann, B.M.; Evans, A.M.; Miller, L.A.D.; Toal, D.R.; Lonergan, S.; Elsea, S.H.; Pappan, K.L. Metabolomics in the Clinic: A Review of the Shared and Unique Features of Untargeted Metabolomics for Clinical Research and Clinical Testing. J. Mass Spectrom. 2018, 53, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- van Outersterp, R.E.; Moons, S.J.; Engelke, U.F.H.; Bentlage, H.; Peters, T.M.A.; van Rooij, A.; Huigen, M.C.D.G.; de Boer, S.; van der Heeft, E.; Kluijtmans, L.A.J.; et al. Amadori Rearrangement Products as Potential Biomarkers for Inborn Errors of Amino-Acid Metabolism. Commun. Biol. 2021, 4, 367. [Google Scholar] [CrossRef] [PubMed]
- Engelke, U.F.G.; van Outersterp, R.E.; Merx, J.; van Geenen, F.A.M.G.; van Rooij, A.; Berden, G.; Huigen, M.C.D.G.; Kluijtmans, L.A.J.; Peters, T.M.A.; Al-Shekaili, H.H.; et al. Untargeted Metabolomics and Infrared Ion Spectroscopy Identify Biomarkers for Pyridoxine-Dependent Epilepsy. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E.; Engelke, U.F.H.; de Boer, S.; Hoegen, B.; Kwast, H.J.T.; van de Vorst, M.; Huigen, M.C.D.G.; Keularts, I.M.L.W.; et al. Next-Generation Metabolic Screening: Targeted and Untargeted Metabolomics for the Diagnosis of Inborn Errors of Metabolism in Individual Patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, B.B.; Mohapatra, S. Tools and Resources for Metabolomics Research Community: A 2017–2018 Update. Electrophoresis 2019, 40, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B. New Software Tools, Databases, and Resources in Metabolomics: Updates from 2020. Metabolomics 2021, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Spicer, R.; Salek, R.M.; Moreno, P.; Cañueto, D.; Steinbeck, C. Navigating Freely-Available Software Tools for Metabolomics Analysis. Metabolomics 2017, 13, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal Chem 2012, 84, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35 (Suppl. 1), 521–526. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, R.; Mallick, P. Data Conversion with {ProteoWizard} MsConvert. Methods Mol. Biol. 2017, 1550, 339–368. [Google Scholar] [PubMed]
- Yoo, A.B.; Jette, M.A.; Grondona, M. SLURM: Simple Linux Utility for Resource Management. In Job Scheduling Strategies for Parallel Processing; Feitelson, D., Rudolph, L., Schwiegelshohn, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; pp. 44–60. [Google Scholar]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific Containers for Mobility of Compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Maurer, C.; Friedberg, R.C. International Organization for Standardization (ISO) 15189. Ann. Lab. Med. 2017, 37, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Feature Mass | Retention Time Delta % | Mean Intensity Patient | ESI− | ESI+ |
|---|---|---|---|---|---|
| Dihydrouracil | 115.0501 | 4.716981 | 6,865,418 | - | ↑ |
| Ornithine | 133.0973 | 0.990099 | 3,652,369.5 | - | ↑ |
| Xanthine | 153.0407 | 6.122449 | 14,056,124.5 | - | ↑ |
| Ornithine | 155.079 | 0.990099 | 426,197.5 | - | ↑ |
| Pimelic acid | 161.0808 | 2.523659 | 1,699,250.5 | - | ↑ |
| L-Phenylalanine | 167.0892 | 2.506964 | 25,365,509 | - | ↑ |
| Xanthine | 175.0227 | 6.122449 | 616,580 | - | ↑ |
| L-Tyrosine | 183.0842 | 3.317535 | 8,101,318.5 | - | ↑ |
| Pimelic acid | 184.0662 | 2.523659 | 1,089,222 | - | ↑ |
| L-Phenylalanine | 188.0678 | 2.506964 | 2,532,136.5 | - | ↑ |
| L-Phenylalanine | 189.0713 | 2.506964 | 268,255 | - | ↑ |
| N-Acetylmannosamine | 244.0796 | 1.470588 | 9,803,499 | - | ↑ |
| gamma-Glutamylphenylalanine | 296.1308 | 2.763385 | 20,148,682.5 | - | ↑ |
| L-Palmitoylcarnitine | 400.3423 | 0 | 19,211,216.5 | - | ↑ |
| L-Palmitoylcarnitine | 401.3458 | 0 | 5,606,300 | - | ↑ |
| L-Palmitoylcarnitine | 422.3247 | 0 | 270,923.5 | - | ↑ |
| Mesaconic acid | 129.0194 | 6.329114 | 19,645,695.5 | ↑ | - |
| Xanthine | 151.026 | 3.255814 | 22,174,103 | ↑ | - |
| Xanthine | 152.0289 | 3.255814 | 1,288,583 | ↑ | - |
| Pimelic acid | 159.0661 | 0.770416 | 28,452,947 | ↑ | - |
| Pimelic acid | 160.0697 | 0.770416 | 7,346,168 | ↑ | - |
| L-Phenylalanine | 164.0714 | 6.376812 | 4,407,907 | ↑ | - |
| L-Phenylalanine | 165.0751 | 2.608696 | 9,234,293.5 | ↑ | - |
| L-Tyrosine | 180.0664 | 5.294118 | 33,613,076 | ↑ | - |
| L-Tyrosine | 180.0661 | 8.823529 | 462,782.5 | ↑ | - |
| N-Acetylmannosamine | 256.0597 | 1.470588 | 5,667,181.5 | ↑ | - |
| gamma-Glutamylphenylalanine | 293.1141 | 3.971119 | 47,200,428 | ↑ | - |
| gamma-Glutamylphenylalanine | 294.1174 | 3.971119 | 7,670,664 | ↑ | - |
| Bar Plot | Feature Name | m/z | Retention Time (min) | Adduct | HMDB ID | Fold Change |
|---|---|---|---|---|---|---|
| (a) | Tetradecenoyl/C14:1 | 370.295 | 13.37 | M + H | HMDB0002014 | 407.936 |
| (b) | Dodecanoyl/C12:0 | 367.264 | 12.24 | M + Na | HMDB0002250 | 51.387 |
| (c) | Tetradecadienyl/C14:2 | 368.279 | 12.82 | M + H | HMDB0013331 | 156.804 |
| (d) | Tetradecenoyl/C14:0 | 372.311 | 13.82 | M + H | HMDB0005066 | 354.890 |
| (e) | Hexadecenoyl/C16:1 | 398.326 | 14.08 | M + H | HMDB0013207 | 1227.147 |
| (f) | Palmitoyl/C16:0 | 401.345 | 14.52 | M + H | HMDB0000222 | 59.362 |
| (g) | Linoleyl/C18:2 | 424.342 | 14.32 | M + H | HMDB0006469 | 66.594 |
| (h) | Octadecenyl/C18:1 | 426.357 | 14.69 | M + H | HMDB0013338 | 74.586 |
| (i) | Stearoyl/C18:0 | 428.373 | 15.08 | M + H | HMDB0000848 | 29.269 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoegen, B.; Zammit, A.; Gerritsen, A.; Engelke, U.F.H.; Castelein, S.; van de Vorst, M.; Kluijtmans, L.A.J.; Huigen, M.C.D.G.; Wevers, R.A.; van Gool, A.J.; et al. Metabolomics-Based Screening of Inborn Errors of Metabolism: Enhancing Clinical Application with a Robust Computational Pipeline. Metabolites 2021, 11, 568. https://doi.org/10.3390/metabo11090568
Hoegen B, Zammit A, Gerritsen A, Engelke UFH, Castelein S, van de Vorst M, Kluijtmans LAJ, Huigen MCDG, Wevers RA, van Gool AJ, et al. Metabolomics-Based Screening of Inborn Errors of Metabolism: Enhancing Clinical Application with a Robust Computational Pipeline. Metabolites. 2021; 11(9):568. https://doi.org/10.3390/metabo11090568
Chicago/Turabian StyleHoegen, Brechtje, Alan Zammit, Albert Gerritsen, Udo F. H. Engelke, Steven Castelein, Maartje van de Vorst, Leo A. J. Kluijtmans, Marleen C. D. G. Huigen, Ron A. Wevers, Alain J. van Gool, and et al. 2021. "Metabolomics-Based Screening of Inborn Errors of Metabolism: Enhancing Clinical Application with a Robust Computational Pipeline" Metabolites 11, no. 9: 568. https://doi.org/10.3390/metabo11090568
APA StyleHoegen, B., Zammit, A., Gerritsen, A., Engelke, U. F. H., Castelein, S., van de Vorst, M., Kluijtmans, L. A. J., Huigen, M. C. D. G., Wevers, R. A., van Gool, A. J., Gilissen, C., Coene, K. L. M., & Kulkarni, P. (2021). Metabolomics-Based Screening of Inborn Errors of Metabolism: Enhancing Clinical Application with a Robust Computational Pipeline. Metabolites, 11(9), 568. https://doi.org/10.3390/metabo11090568